
Clustering of Aromatic Amino Acid Residues around Methionine in Proteins


Abstract
:1. Introduction
2. Materials and Methods
2.1. Identification of “3-Bridge” Clusters in Protein Structures
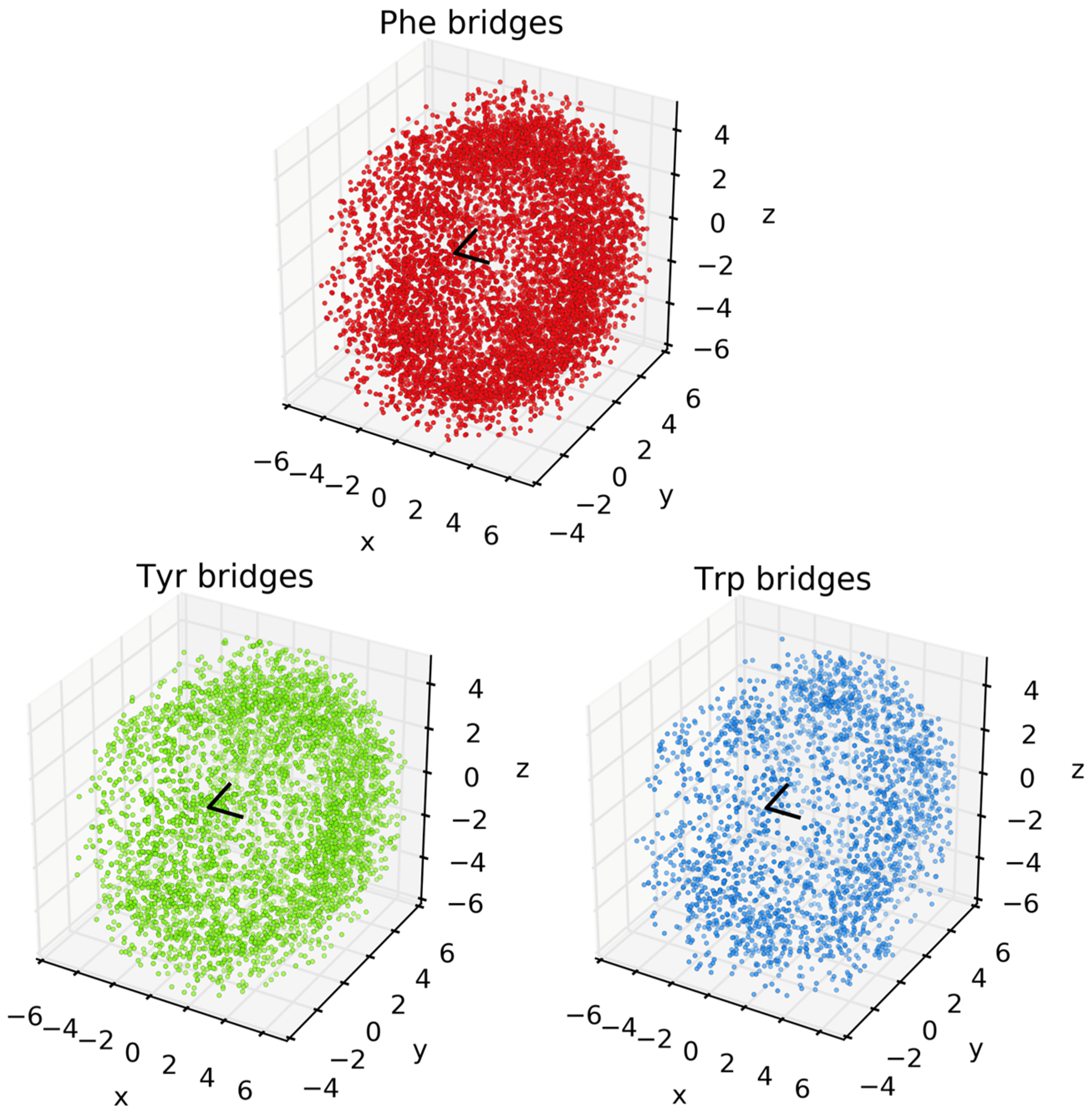
2.2. Assessing the Position of Aromatic Residues about Methionine
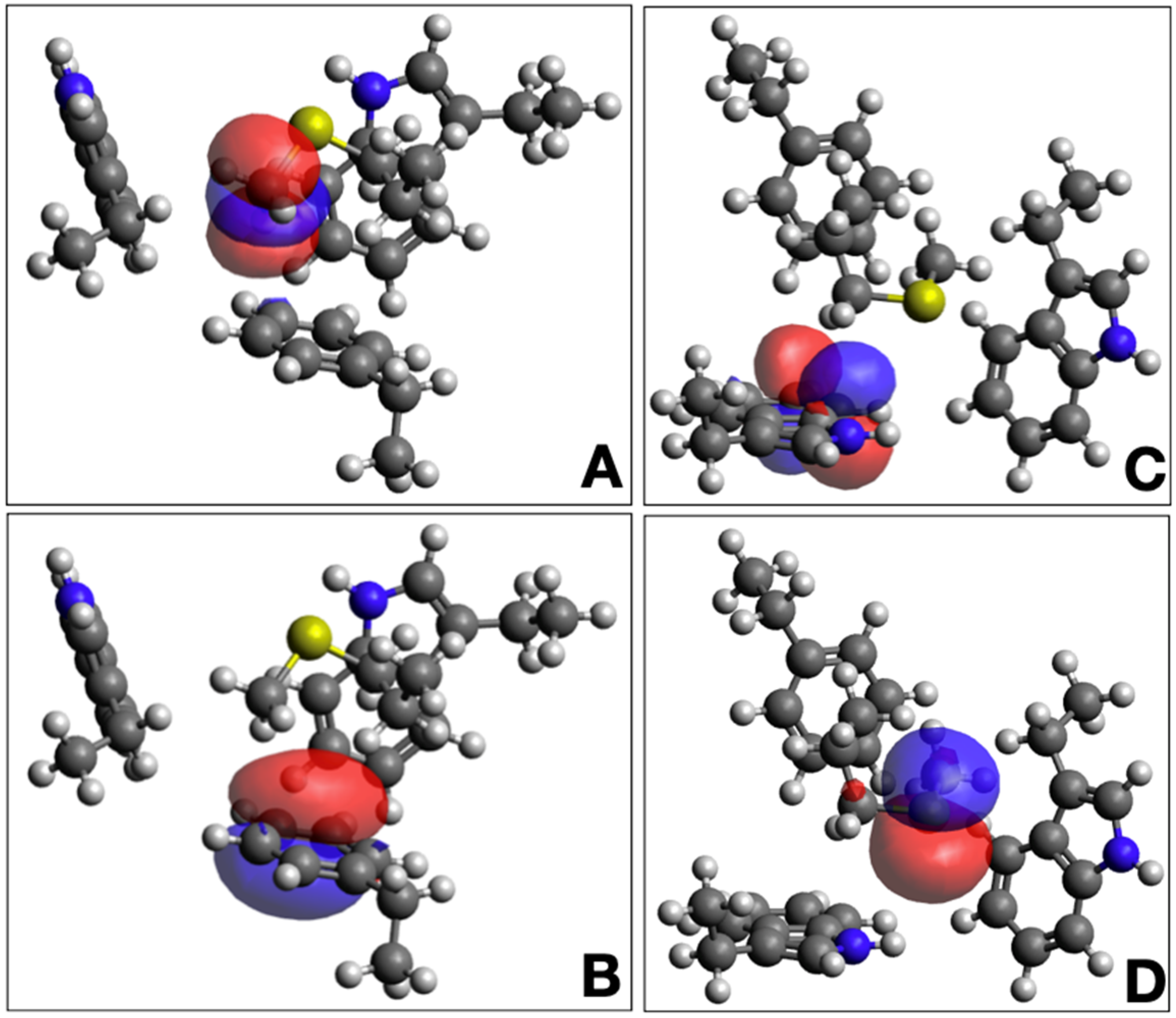
2.3. Density Functional Calculations
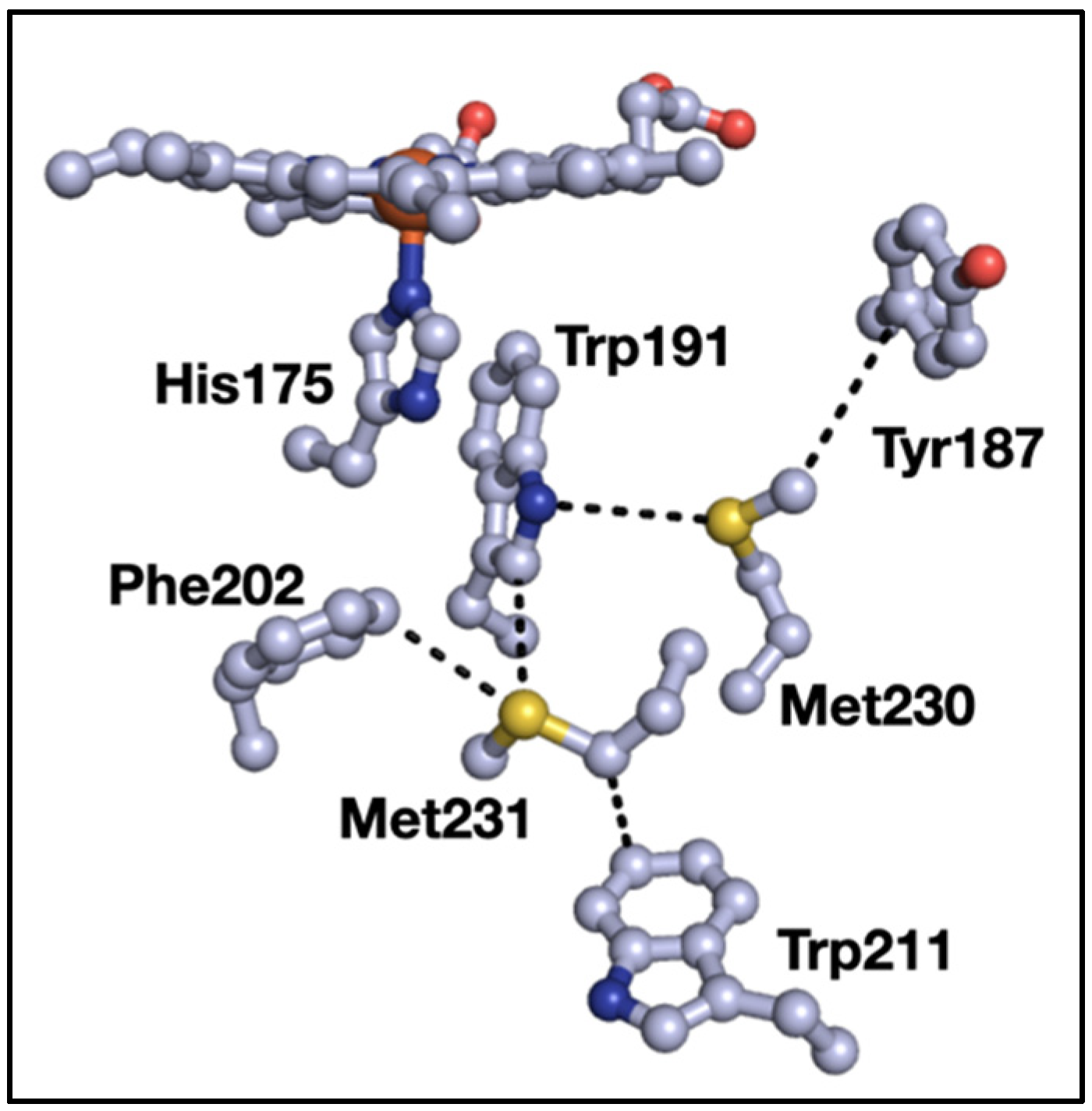
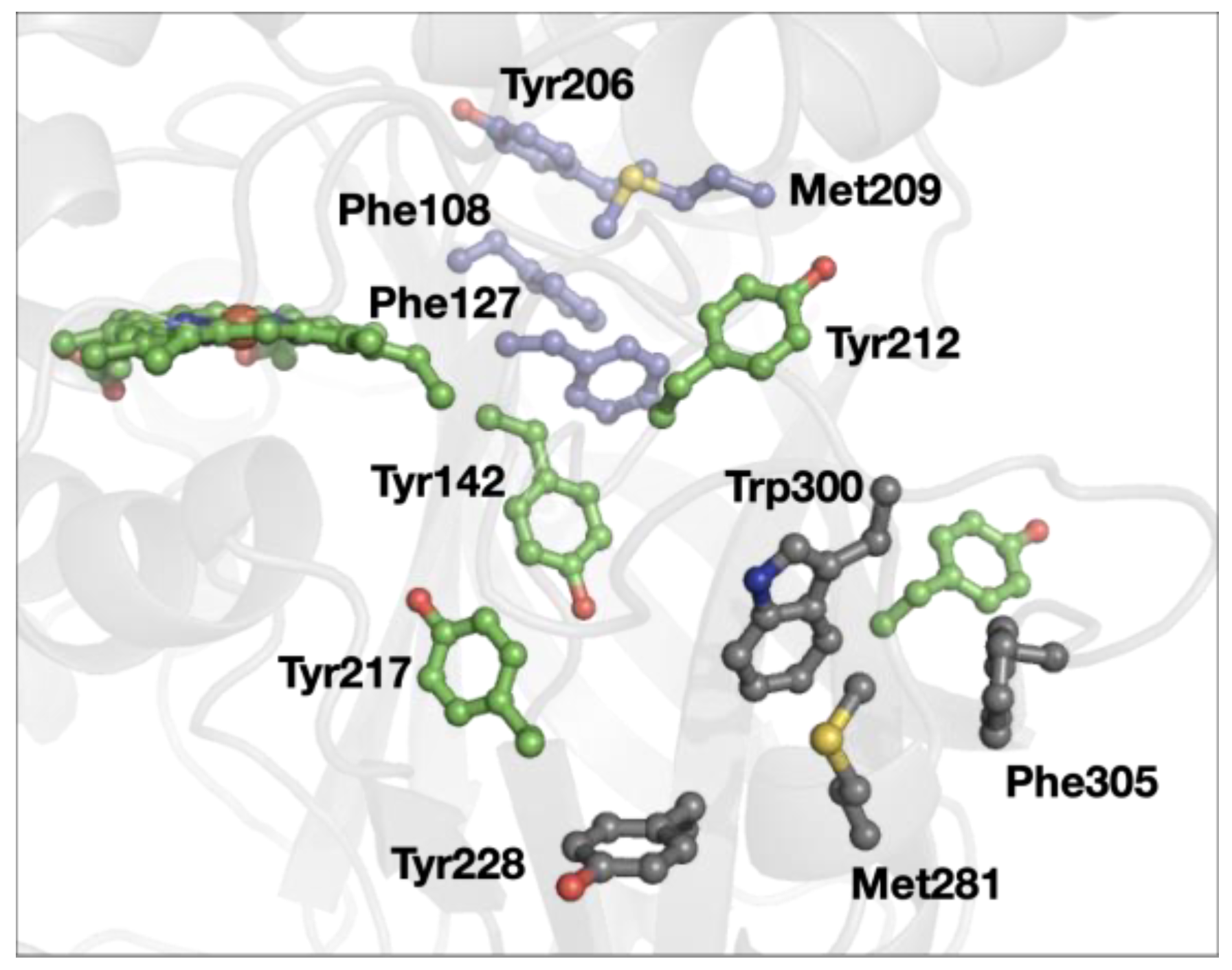
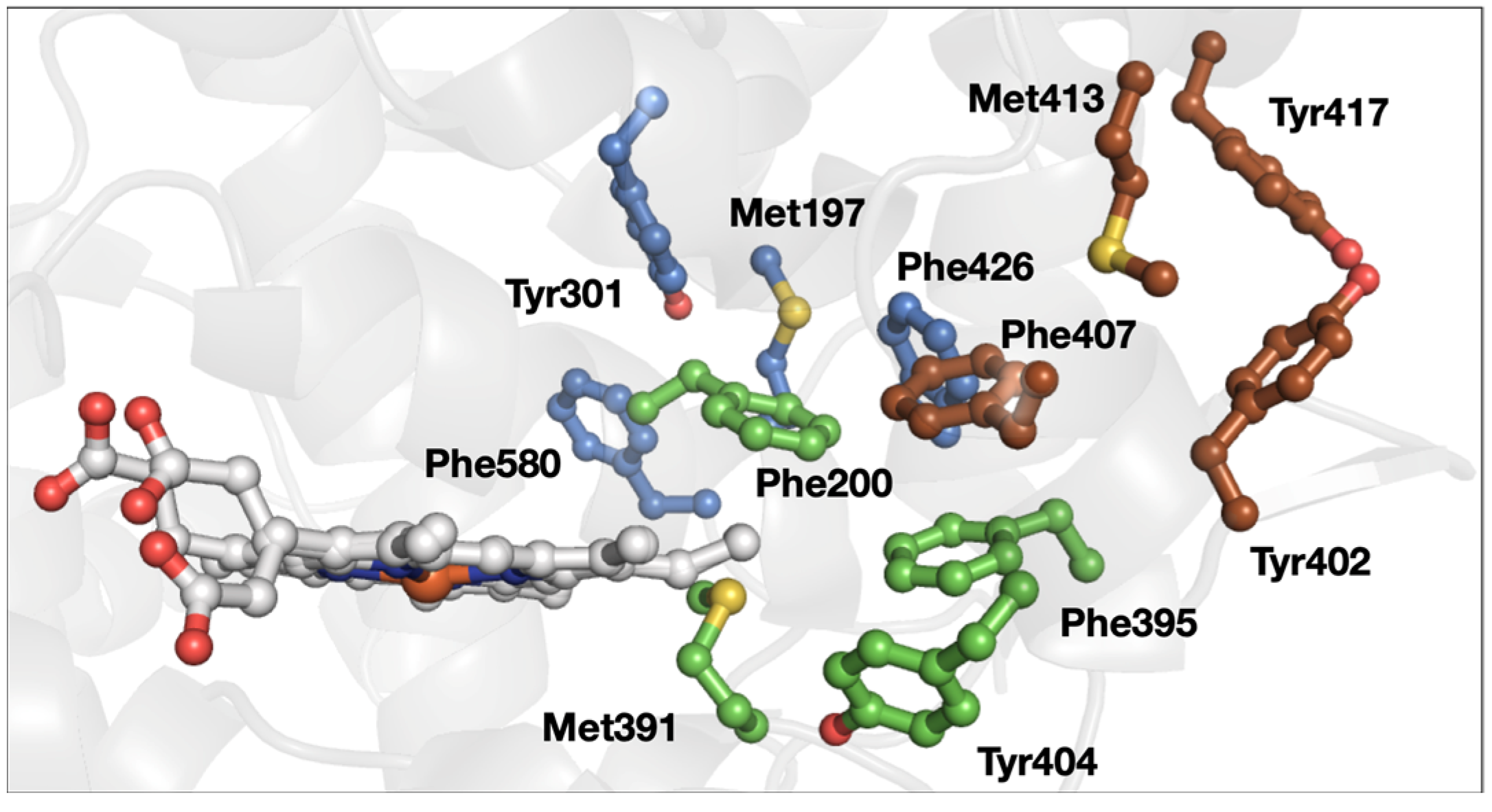
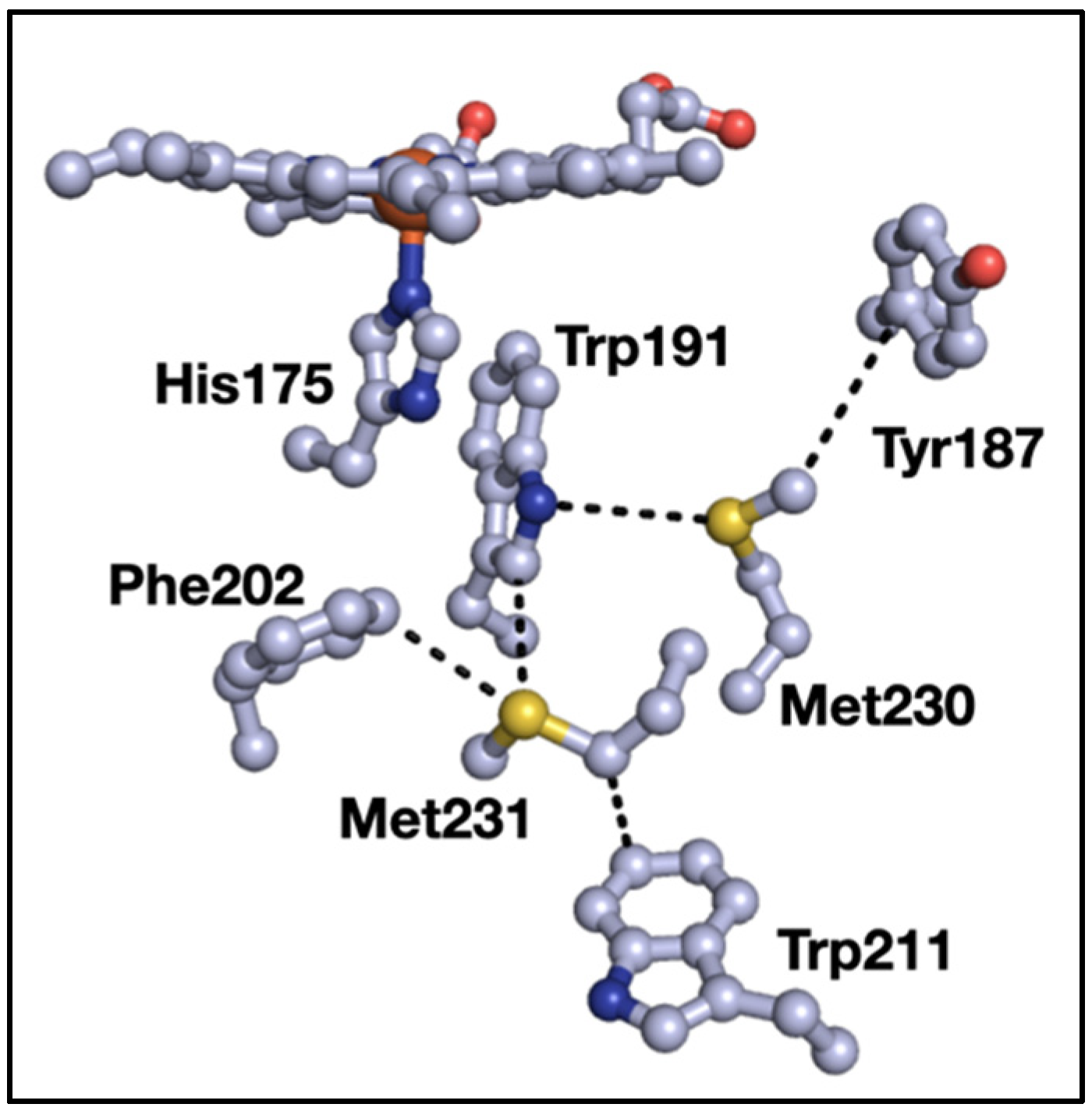
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gallivan, J.P.; Dougherty, D.A. Cation-π Interactions in Structural Biology. Proc. Natl. Acad. Sci. USA 1999, 96, 9459–9464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.C.; Dougherty, D.A. The Cation−π Interaction. Chem. Rev. 1997, 97, 1303–1324. [Google Scholar] [CrossRef] [PubMed]
- Keskin, O.; Gursoy, A.; Ma, B.; Nussinov, R. Principles of Protein−Protein Interactions: What Are the Preferred Ways For Proteins To Interact? Chem. Rev. 2008, 108, 1225–1244. [Google Scholar] [CrossRef] [PubMed]
- Babine, R.E.; Bender, S.L. Molecular Recognition of Protein−Ligand Complexes: Applications to Drug Design. Chem. Rev. 1997, 97, 1359–1472. [Google Scholar] [CrossRef]
- Martinez, R.C.; Iverson, L.B. Rethinking the Term “Pi-Stacking”. Chem. Sci. 2012, 3, 2191–2201. [Google Scholar] [CrossRef] [Green Version]
- Dougherty, D.A. The Cation−π Interaction. Acc. Chem. Res. 2013, 46, 885–893. [Google Scholar] [CrossRef] [Green Version]
- Schottel, B.L.; Chifotides, H.T.; Dunbar, K.R. Anion-π Interactions. Chem. Soc. Rev. 2008, 37, 68–83. [Google Scholar] [CrossRef]
- Zauhar, R.J.; Colbert, C.L.; Morgan, R.S.; Welsh, W.J. Evidence for a Strong Sulfur–Aromatic Interaction Derived from Crystallographic Data. Biopolymers 2000, 53, 233–248. [Google Scholar] [CrossRef]
- Weber, D.S.; Warren, J.J. A Survey of Methionine-Aromatic Interaction Geometries in the Oxidoreductase Class of Enzymes: What Could Met-Aromatic Interactions Be Doing near Metal Sites? J. Inorg. Biochem. 2018, 186, 34–41. [Google Scholar] [CrossRef]
- Weber, D.S.; Warren, J.J. The Interaction between Methionine and Two Aromatic Amino Acids Is an Abundant and Multifunctional Motif in Proteins. Arch. Biochem. Biophys. 2019, 672, 108053. [Google Scholar] [CrossRef]
- Finzel, B.C.; Poulos, T.L.; Kraut, J. Crystal Structure of Yeast Cytochrome c Peroxidase Refined at 1.7-A Resolution. J. Biol. Chem. 1984, 259, 13027–13036. [Google Scholar] [CrossRef]
- Kim, K.; Erman, J.E. Methionine Modification in Cytochrome-c Peroxidase. Biochim. Biophys. Acta 1988, 954, 95–107. [Google Scholar] [CrossRef]
- Fishel, L.A.; Farnum, M.F.; Mauro, J.M.; Miller, M.A.; Kraut, J.; Liu, Y.; Tan, X.L.; Scholes, C.P. Compound I Radical in Site-Directed Mutants of Cytochrome c Peroxidase as Probed by Electron Paramagnetic Resonance and Electron-Nuclear Double Resonance. Biochemistry 1991, 30, 1986–1996. [Google Scholar] [CrossRef]
- Barrows, T.P.; Bhaskar, B.; Poulos, T.L. Electrostatic Control of the Tryptophan Radical in Cytochrome c Peroxidase. Biochemistry 2004, 43, 8826–8834. [Google Scholar] [CrossRef] [PubMed]
- Reid, K.S.C.; Lindley, P.F.; Thornton, J.M. Sulphur-Aromatic Interactions in Proteins. FEBS Lett. 1985, 190, 209–213. [Google Scholar] [CrossRef] [Green Version]
- Warme, P.K.; Morgan, R.S. A Survey of Amino Acid Side-Chain Interactions in 21 Proteins. J. Mol. Biol. 1978, 118, 289–304. [Google Scholar] [CrossRef]
- Morgan, R.S.; Tatsch, C.E.; Gushard, R.H.; Mcadon, J.M.; Warme, P.K. Chains Of Alternating Sulfur And π-Bonded Atoms In Eight Small Proteins. Int. J. Pept. Prot. Res. 1978, 11, 209–217. [Google Scholar] [CrossRef]
- Tatko, C.D.; Waters, M.L. Investigation of the Nature of the Methionine–π Interaction in β-Hairpin Peptide Model Systems. Protein Sci. 2004, 13, 2515–2522. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Tamayo, J.C.; Cordomí, A.; Olivella, M.; Mayol, E.; Fourmy, D.; Pardo, L. Analysis of the Interactions of Sulfur-Containing Amino Acids in Membrane Proteins. Protein Sci. 2016, 25, 1517–1524. [Google Scholar] [CrossRef] [Green Version]
- Bodner, B.L.; Jackman, L.M.; Morgan, R.S. NMR Study of 1:1 Complexes between Divalent Sulfur and Aromatic Compounds: A Model for Interactions in Globular Proteins. Biochem. Biophys. Res. Commun. 1980, 94, 807–813. [Google Scholar] [CrossRef]
- Chung, W.J.; Ammam, M.; Gruhn, N.E.; Nichol, G.S.; Singh, W.P.; Wilson, G.S.; Glass, R.S. Interactions of Arenes and Thioethers Resulting in Facilitated Oxidation. Org. Lett. 2009, 11, 397–400. [Google Scholar] [CrossRef]
- Monney, N.P.-A.; Bally, T.; Bhagavathy, G.S.; Glass, R.S. Spectroscopic Evidence for a New Type of Bonding between a Thioether Radical Cation and a Phenyl Group. Org. Lett. 2013, 15, 4932–4935. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Neese, F. The ORCA Program System. WIREs: Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software Update: The ORCA Program System, Version 4.0. WIREs: Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-Fitting Basis Sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. NBO 7.0: New Vistas in Localized and Delocalized Chemical Bonding Theory. J. Comput. Chem. 2019, 40, 2234–2241. [Google Scholar] [CrossRef]
- Schneider, W.B.; Bistoni, G.; Sparta, M.; Saitow, M.; Riplinger, C.; Auer, A.A.; Neese, F. Decomposition of Intermolecular Interaction Energies within the Local Pair Natural Orbital Coupled Cluster Framework. J. Chem. Theory Comput. 2016, 12, 4778–4792. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- ProtScale Tool: Amino Acid Composition (%) in the UniProtKB/Swiss-Prot Data Bank. Available online: https://web.expasy.org/protscale/pscale/A.A.Swiss-Prot.html (accessed on 18 February 2021).
- Brandl, M.; Weiss, M.S.; Jabs, A.; Sühnel, J.; Hilgenfeld, R. C-H⋯π-Interactions in Proteins. J. Mol. Biol. 2001, 307, 357–377. [Google Scholar] [CrossRef]
- Gober, J.G.; Ghodge, S.V.; Bogart, J.W.; Wever, W.J.; Watkins, R.R.; Brustad, E.M.; Bowers, A.A. P450-Mediated Non-Natural Cyclopropanation of Dehydroalanine-Containing Thiopeptides. ACS Chem. Biol. 2017, 12, 1726–1731. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC. Available online: https://pymol.org/sites/default/files/pymol.bib (accessed on 11 January 2019).
- Galgonek, J.; Vymětal, J.; Jakubec, D.; Vondrášek, J. Amino Acid Interaction (INTAA) Web Server. Nucleic Acids Res. 2017, 45, W388–W392. [Google Scholar] [CrossRef] [Green Version]
- Scrutton, N.S.; Raine, A.R. Cation-Pi Bonding and Amino-Aromatic Interactions in the Biomolecular Recognition of Substituted Ammonium Ligands. Biochem. J. 1996, 319, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Gallivan, J.P.; Zhang, Y.; Li, L.; Lester, H.A.; Dougherty, D.A. From Ab Initio Quantum Mechanics to Molecular Neurobiology: A Cation–π Binding Site in the Nicotinic Receptor. Proc. Natl. Acad. Sci. USA 1998, 95, 12088–12093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burley, S.K.; Petsko, G.A. Amino-Aromatic Interactions in Proteins. FEBS Lett. 1986, 203, 139–143. [Google Scholar] [CrossRef] [Green Version]
- Tsuzuki, S. CH/π Interactions. Annual Reports Section “C”. Phys. Chem. 2012, 108, 69–95. [Google Scholar] [CrossRef]
- Jiménez-Moreno, E.; Jiménez-Osés, G.; Gómez, A.M.; Santana, A.G.; Corzana, F.; Bastida, A.; Jiménez-Barbero, J.; Asensio, J.L. A Thorough Experimental Study of CH/π Interactions in Water: Quantitative Structure–Stability Relationships for Carbohydrate/Aromatic Complexes. Chem. Sci. 2015, 6, 6076–6085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishio, M.; Umezawa, Y.; Honda, K.; Tsuboyama, S.; Suezawa, H. CH/π Hydrogen Bonds in Organic and Organometallic Chemistry. CrystEngComm 2009, 11, 1757–1788. [Google Scholar] [CrossRef]
- Winkler, J.R.; Gray, H.B. Electron Flow through Biological Molecules: Does Hole Hopping Protect Proteins from Oxidative Damage? Q. Rev. Biophys. 2015, 48, 411–420. [Google Scholar] [CrossRef] [Green Version]
- Gray, H.B.; Winkler, J.R. Hole Hopping through Tyrosine/Tryptophan Chains Protects Proteins from Oxidative Damage. Proc. Natl. Acad. Sci. USA 2015, 112, 10920–10925. [Google Scholar] [CrossRef] [Green Version]
- Polizzi, N.F.; Migliore, A.; Therien, M.J.; Beratan, D.N. Defusing Redox Bombs? Proc. Natl. Acad. Sci. USA 2015, 112, 10821–10822. [Google Scholar] [CrossRef] [Green Version]
- Teo, R.D.; Wang, R.; Smithwick, E.R.; Migliore, A.; Therien, M.J.; Beratan, D.N. Mapping Hole Hopping Escape Routes in Proteins. Proc. Natl. Acad. Sci. USA 2019, 116, 15811–15816. [Google Scholar] [CrossRef] [Green Version]
- Kathiresan, M.; English, A.M. LC-MS/MS Suggests That Hole Hopping in Cytochrome c Peroxidase Protects Its Heme from Oxidative Modification by Excess H2O2. Chem. Sci. 2017, 8, 1152–1162. [Google Scholar] [CrossRef] [Green Version]
- Kathiresan, M.; English, A.M. LC-MS/MS Proteoform Profiling Exposes Cytochrome c Peroxidase Self-Oxidation in Mitochondria and Functionally Important Hole Hopping from Its Heme. J. Am. Chem. Soc. 2018, 140, 12033–12039. [Google Scholar] [CrossRef] [PubMed]
- Beratan, D.N.; Skourtis, S.S.; Balabin, I.A.; Balaeff, A.; Keinan, S.; Venkatramani, R.; Xiao, D. Steering Electrons on Moving Pathways. Acc. Chem. Res. 2009, 42, 1669–1678. [Google Scholar] [CrossRef] [Green Version]
- Skourtis, S.S.; Waldeck, D.H.; Beratan, D.N. Fluctuations in Biological and Bioinspired Electron-Transfer Reactions. Annu. Rev. Phys. Chem. 2010, 61, 461–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maté, M.J.; Zamocky, M.; Nykyri, L.M.; Herzog, C.; Alzari, P.M.; Betzel, C.; Koller, F.; Fita, I. Structure of Catalase-A from Saccharomyces Cerevisiae. J. Mol. Biol. 1999, 286, 135–149. [Google Scholar] [CrossRef]
- Gupta, K.; Selinsky, B.S.; Kaub, C.J.; Katz, A.K.; Loll, P.J. The 2.0Å Resolution Crystal Structure of Prostaglandin H2 Synthase-1: Structural Insights into an Unusual Peroxidase. J. Mol. Biol. 2004, 335, 503–518. [Google Scholar] [CrossRef] [PubMed]
- Ridder, I.S.; Rozeboom, H.J.; Dijkstra, B.W. Haloalkane Dehalogenase from Xanthobacter autotrophicus GJ10 Refined at 1.15 Å Resolution. Acta Cryst. D 1999, 55, 1273–1290. [Google Scholar] [CrossRef] [PubMed]
- Schanstra, J.P.; Janssen, D.B. Kinetics of Halide Release of Haloalkane Dehalogenase: Evidence for a Slow Conformational Change. Biochemistry 1996, 35, 5624–5632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krooshof, G.H.; Ridder, I.S.; Tepper, A.W.J.W.; Vos, G.J.; Rozeboom, H.J.; Kalk, K.H.; Dijkstra, B.W.; Janssen, D.B. Kinetic Analysis and X-Ray Structure of Haloalkane Dehalogenase with a Modified Halide-Binding Site. Biochemistry 1998, 37, 15013–15023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otyepka, M.; Damborský, J. Functionally Relevant Motions of Haloalkane Dehalogenases Occur in the Specificity-Modulating Cap Domains. Protein Sci. 2002, 11, 1206–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touw, W.G.; Vriend, G. BDB: Databank of PDB Files with Consistent B-Factors. Protein Eng. Des. Sel. 2014, 27, 457–462. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Liu, Q.; Qu, G.; Feng, Y.; Reetz, M.T. Utility of B-Factors in Protein Science: Interpreting Rigidity, Flexibility, and Internal Motion and Engineering Thermostability. Chem. Rev. 2019, 119, 1626–1665. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Met24 b | Trp20 c | Trp31 c | Phe41 c | |
|---|---|---|---|---|
| Wild Type | −10.9 | −3.75 | −2.02 | −3.11 |
| Trp20Ala | −7.44 | −0.12 | −2.01 | −3.16 |
| Trp31Ala | −9.12 | −3.72 | −0.31 | −3.13 |
| Phe41Ala | −7.54 | −3.59 | −1.87 | −0.22 |
| Trp20Ala/Trp31Ala | −4.21 | −0.12 | −1.86 | −0.22 |
| Trp20Ala/Phe41Ala | −5.68 | −0.12 | −0.31 | −3.16 |
| Trp31Ala/Phe41Ala | −5.98 | −3.56 | −0.37 | −0.22 |
| Trp20Ala/Trp31Ala/Phe41Ala | −2.68 | −0.12 | −0.37 | −0.22 |
| Met24Ala | −2.79 | −0.65 | −0.08 | −1.23 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gibbs, C.A.; Weber, D.S.; Warren, J.J. Clustering of Aromatic Amino Acid Residues around Methionine in Proteins. Biomolecules 2022, 12, 6. https://doi.org/10.3390/biom12010006
Gibbs CA, Weber DS, Warren JJ. Clustering of Aromatic Amino Acid Residues around Methionine in Proteins. Biomolecules. 2022; 12(1):6. https://doi.org/10.3390/biom12010006
Chicago/Turabian StyleGibbs, Curtis A., David S. Weber, and Jeffrey J. Warren. 2022. "Clustering of Aromatic Amino Acid Residues around Methionine in Proteins" Biomolecules 12, no. 1: 6. https://doi.org/10.3390/biom12010006
APA StyleGibbs, C. A., Weber, D. S., & Warren, J. J. (2022). Clustering of Aromatic Amino Acid Residues around Methionine in Proteins. Biomolecules, 12(1), 6. https://doi.org/10.3390/biom12010006