
Lignin-Based Nonviral Gene Carriers Functionalized by Poly[2-(Dimethylamino)ethyl Methacrylate]: Effect of Grafting Degree and Cationic Chain Length on Transfection Efficiency



Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Quantification of Total Hydroxyl Content in Lignin via Acetylation
2.3. Synthesis of Lignin-Based Macroinitiators
2.3.1. Synthesis of LnMI-a (MD 3.0%)
2.3.2. Synthesis of LnMI-h (MD 100.0%)
2.4. Synthesis of Lignin-PDMAEMA Graft Copolymers
2.5. In Vitro Transfection and Luciferase Assay
3. Results and Discussion
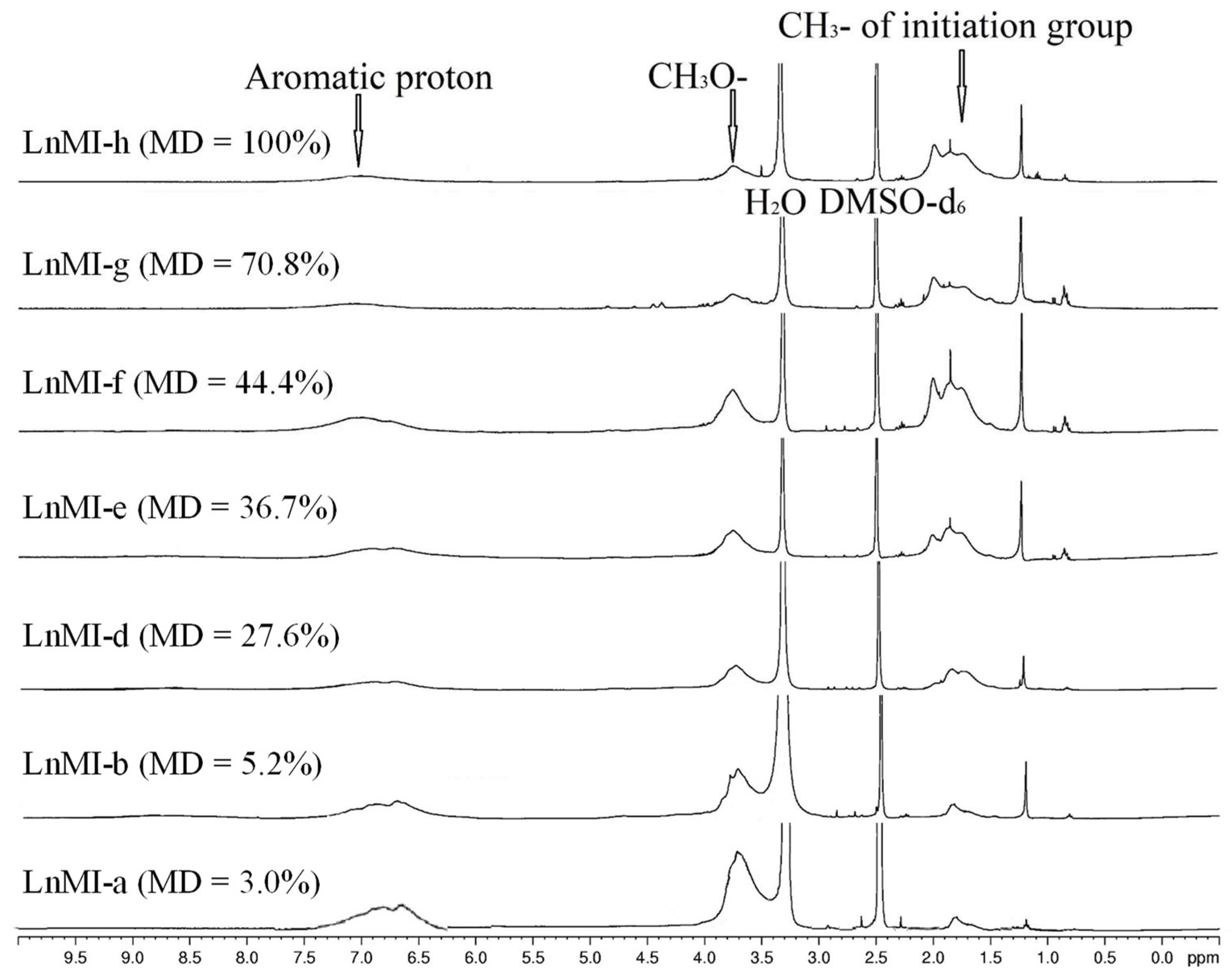
3.1. Preparation of ATRP-Initiator Modified Lignin with Various Modification Degree
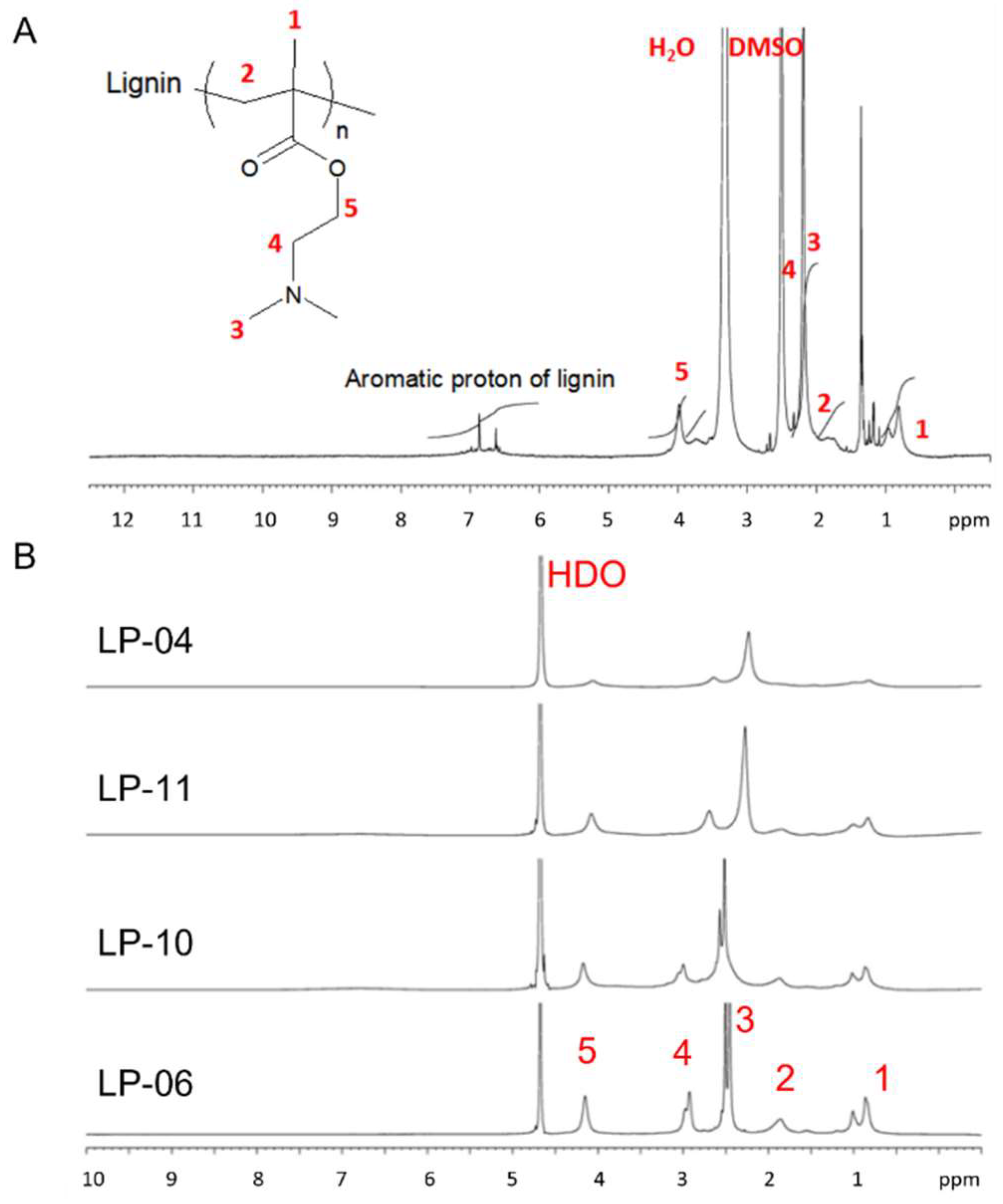
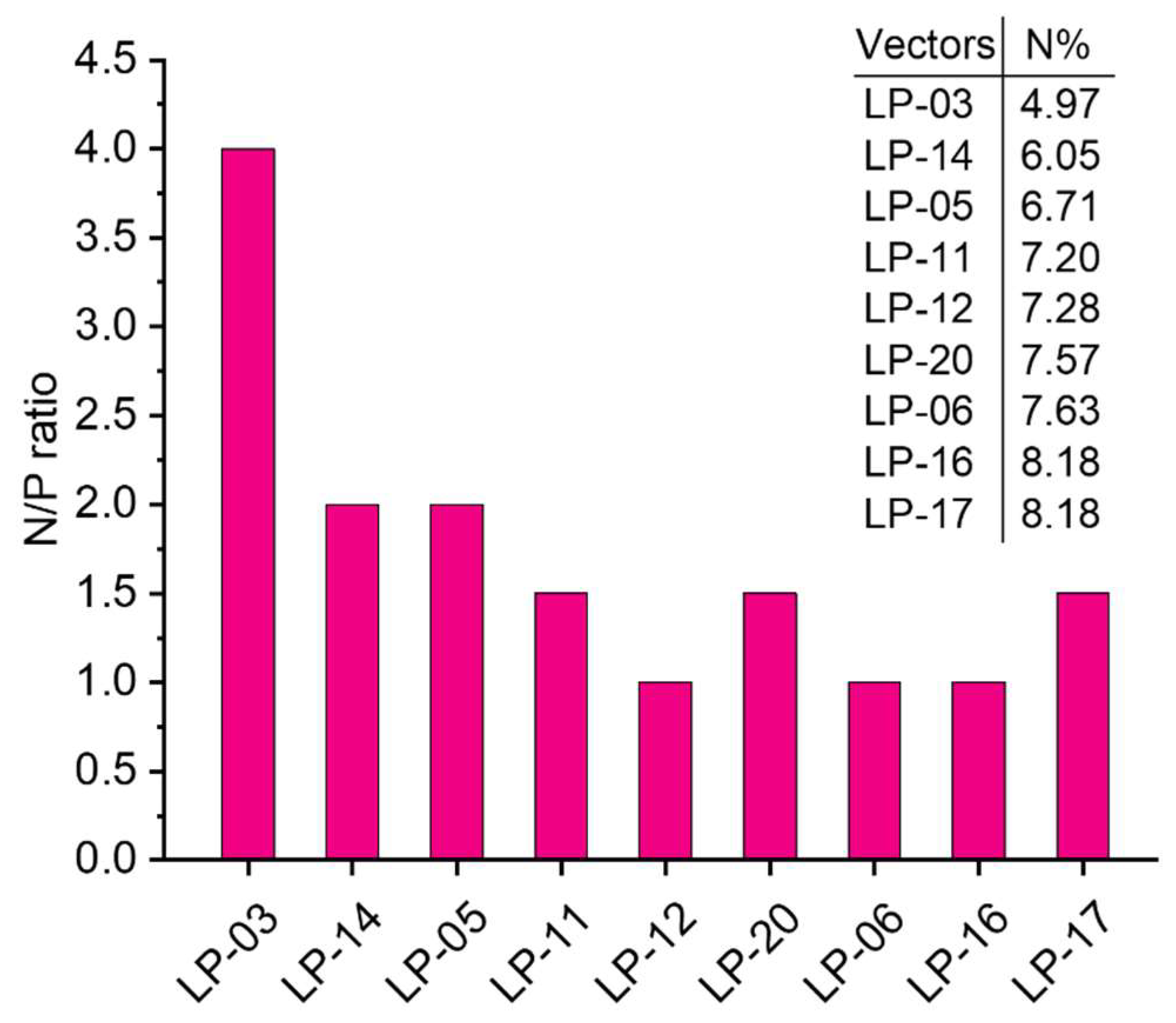
3.2. Preparation of Lignin-PDMAEMA Graft Copolymers with Varied Grafting Degree and Cationic Chain Length through ATRP
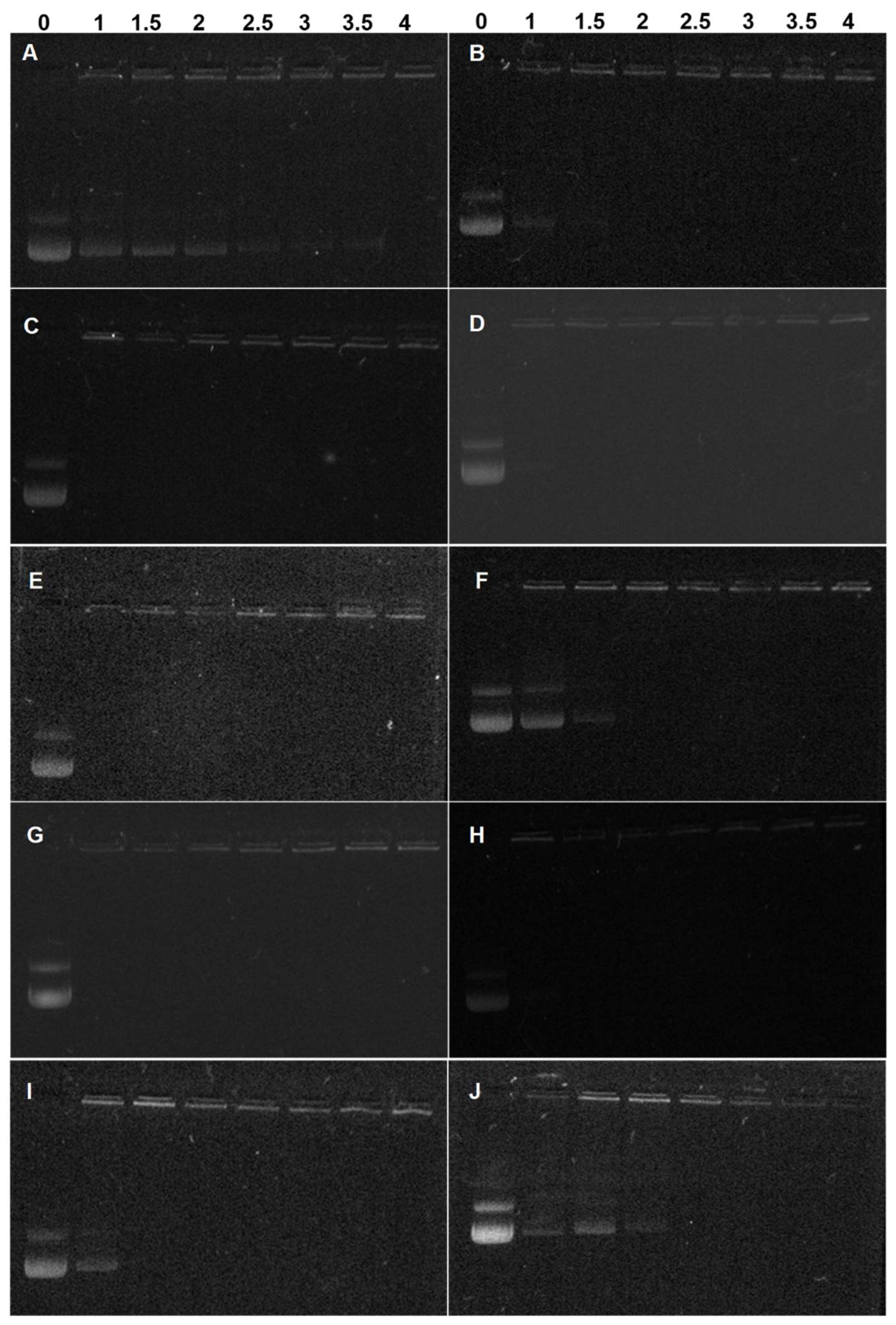
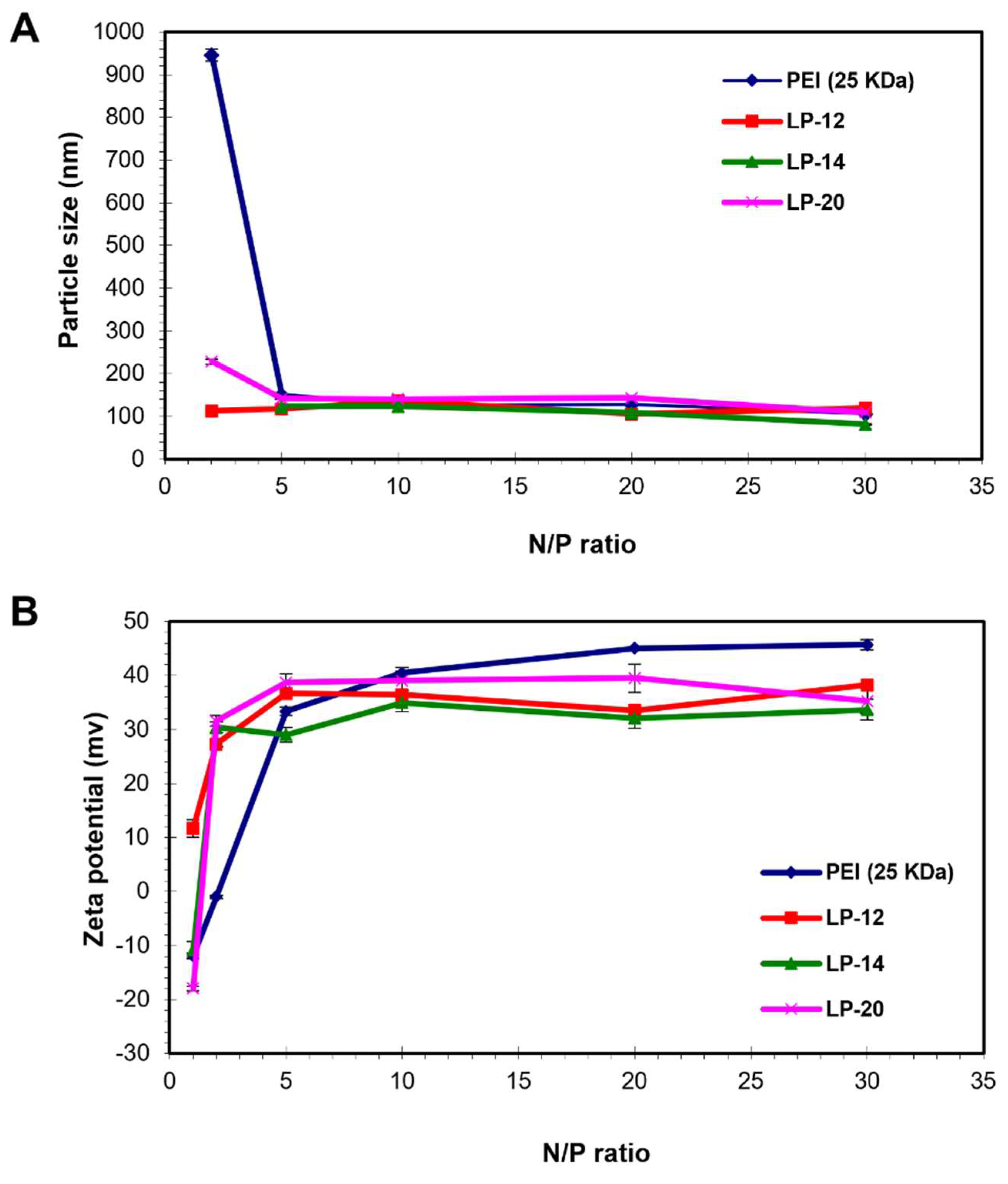
3.3. pDNA Condensation Capability of Lignin Graft Copolymers
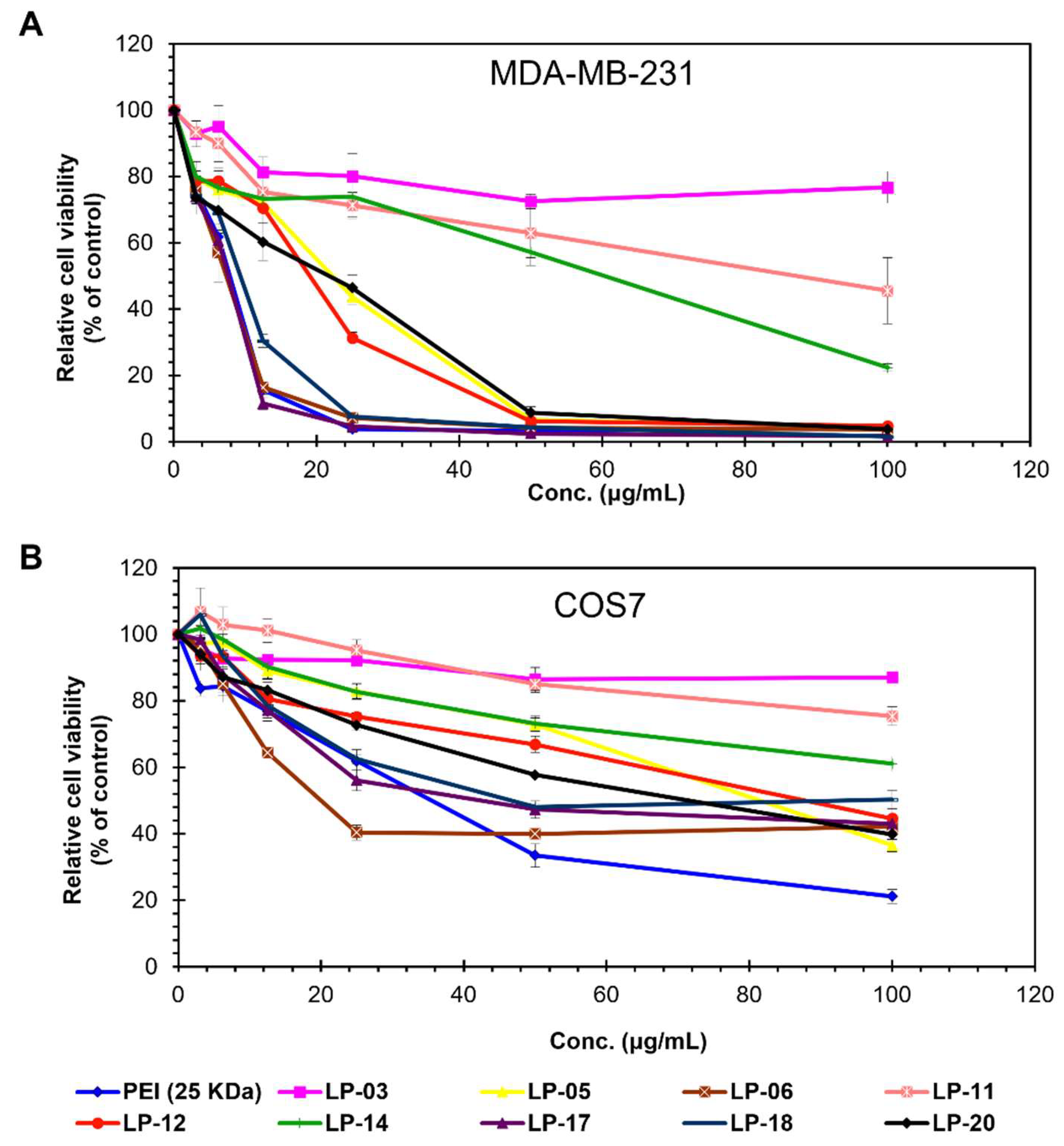
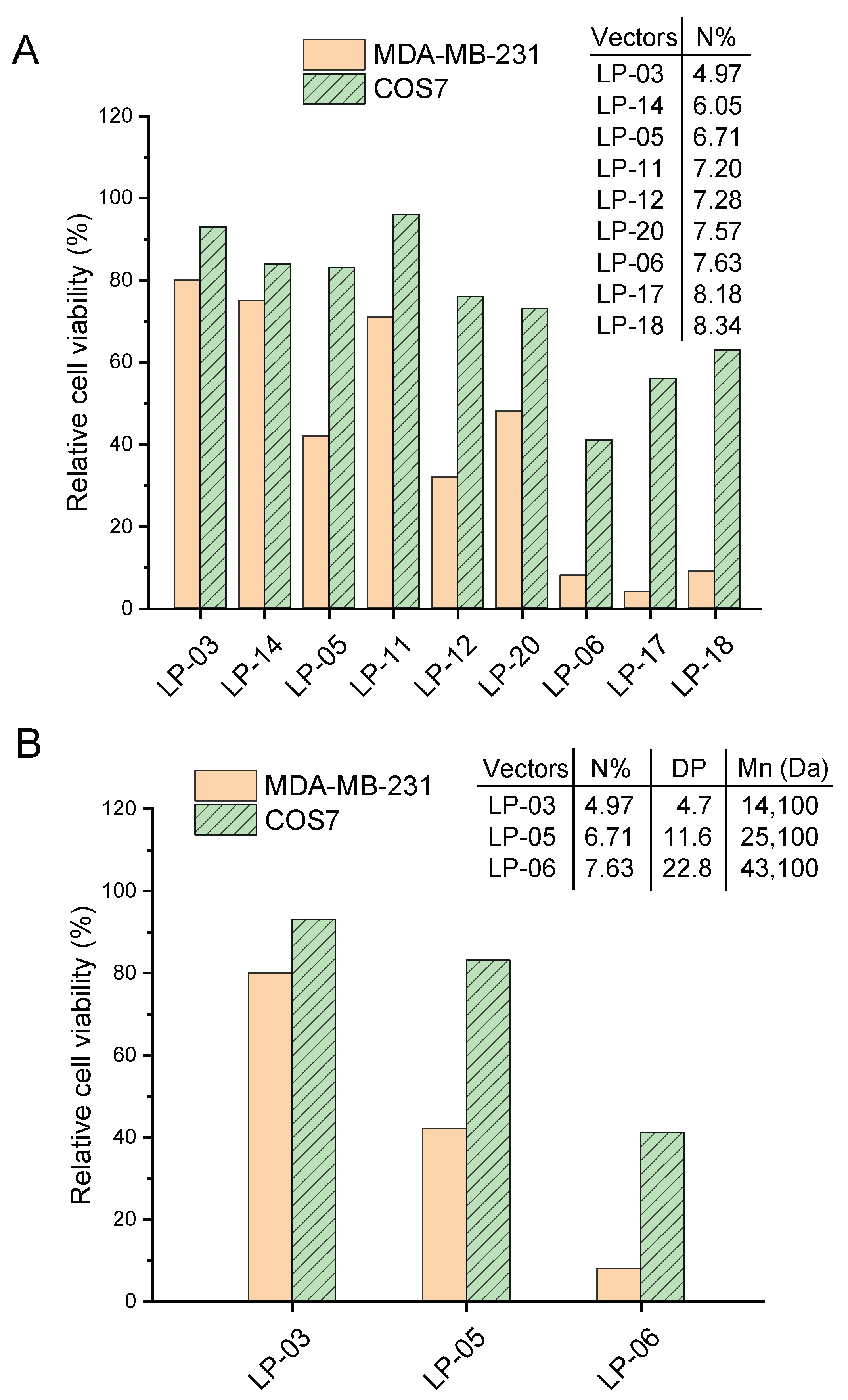
3.4. Cytotoxic Effect of Lignin-PDMAEMA Copolymers on MDA-MB-231 and COS7 Cells
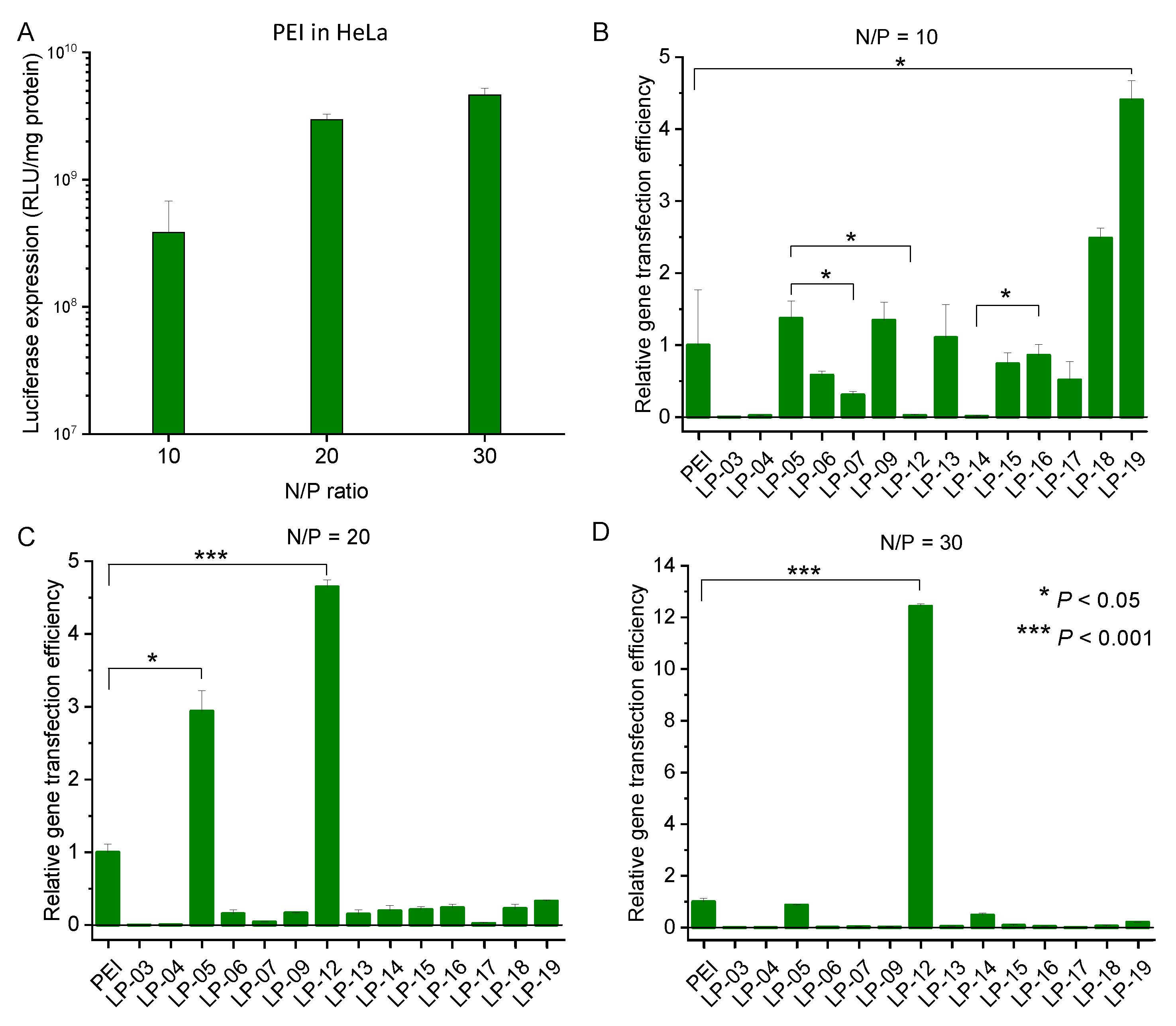
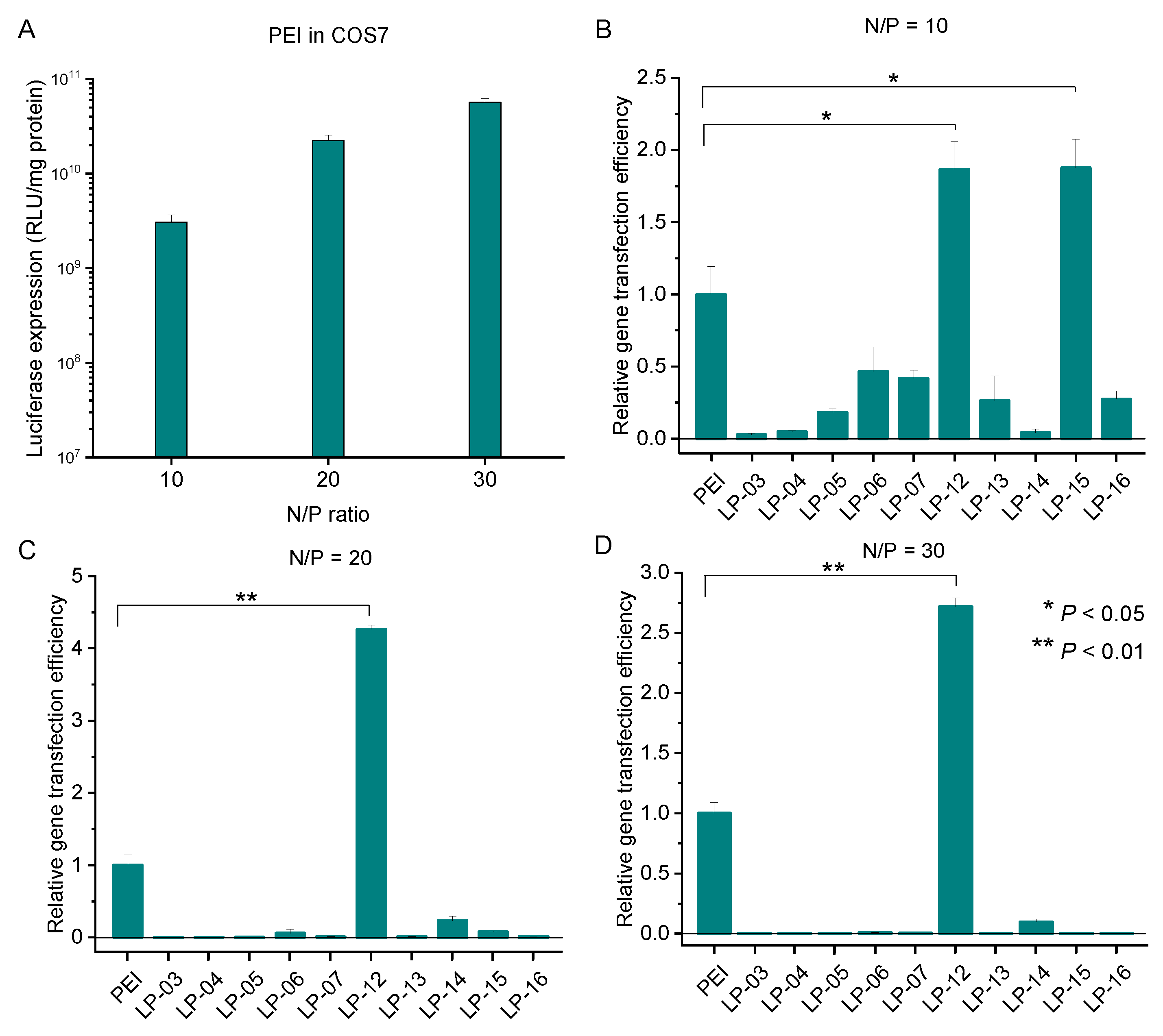
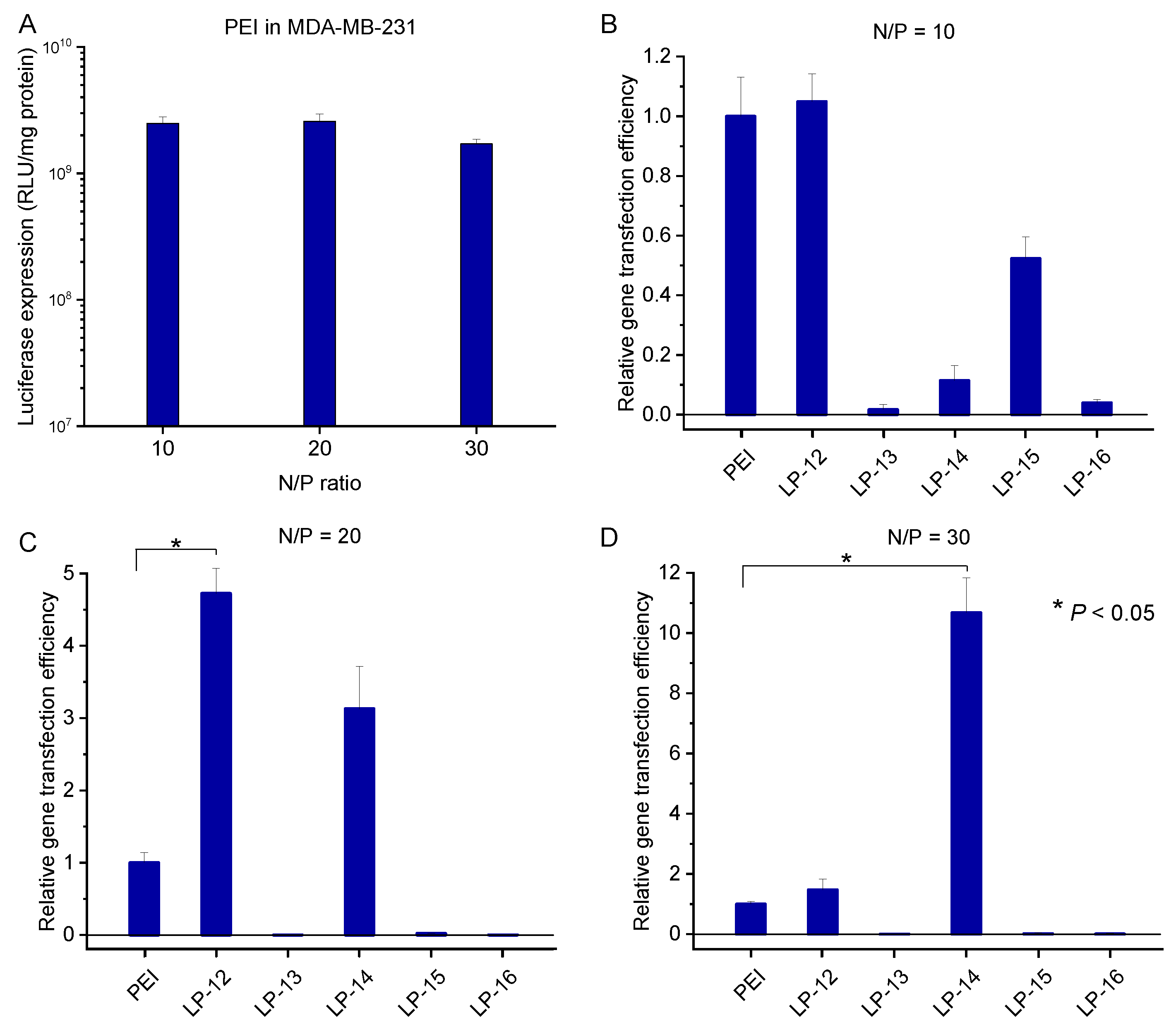
3.5. In Vitro Transfection Efficiency of Lignin-PDMAEMA Copolymers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Rahimi, A.; Ulbrich, A.; Coon, J.J.; Stahl, S.S. Formic-acid-induced depolymerization of oxidized lignin to aromatics. Nature 2014, 515, 249–252. [Google Scholar] [CrossRef]
- Thakur, V.K.; Thakur, M.K.; Raghavan, P.; Kessler, M.R. Progress in Green Polymer Composites from Lignin for Multifunctional Applications: A Review. ACS Sustain. Chem. Eng. 2014, 2, 1072–1092. [Google Scholar] [CrossRef]
- Katahira, R.; Elder, T.J.; Beckham, G.T. Chapter 1 A Brief Introduction to Lignin Structure. In Lignin Valorization: Emerging Approaches; The Royal Society of Chemistry: London, UK, 2018; pp. 1–20. [Google Scholar]
- Kumar, R.; Butreddy, A.; Kommineni, N.; Reddy, P.G.; Bunekar, N.; Sarkar, C.; Dutt, S.; Mishra, V.K.; Aadil, K.R.; Mishra, Y.K.; et al. Lignin: Drug/Gene Delivery and Tissue Engineering Applications. Int. J. Nanomed. 2021, 16, 2419–2441. [Google Scholar] [CrossRef]
- Liu, R.; Dai, L.; Xu, C.L.; Wang, K.; Zheng, C.Y.; Si, C.L. Lignin-Based Micro- and Nanomaterials and their Composites in Biomedical Applications. ChemSusChem 2020, 13, 4266–4283. [Google Scholar] [CrossRef]
- Dominguez-Robles, J.; Carcamo-Martinez, A.; Stewart, S.A.; Donnelly, R.F.; Larraneta, E.; Borrega, M. Lignin for pharmaceutical and biomedical applications-Could this become a reality? Sustain. Chem. Pharm. 2020, 18, 8. [Google Scholar] [CrossRef]
- Figueiredo, P.; Lintinen, K.; Hirvonen, J.T.; Kostiainen, M.A.; Santos, H.A. Properties and chemical modifications of lignin: Towards lignin-based nanomaterials for biomedical applications. Prog. Mater. Sci. 2018, 93, 233–269. [Google Scholar] [CrossRef]
- Behr, J.P. Synthetic gene-transfer vectors. Accounts Chem. Res. 1993, 26, 274–278. [Google Scholar] [CrossRef]
- Wang, J.H.; Ye, X.L.; Ni, H.B.; Zhang, J.F.; Ju, S.Q.; Ding, W.F. Transfection Efficiency Evaluation and Endocytosis Exploration of Different Polymer Condensed Agents. DNA Cell Biol. 2019, 38, 1048–1055. [Google Scholar] [CrossRef]
- Zhang, Z.X.; Wen, Y.T.; Song, X.; Zhu, J.L.; Li, J. Nonviral DNA Delivery System with Supramolecular PEGylation Formed by Host-Guest Pseudo-Block Copolymers. ACS Appl. Biol. Mater. 2021, 4, 5057–5070. [Google Scholar] [CrossRef]
- Li, H.; Peng, E.; Zhao, F.; Li, J.; Xue, J. Supramolecular Surface Functionalization of Iron Oxide Nanoparticles with α-Cyclodextrin-Based Cationic Star Polymer for Magnetically-Enhanced Gene Delivery. Pharmaceutics 2021, 13, 1884. [Google Scholar] [CrossRef]
- Wen, Y.T.; Bai, H.Z.; Zhu, J.L.; Song, X.; Tang, G.P.; Li, J. A supramolecular platform for controlling and optimizing molecular architectures of siRNA targeted delivery vehicles. Sci. Adv. 2020, 6, eabc2148. [Google Scholar] [CrossRef]
- Ooi, Y.J.; Wen, Y.T.; Zhu, J.L.; Song, X.; Li, J. Surface Charge Switchable Polymer/DNA Nanoparticles Responsive to Tumor Extracellular pH for Tumor-Triggered Enhanced Gene Delivery. Biomacromolecules 2020, 21, 1136–1148. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Zhao, F.; Zhang, D.H.; Li, J. Hyaluronic acid conjugated beta-cyclodextrin-oligoethylenimine star polymer for CD44-targeted gene delivery. Int. J. Pharm. 2015, 483, 169–179. [Google Scholar] [CrossRef]
- Zhao, F.; Yin, H.; Li, J. Supramolecular self-assembly forming a multifunctional synergistic system for targeted co-delivery of gene and drug. Biomaterials 2014, 35, 1050–1062. [Google Scholar] [CrossRef]
- Wen, Y.T.; Zhang, Z.X.; Li, J. Highly Efficient Multifunctional Supramolecular Gene Carrier System Self-Assembled from Redox-Sensitive and Zwitterionic Polymer Blocks. Adv. Funct. Mater. 2014, 24, 3874–3884. [Google Scholar] [CrossRef]
- Zhao, F.; Yin, H.; Zhang, Z.X.; Li, J. Folic Acid Modified Cationic gamma-Cyclodextrin-oligoethylenimine Star Polymer with Bioreducible Disulfide Linker for Efficient Targeted Gene Delivery. Biomacromolecules 2013, 14, 476–484. [Google Scholar] [CrossRef]
- Liu, X.H.; Yin, H.; Zhang, Z.X.; Diao, B.S.; Li, J. Functionalization of lignin through ATRP grafting of poly(2-dimethylaminoethyl methacrylate) for gene delivery. Colloid Surf. B-Biointerfaces 2015, 125, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Diao, B.; Zhang, Z.; Zhu, J.; Li, J. Biomass-based thermogelling copolymers consisting of lignin and grafted poly(N-isopropylacrylamide), poly(ethylene glycol), and poly(propylene glycol). RSC Adv. 2014, 4, 42996–43003. [Google Scholar] [CrossRef]
- Ten, E.; Ling, C.; Wang, Y.; Srivastava, A.; Dempere, L.A.; Vermerris, W. Lignin Nanotubes As Vehicles for Gene Delivery into Human Cells. Biomacromolecules 2014, 15, 327–338. [Google Scholar] [CrossRef]
- Naron, D.R.; Collard, F.X.; Tyhoda, L.; Gorgens, J.F. Characterisation of lignins from different sources by appropriate analytical methods: Introducing thermogravimetric analysis-thermal desorption-gas chromatography-mass spectroscopy. Ind. Crop. Prod. 2017, 101, 61–74. [Google Scholar] [CrossRef]
- Rico-Garcia, D.; Ruiz-Rubio, L.; Perez-Alvarez, L.; Hernandez-Olmos, S.L.; Guerrero-Ramirez, G.L.; Vilas-Vilela, J.L. Lignin-Based Hydrogels: Synthesis and Applications. Polymers 2020, 12, 23. [Google Scholar] [CrossRef] [Green Version]
- Meng, Y.; Lu, J.; Cheng, Y.; Li, Q.; Wang, H.S. Lignin-based hydrogels: A review of preparation, properties, and application. Int. J. Biol. Macromol. 2019, 135, 1006–1019. [Google Scholar] [CrossRef]
- Sen, S.; Patil, S.; Argyropoulos, D.S. Thermal properties of lignin in copolymers, blends, and composites: A review. Green Chem. 2015, 17, 4862–4887. [Google Scholar] [CrossRef]
- Kazzaz, A.E.; Feizi, Z.H.; Fatehi, P. Grafting strategies for hydroxy groups of lignin for producing materials. Green Chem. 2019, 21, 5714–5752. [Google Scholar] [CrossRef] [Green Version]
- Laurichesse, S.; Averous, L. Chemical modification of lignins: Towards biobased polymers. Prog. Polym. Sci. 2014, 39, 1266–1290. [Google Scholar] [CrossRef]
- Zhang, Z.X.; Liu, X.; Xu, F.J.; Loh, X.J.; Kang, E.T.; Neoh, K.G.; Li, J. Pseudo-block copolymer based on star-shaped poly(N-isopropylacrylamide) with a beta-cyclodextrin core and guest-bearing PEG: Controlling thermoresponsivity through supramolecular self-assembly. Macromolecules 2008, 41, 5967–5970. [Google Scholar] [CrossRef]
- Zhang, Z.X.; Liu, K.L.; Li, J. Self-Assembly and Micellization of a Dual Thermoresponsive Supramolecular Pseudo-Block Copolymer. Macromolecules 2011, 44, 1182–1193. [Google Scholar] [CrossRef]
- Zhang, Z.X.; Liu, K.L.; Li, J. A Thermoresponsive Hydrogel Formed from a Star-Star Supramolecular Architecture. Angew. Chem.-Int. Edit. 2013, 52, 6180–6184. [Google Scholar] [CrossRef] [PubMed]
- Tiera, M.J.; Shi, Q.; Winnik, F.M.; Fernandes, J.C. Polycation-based gene therapy: Current knowledge and new perspectives. Curr. Gene Ther. 2011, 11, 288–306. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lignin-Based Macroinitiators 2 | The Initial Molar Ratios of BiBB/Lignin | Reaction Medium 3 | TEA (mmol) 4 | MD (%) 5 |
|---|---|---|---|---|
| LnMI-a | 24.7 | DMA | \ | 3.0 |
| LnMI-b | 34.6 | DMA | \ | 5.2 |
| LnMI-c | 36.2 | DMA | \ | 20.2 |
| LnMI-d | 49.4 | DMA | \ | 27.6 |
| LnMI-e | 64.8 | DMA | \ | 36.7 |
| LnMI-f | 114.5 | DMA | \ | 44.4 |
| LnMI-g | 229.0 | DMA | \ | 70.8 |
| LnMI-h | 111.2 | THF | 98.8 | 100 |
| Lignin-Based Macroinitiators 1 | MD (%) 2 | Number of -Br per LnMI 3 | Mn (Da) 4 | EA Results (%) | Yield (%) | ||
|---|---|---|---|---|---|---|---|
| C | N | H | |||||
| LnMI-a | 3.0 | 1.1 | 5166 | 56.09 | 0.73 | 5.02 | 45.4 |
| LnMI-b | 5.2 | 1.9 | 5286 | 63.03 | 0.28 | 5.48 | 46.1 |
| LnMI-c | 20.2 | 7.5 | 6115 | 57.69 | 0.59 | 5.16 | 50.1 |
| LnMI-d | 27.6 | 10.2 | 6523 | 55.30 | 0.40 | 4.40 | 47.8 |
| LnMI-e | 36.7 | 13.6 | 7025 | 54.89 | 0.49 | 4.96 | 53.4 |
| LnMI-f | 44.4 | 16.5 | 7451 | 53.46 | 0.51 | 4.83 | 63.8 |
| LnMI-g | 70.8 | 26.2 | 8908 | 52.04 | 0.94 | 4.96 | 63.5 |
| LnMI-h | 100 | 37.1 | 10,519 | 49.91 | 0.85 | 4.75 | 84.8 |
| Lignin PDMAEMA | Initiator | Initial [M]0/[I]0/[Cu]0/[L]0 4 | Monomer (mmol) | Reaction Medium | Yield (%) |
|---|---|---|---|---|---|
| LP-01 | LnMI-a (MD = 3.0%) | 100:1:1:1 | 5.0 | DMF/3 mL | 23 |
| LP-02 | 200:1:1:1 | 10.0 | DMF/3 mL | 17 | |
| LP-03 | LnMI-d (MD = 27.6%) | 10:1:1:1 | 1.3 | THF/5 mL | 42 |
| LP-04 | 20:1:1:1 | 2.6 | THF/5 mL | 38 | |
| LP-05 | 30:1:1:1 | 3.9 | THF/5 mL | 42 | |
| LP-06 | 50:1:1:1 | 6.5 | THF/5 mL | 46 | |
| LP-07 | 100:1:1:1 | 13.0 | THF/5 mL | 26 | |
| LP-08 2 | 100:1:1:1 | 13.0 | Dioxane/3 mL | / | |
| LP-09 2 | 100:1:1:1 | 13.0 | Dioxane/4 mL | 57 | |
| LP-10 | LnMI-b (MD = 5.2%) | 50:1:1:1 | 1.45 | Dioxane/2 mL | 36 |
| LP-11 | 100:1:1:1 | 2.9 | Dioxane/2 mL | 56 | |
| LP-12 | LnMI-e (MD = 36.7%) | 50:1:1:1 | 6.5 | Dioxane/4 mL | 38 |
| LP-13 | 100:1:1:1 | 13.0 | Dioxane/4 mL | 59 | |
| LP-14 3 | LnMI-f (MD = 44.4%) | 50:1:1:1 | 6.5 | Dioxane/4 mL | 21 |
| LP-15 3 | 100:1:1:1 | 13.0 | THF/4 mL | 28 | |
| LP-16 3 | 100:1:1:1 | 13.0 | Dioxane/4 mL | 44 | |
| LP-17 | LnMI-g (MD = 70.8%) | 100:1:1:1 | 13.0 | Dioxane/4 mL | 38 |
| LP-18 | LnMI-h (MD = 100%) | 50:1:1:1 | 6.5 | Dioxane/4 mL | 43 |
| LP-19 2 | 100:1:1:1 | 18.0 | Dioxane/3 mL | 49 | |
| LP-20 | LnMI-c (MD = 20.2%) | 70:1:1:1 | 18.0 | Dioxane/3 mL | 55 |
| Lignin-PDMAEMA | EA Results (%) | Mn (Da) 1 | Number of DMAEMA Unit Per Arm 1 | ||
|---|---|---|---|---|---|
| N | C | H | |||
| LP-01 | 3.93 | 55.45 | 6.57 | 8300 | 18.2 |
| LP-02 | 6.81 | 59.13 | 8.54 | 20,500 | 88.9 |
| LP-03 | 4.97 | 57.6 | 7.41 | 14,100 | 4.7 |
| LP-04 | 6.07 | 59.47 | 8.04 | 19,200 | 7.9 |
| LP-05 | 6.71 | 57.82 | 8.25 | 25,100 | 11.6 |
| LP-06 | 7.63 | 49.82 | 8.19 | 43,100 | 22.8 |
| LP-07 | 7.80 | 56.62 | 8.36 | 49,700 | 26.9 |
| LP-08 2 | 7.07 | 50.77 | 8.22 | 30,100 | 14.7 |
| LP-09 2 | 6.55 | 46.86 | 8.07 | 23,500 | 10.6 |
| LP-10 | 5.24 | 52.85 | 6.74 | 12,400 | 23.9 |
| LP-11 | 7.20 | 57.42 | 8.02 | 26,600 | 71.2 |
| LP-12 | 7.28 | 58.76 | 8.79 | 36,100 | 13.6 |
| LP-13 | 8.19 | 56.69 | 9.21 | 80,800 | 34.5 |
| LP-14 3 | 6.05 | 55.16 | 7.79 | 21,700 | 5.5 |
| LP-15 3 | 7.84 | 58.98 | 9.16 | 58,300 | 19.6 |
| LP-16 3 | 8.18 | 58.97 | 10.27 | 85,500 | 30.1 |
| LP-17 | 8.18 | 59.09 | 9.30 | 95,800 | 21.1 |
| LP-18 | 8.34 | 58.94 | 10.81 | 146,200 | 23.3 |
| LP-19 2 | 7.06 | 47.27 | 8.39 | 45,500 | 6.0 |
| LP-20 | 7.57 | 59.28 | 8.67 | 39,700 | 28.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Yin, H.; Song, X.; Zhang, Z.; Li, J. Lignin-Based Nonviral Gene Carriers Functionalized by Poly[2-(Dimethylamino)ethyl Methacrylate]: Effect of Grafting Degree and Cationic Chain Length on Transfection Efficiency. Biomolecules 2022, 12, 102. https://doi.org/10.3390/biom12010102
Liu X, Yin H, Song X, Zhang Z, Li J. Lignin-Based Nonviral Gene Carriers Functionalized by Poly[2-(Dimethylamino)ethyl Methacrylate]: Effect of Grafting Degree and Cationic Chain Length on Transfection Efficiency. Biomolecules. 2022; 12(1):102. https://doi.org/10.3390/biom12010102
Chicago/Turabian StyleLiu, Xiaohong, Hui Yin, Xia Song, Zhongxing Zhang, and Jun Li. 2022. "Lignin-Based Nonviral Gene Carriers Functionalized by Poly[2-(Dimethylamino)ethyl Methacrylate]: Effect of Grafting Degree and Cationic Chain Length on Transfection Efficiency" Biomolecules 12, no. 1: 102. https://doi.org/10.3390/biom12010102
APA StyleLiu, X., Yin, H., Song, X., Zhang, Z., & Li, J. (2022). Lignin-Based Nonviral Gene Carriers Functionalized by Poly[2-(Dimethylamino)ethyl Methacrylate]: Effect of Grafting Degree and Cationic Chain Length on Transfection Efficiency. Biomolecules, 12(1), 102. https://doi.org/10.3390/biom12010102