
The Role of Proteases and Serpin Protease Inhibitors in β-Cell Biology and Diabetes


Abstract
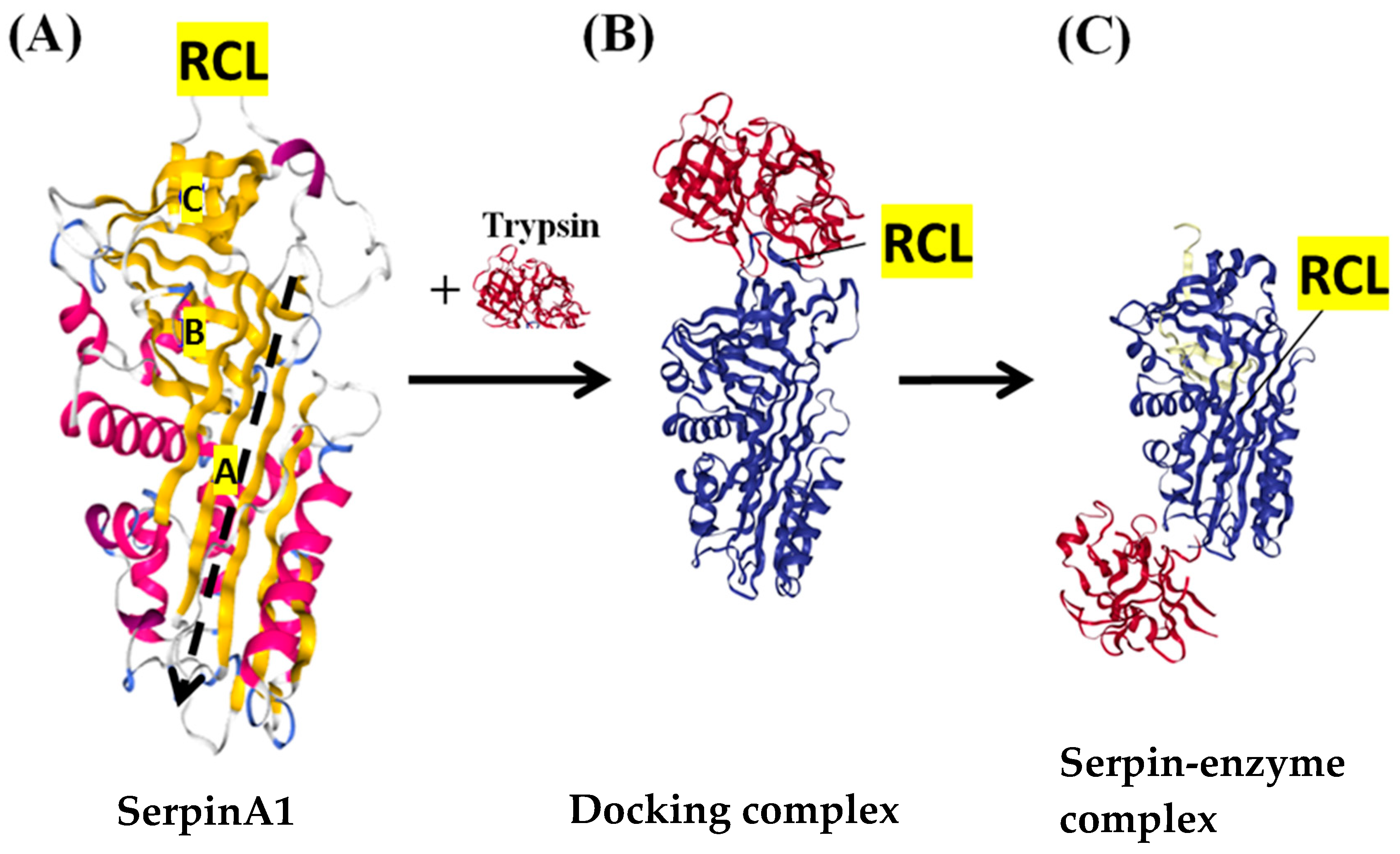
:1. Introduction
2. Preclinical and Clinical Attempts to Treat Type 1 Diabetes with Proteases
3. Preclinical and Clinical Attempts to Treat Type 1 Diabetes with Antiproteases
4. The Role of Proteases Inhibited by Clade B Serpins in Diabetes
5. The Role of Clade B Serpins in Diabetes
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Silverman, G.A.; Bird, P.I.; Carrell, R.W.; Church, F.C.; Coughlin, P.B.; Gettins, P.G.; Irving, J.A.; Lomas, D.A.; Luke, C.J.; Moyer, R.W.; et al. The serpins are an expanding superfamily of structurally similar but functionally diverse proteins. Evolution, mechanism of inhibition, novel functions, and a revised nomenclature. J. Biol. Chem. 2001, 276, 33293–33296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, R.H.P.; Zhang, Q.; McGowan, S.; Buckle, A.M.; Silverman, G.A.; Wong, W.; Rosado, C.J.; Langendorf, C.G.; Pike, N.P.; Bird, P.I.; et al. An overview of the serpin superfamily. Genome Biol. 2006, 7, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belorgey, D.; Hagglof, P.; Karlsson-Li, S.; Lomas, D.A. Protein misfolding and the serpinopathies. Prion 2007, 1, 15–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatto, M.; Iaccarino, L.; Ghirardello, A.; Bassi, N.; Pontisso, P.; Punzi, L.; Shoenfeld, Y.; Doria, A. Serpins, immunity and autoimmunity: Old molecules, new functions. Clin. Rev. Allergy Immunol. 2013, 45, 267–280. [Google Scholar] [CrossRef]
- Elliott, P.R.; Lomas, D.A.; Carrell, R.W.; Abrahams, J.P. Inhibitory conformation of the reactive loop of alpha 1-antitrypsin. Nat. Struct. Biol. 1996, 3, 676–681. [Google Scholar] [CrossRef]
- Carrell, R.W.; Owen, M.C. Plakalbumin, alpha 1-antitrypsin, antithrombin and the mechanism of inflammatory thrombosis. Nature 1985, 317, 730–732. [Google Scholar] [CrossRef]
- Gettins, P.G. Serpin structure, mechanism, and function. Chem. Rev. 2002, 102, 4751–4804. [Google Scholar] [CrossRef]
- Janciauskiene, S.M.; Bals, R.; Koczulla, R.; Vogelmeier, C.; Kohnlein, T.; Welte, T. The discovery of alpha1-antitrypsin and its role in health and disease. Respir. Med. 2011, 105, 1129–1139. [Google Scholar] [CrossRef] [Green Version]
- Lomas, D.A.; Evans, D.L.; Finch, J.T.; Carrell, R.W. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature 1992, 357, 605–607. [Google Scholar] [CrossRef]
- Elliott, P.R.; Bilton, D.; Lomas, D.A. Lung polymers in Z alpha1-antitrypsin deficiency-related emphysema. Am. J. Respir. Cell Mol. Biol. 1998, 18, 670–674. [Google Scholar] [CrossRef]
- Pappalardo, E.; Zingale, L.C.; Terlizzi, A.; Zanichelli, A.; Folcioni, A.; Cicardi, M. Mechanisms of C1-inhibitor deficiency. Immunobiology 2002, 205, 542–551. [Google Scholar] [CrossRef]
- Silverman, G.A.; Whisstock, J.C.; Askew, D.J.; Pak, S.C.; Luke, C.J.; Cataltepe, S.; Irving, J.A.; Bird, P.I. Human clade B serpins (ov-serpins) belong to a cohort of evolutionarily dispersed intracellular proteinase inhibitor clades that protect cells from promiscuous proteolysis. Cell. Mol. Life Sci. 2004, 61, 301–325. [Google Scholar] [PubMed]
- Izuhara, K.; Ohta, S.; Kanaji, S.; Shiraishi, H.; Arima, K. Recent progress in understanding the diversity of the human ov-serpin/clade B serpin family. Cell. Mol. Life Sci. 2008, 65, 2541–2553. [Google Scholar] [CrossRef]
- Bird, P.I. Regulation of pro-apoptotic leucocyte granule serine proteinases by intracellular serpins. Immunol. Cell Biol. 1999, 77, 47–57. [Google Scholar] [CrossRef]
- Pike, R.N.; Bottomley, S.P.; Irving, J.A.; Bird, P.I.; Whisstock, J.C. Serpins: Finely balanced conformational traps. IUBMB Life 2002, 54, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kaiserman, D.; Bird, P.I. Control of granzymes by serpins. Cell Death Differ. 2010, 17, 586–595. [Google Scholar] [CrossRef] [Green Version]
- Vidalino, L.; Doria, A.; Qarta, S.M.; Crescenzi, M.; Ruvoletto, M.; Frezzato, F.; Trentin, L.; Turato, C.; Parolin, M.C.; Ghirardello, A.; et al. SERPINB3 expression on B-cell surface in autoimmune diseases and hepatitis C virus-related chronic liver infection. Exp. Biol. Med. 2012, 237, 793–802. [Google Scholar] [CrossRef]
- El Ouaamari, A.; Dirice, E.; Gedeon, N.; Hu, J.; Zhou, J.-Y.; Shirakawa, J.; Hou, L.; Goodman, J.; Karampelias, C.; Qiang, G.; et al. SerpinB1 promotes pancreatic beta cell proliferation. Cell Metab. 2016, 23, 194–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czyzyk, J.; Henegariu, O.; Preston-Hurlburt, P.; Baldzizhar, R.; Fedorchuk, C.; Esplugues, E.; Bottomly, K.; Gorus, F.K.; Herold, K.; Flavell, R.A. Enhanced anti-serpin antibody activity inhibits autoimmune inflammation in type 1 diabetes. J. Immunol. 2012, 188, 6319–6327. [Google Scholar] [CrossRef] [Green Version]
- Kryvalap, Y.; Jiang, M.L.; Kryvalap, N.; Hendrickson, C.; Czyzyk, J. SerpinB13 antibodies promote β cell development and resistance to type 1 diabetes. Sci. Transl. Med. 2012, 13, eabf1787. [Google Scholar] [CrossRef]
- Shellenberger, T.D.; Mazumdar, A.; Henderson, Y.; Briggs, K.; Wang, M.; Chattopadhyay, C.; Jayakumar, A.; Frederick, M.; Clayman, G.L. Headpin: A serpin with endogenous and exogenous suppression of angiogenesis. Cancer Res. 2005, 65, 11501–11509. [Google Scholar] [CrossRef] [Green Version]
- Nakazawa, M.; Emancipator, S.N.; Lamm, M.E. Removal of glomerular immune complezes in passive serum sickness nephritis by treatment in vivo with proteolytic enzymes. Lab. Investig. 1986, 55, 551–556. [Google Scholar]
- White, R.B.; Lowrie, L.; Stork, J.E.; Iskandar, S.S.; Lamm, M.E.; Emancipator, S.N. Targeted enzyme therapy of experimental glomerulonephritis in rats. J. Clin. Investig. 1991, 87, 1819–1827. [Google Scholar] [CrossRef]
- Gaciong, Z.; Paczek, L.; Bojakowski, K.; Socha, K.; Wisniewski, M.; Heidland, A. Beneficial effect of proteases on allogrfat arterioscleorosis in a rat aurtic model. Nephrol. Dial. Transplant. 1996, 11, 987–989. [Google Scholar] [CrossRef] [PubMed]
- Targoni, O.S.; Tary-Lehmann, M.; Lehmann, P.V. Prevention of murine EAE by oral hydrolytic enyzme treatment. J. Autoimmunity 1999, 12, 191–198. [Google Scholar] [CrossRef] [Green Version]
- Wiest-Ladenburger, U.; Richter, W.; Moeller, P.; Boehm, B.O. Protease treament delays diabetes onset in diuabetes-prone nonobese diabetic (NOD) mice. Int. J. Immunother. 1997, 13, 75–78. [Google Scholar]
- Roep, B.O.; van den Engel, N.K.; van Halteren, A.G.S.; Duinkererken, G.; Martin, S. Modulation of autoimmunity to beta-cell antigens by proteases. Diabetologia 2002, 45, 686–692. [Google Scholar] [CrossRef] [Green Version]
- Baldzizhar, R.; Fedorchuk, C.; Iha, M.; Rathinam, C.; Henegariu, O.; Czyzyk, J. Anti-serpin antibody-mediated regulation of proteases in autpimmune diabetes. J. Biol. Chem. 2013, 288, 1612–1619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kempf, K.; Manzo, G.; Hanifi-Moghaddam, P.; Kappler, S.; Seissler, J.; Jaeger, C.; Boehm, B.; Roden, M.; Kolb, H.; Martin, S.; et al. Effect of combined oral proteases and flavonoid treatment in subjects at risk of Type 1 diabetes. Diabet. Med. 2009, 26, 1309–1310. [Google Scholar] [CrossRef]
- Ganrot, P.O.; Gydell, K.; Ekelund, H. Serum concentrations of α2-macroglobulin, haptoglobin and α1-anti-trypsin in diabetes mellitus. Acta Endocrinol. 1967, 55, 537–544. [Google Scholar] [CrossRef]
- Tsianos, E.B.; Stahakis, N.E. Soluble fibrin complexes and fibrionegn heterogeneity in diabetes mellitus. Thromb. Haemost. 1980, 44, 130–134. [Google Scholar]
- Dornan, T.L.; Rhymes, I.L.; Cederholm-Williams, S.A.; Rizza, C.R.; Pepys, M.B.; Bron, A.J.; Turner, R.C. Plasma haemostatic factors and diabetic retnopathy. Eur. J. Clin. Investig. 1983, 13, 231–235. [Google Scholar] [CrossRef]
- Finotti, P.; Piccoli, A.; Carraro, P. Alteration of plasma protreinase-antiproteinase system in type 1 diabetic patients. Influence of sex and relationship with metabolic control. Diabetes Res. Clin. Pract. 1992, 18, 35–42. [Google Scholar] [CrossRef]
- Sandler, M.; Stewart, R.I.; Gemperli, B.M.; Hanekom, C.; Kuhn, S.H. Serum α1-protease inhibitor in diabetes mellitus: Reduced concentration and impaired activity. Diabetes Res. Clin. Pract. 1988, 5, 249–255. [Google Scholar] [CrossRef]
- Song, S.; Goudy, K.; Campbell-Thompson, M.; Wasserfall, C.; Scott-Jorgensen, M.; Wang, J.; Tang, Q.; Crawford, J.M.; Ellis, T.M.; Atkinson, M.A. Recombinant adeno-associated virus-mediated alpha-1 antitrypson gene therapy prevents type 1 diabetes in NOD mice. Gene Ther. 2004, 11, 181–186. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Tang, M.; Wasserfall, C.; Kou, Z.; Campbell-Thompson, M.; Gardemann, T.; Crawford, J.; Atkinson, M.; Song, S. α1-Antitrypsin gene therapy modulates cellular immunity and efficienctly prevnets type 1diabetes in nonobese diabetic mice. Hum. Gene Ther. 2006, 17, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Koulmanda, M.; Bhasin, M.; Hoffman, L.; Fan, Z.; Qipo, A.; Shi, H.; Nonner-Weir, S.; Putheti, P.; Degauque, N.; Libermann, T.A.; et al. Curative and β cell regenerative effects of α1-antitrypsin treatment in autoimmune diabetic NOD mice. Proc. Natl. Acad. Sci. USA 2008, 105, 16242–16247. [Google Scholar] [CrossRef] [Green Version]
- Guttman, O.; Yossef, R.; Freixo-Lima, G.; Rider, P.; Porgador, A.; Lewis, E.C. Alpha1-Antitrypsin modifies general NK cell interactions with dendritic cells and specific interactions with istel beta-cells in favor of protectin from autoimmune diabetes. Immunology 2014, 144, 530–539. [Google Scholar] [CrossRef]
- Gottlieb, P.A.; Alkanani, A.K.; Michels, A.W.; Lewis, E.C.; Shapiro, L.; Dinarello, C.A.; Zipris, D. alpha1-Antitrypsin therapy downregulates toll-like receptor-induced IL-1 responses in monocytes and myeloid dendritic cells and may improve islet function in recently diagnosed patinets with type 1 diabetes. J. Clin. Endocrinol. Metab. 2014, 99, E1418–E1426. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Lu, Y.; Campbell-Thompson, M.; Spencer, T.; Wasserfall, C.; Atkinson, M.; Song, S. α1-Antitrypsin protects β-cells from apoptosis. Diabetes 2007, 56, 1316–1323. [Google Scholar] [CrossRef] [Green Version]
- Lewis, E.C.; Shapiro, L.; Bowers, O.J.; Dinarello, C.A. α1-Antitrypsin monotherapy prolongs islet allograft survival in mice. Proc. Natl. Acad. Sci. USA 2005, 102, 12153–12158. [Google Scholar] [CrossRef] [Green Version]
- Lewis, E.X.; Mizrahi, M.; Toledano, M.; DeFelice, N.; Wright, J.L.; Churg, A.; Shapiro, L.; Dinarello, C.A. α1-Antitrypsin monotherapy induces immune tolerance during islet allogrfat transplantation in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 16236–16241. [Google Scholar] [CrossRef] [Green Version]
- Koulmanda, M.; Bhasin, M.; Fan, Z.; Hanidziar, D.; Goel, N.; Putheti, P.; Movahedi, M.; Libermann, T.; Strom, T.B. Alpha 1-antitrypsin reduces inflammation and enhnaces mouse pancreatic iselt transplant survival. Proc. Natl. Acad. Sci. USA 2012, 109, 15443–15548. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Liao, Y.-T.; Jian, Y.-Q.; Zhang, X.-D.; Wei, P.; Qi, H.; Deng, C.-Y.; Li, F.-R. Alpha-1-antitrypsin for the improvement of autopimmunity and alogrfat rejection in beta cell transplantation. Immunol. Lett. 2013, 150, 61–68. [Google Scholar] [CrossRef]
- Rachmiel, M.; Strauss, P.; Dror, N.; Benzaquen, H.; Horesh, O.; Tov, N.; Weintrob, N.; Landau, Z.; Ben-Ami, M.; Haim, A. Alpha-1 antitrypsin therapy is safe and well tolerated in children and adolescents with recent onset type 1 diabetes mellitus. Pediatr. Diabetes 2016, 17, 351–359. [Google Scholar] [CrossRef]
- Lebenthal, Y.; Brener, A.; Hershkovitz, E.; Shehadeh, N.; Shalitin, S.; Lewis, E.C.; Elias, D.; Haim, A.; Barash, G.; Loewenthal, N.; et al. A phase II, double-blind, randomized, placebo-controlled, multicenter study evaluation the efficacy and safety of alpha-1 antitrypsin (AAT) (Glassai) in the treatment of recent-onset type 1 diabetes. Int. J. Mol. Sci. 2019, 20, 6032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, T.; Roth, W.; Wong, P.; Nelson, A.; Farr, A.; Deussing, J.; Villadangos, J.A.; Ploegh, H.; Peters, C.; Rudensky, A.Y. Cathepsin L: Critical role in Ii degradation and CD4 T cell selection in the thymus. Science 1998, 280, 450–453. [Google Scholar] [CrossRef] [PubMed]
- Maehr, R.; Mintern, J.D.; Herman, A.E.; Lennon-Dumemil, A.-M.; Mathis, D.; Benoist, C.; Ploegh, H.L. Cathepsin L is essential for onset of autimmune diabetes in Nod mice. J. Clin. Investig. 2005, 115, 2934–2943. [Google Scholar] [CrossRef] [Green Version]
- Hsing, L.C.; Kirk, E.A.; McMillen, T.S.; Hsiao, S.-H.; Caldwell, M.; Houston, B.; Rudensky, A.Y.; JeBoeuf, R.C. Role for cathepsin S, L, and B in insulitis and diabetes in the NOD mouse. J. Autoimmun. 2010, 34, 96–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korpos, E.; Kadri, N.; Kappelhoff, R.; Wegner, J.; Overall, C.M.; Weber, E.; Holmberg, D.; Cardell, S.; Sorokin, L. The peri-islet basmenet membrane, a barrier to infiltrating leukocytes in type 1 diabetes in mouse and human. Diabetes 2013, 62, 531–542. [Google Scholar] [CrossRef] [Green Version]
- Yamada, A.; Ishimaru, N.; Arakaki, R.; Katunuma, N.; Hayashi, Y. Cathepsin L inhibition prevents murine autoimmune diabeets via suppression of CD8+ T cell activity. PLoS ONE 2010, 5, e12894. [Google Scholar] [CrossRef] [Green Version]
- Ishimaru, N.; Arakaki, R.; Katunuma, N.; Hayashi, Y. Critical role of cathepsin-inhibitors for autoantigen processing and autoimmunity. Adv. Enzyme Regul. 2003, 44, 309–320. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Cooley, J.; Swanson, R.; Ong, P.C.; Pike, R.N.; Bogyo, M.; Olson, S.T.; Remold-O’Donnell, E. The protease cathepsin L regulates Th17 cell differentiation. J. Autoimmun. 2015, 65, 56–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedoya, S.K.; Lam, B.; Lau, K.; Larkin, J., 3rd. Th17 cells in immunity ad autoimmunity. Clin. Dev. Immunol. 2013, 2013, 986789. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Schwartz, J.A.; Sandrock, C.; Bellemore, S.M.; Nikoopour, E. Modulation of autoimmune duseases by interleukin (IL)-17 producing regulatory T helper (Th17) cells. Indian J. Med. Res. 2013, 138, 591–594. [Google Scholar]
- Fu, W.; Wojtkiewicz, G.; Weissleder, R.; Benoist, C.; Mathis, D. Early window of diabetes determinism in NOD mice, dependent on the complement receptor CRIg, identified by noninvasive imaging. Nat. Immunol. 2012, 13, 361–368. [Google Scholar] [CrossRef]
- Lo, C.-W.; Kryvalap, Y.; Sheu, T.; Chang, C.-H.; Czyzyk, J. Cellular proliferation in mouse and human pancreatic islets is regulated by serpin B13 inhibition and downstream targeting of E-cadherin by cathepsin L. Diabetologia 2019, 62, 822–834. [Google Scholar] [CrossRef] [Green Version]
- Remold-O’Donnell, E.; Chin, J.; Alberts, M. Sequence and molecular characterization of human monocyte/neutrophil elastase inhibitor. Proc. Natl. Acad. Sci. USA 1992, 89, 5635–5639. [Google Scholar] [CrossRef] [Green Version]
- Benarafa, C.; Priebe, G.P.; Remold-O’Donnell, E. The neutrophil serine protease inhibitor serpinb1 preserves lung defense functions in Pseudomona aeruginosa infection. J. Exp. Med. 2007, 204, 1901–1909. [Google Scholar] [CrossRef] [Green Version]
- Takebayashi, K.; Hara, K.; Terasawa, T.; Naruse, R.; Suetsugu, M.; Tsuchiya, T.; Inukai, T. Circulating serpinB1 levels and clinical features in patients with type 2 diabetes. BMJ Open Diabetes Res. Care 2016, 4, e000274. [Google Scholar] [CrossRef]
- Hostelley, T.L.; Nesmith, J.E.; Larkin, E.; Jones, A.; Boyes, D.; Leitch, C.C.; Fontaine, M.; Zaghloul, N.A. Exocrine pancreas proteases regulate β-cell proliferation in zebrafish ciliopathy models and in murine systems. Biol. Open 2021, 10, bio046839. [Google Scholar] [CrossRef]
- Kassem, D.H.; Adel, A.; Sayed, G.H.; Kamal, M.M. A novel SERPINB1 single-nucleotide polymorphism associated with glycemic control and β-cel functin in Egyptian type 2 diabetic patients. Front. Endocrinol. 2020, 11, 450. [Google Scholar] [CrossRef] [PubMed]
- Sprecher, C.A.; Morgenstern, K.A.; Mathewes, S.; Dahlen, J.R.; Schrader, S.K.; Foster, D.C.; Kisiel, W. Molecular cloning, expression, and partial characterization of two novel members of the ovalbumin family of serine proteinase inhibitors. J. Biol. Chem. 1995, 270, 29854–29861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strik, M.C.; Bladergroen, B.A.; Wouters, D.; Kisiel, W.; Hooijberg, J.H.; Verlaan, A.R.; Hordijk, P.L.; Schneider, P.; Hack, C.E.; Kummer, J.A. Distribution of the human intracellular serpin protease inhibitor 8 in human tissues. J. Hitochem. Cytochem. 2002, 50, 1443–1453. [Google Scholar] [CrossRef] [Green Version]
- De Koning, P.J.A.; Bovenschen, N.; Broekhuizen, R.; Lips, C.J.M.; Kummer, J.A. Serine protease inhibitor 8 is a novel immunohistochemical marker for neuroendocrine tumors of the pancreas. Pancreas 2009, 38, 461–467. [Google Scholar] [CrossRef]
- Dahlen, J.R.; Jean, F.; Thomas, G.; Foster, D.C.; Kisiel, W. Inhibition of soluble recombinant furin by human proteinase inhibitor 8. J. Biol. Chem. 1998, 273, 1851–1854. [Google Scholar] [CrossRef] [Green Version]
- Kayo, T.; Sawada, Y.; Suda, M.; Konda, Y.; Izumi, T.; Tanak, S.; Shibata, H.; Takeuchi, T. Proprotein-processing endoprotease furin controls growth of pancreatic beta-cells. Diabetes 1996, 46, 53442–53450. [Google Scholar] [CrossRef]
- Louagie, E.; Taylor, N.A.; Flamez, D.; Roebroek, A.J.; Bright, N.A.; Meulemans, S.; Quintens, R.; Herrera, P.J.; Schuit, F.; Van de Ven, W.J.; et al. Role of furin in granular acidification in the endocrine pancreas: Identification of the V-ATPase subunit Ac45 as a candidate substrate. Proc. Natl. Acad. Sci. USA 2008, 105, 12319–12324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppola, I.; Brouwers, B.; Meulemans, S.; Ramos-Molina, B.; Creemers, J.W.M. Differential effects of furin deficincy on insulin receptor processing and glucose control in liver and pancreas β cells of mice. Int. J. Mol. Sci. 2021, 22, 6344. [Google Scholar] [CrossRef]
- Brouwers, B.; Coppola, I.; Vints, K.; Dislich, B.; Jouvet, N.; Van Lommel, L.; Segers, C.; Gounko, N.V.; Thorrez, L.; Schuit, F.; et al. Loss of furin in β-cells induces an mTORC1-ATF4 anabolic pathway that leads to β-cell dysfucntion. Diabetes 2021, 70, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Abts, H.F.; Wells, T.; Mirmohammadsadegh, A.; Kohrer, K.; Michel, H.; Ruzicka, T. Cloning and characterization of hurpin (protease inhibitor 13): A new skin-specific, UV-repressible serine proteinase inhibitor of the ovalbumin serpin family. J. Mol. Biol. 1999, 293, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Wells, T.; Sun, J.; Irving, J.A.; Blum, R.; Smith, A.I.; Whisstock, J.C.; Pike, R.N.; von Mikecz, A.; Ruzicka, T.; Bird, P.I.; et al. Hurpin is a selective inhibitor of lysosomal cathepsin L and protects keratinocytes from ultraviolet-induced apoptosis. Biochemistry 2003, 42, 7381–7389. [Google Scholar] [CrossRef] [PubMed]
- Kryvalap, Y.; Lo, C.-W.; Manuylova, E.; Baldzizhar, R.; Jospe, N.; Czyzyk, J. Antibody response to serpin B13 induces adaptive changes in mouse pancreatic islets and slows down the decline in the residual beta-cell function in children with recent-onset type 1 diabetes mellitus. J. Biol. Chem. 2016, 291, 266–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| Serpin | Alternative Name | Inhibitory Function | Potential Protein Targets |
|---|---|---|---|
| SerpinB1 | PI-2, Neutrophil elastase inhibitor | + | Elastase, Chymotrypsin, Cathepsin G, Protease 3 |
| SerpinB2 | PAI-2 | + | uPA, tPA |
| SerpinB3 | SCCA1 | + | Papain, Cathepsin L, K, S |
| SerpinB4 | SCCA2, Leupin | + | Cathepsin G, Chymase |
| SerpinB5 | Maspin | − | - |
| SerpinB6 | CAP1, PI6 | + | Thrombin, Trypsin, Factor Xa, Cathepsin G, u-PA |
| SerpinB7 | Megsin | + | Plasmin |
| SerpinB8 | CAP2, PI8 | + | Furin, Trypsin, Factor Xa, Thrombin, Chymotrypsin, Subtilisin A |
| SerpinB9 | CAP3, PI9 | + | Granzyme B, Subtilisin A, Caspase 1, 4, 8, 10 |
| SerpinB10 | Bomapin, PI10 | + | Thrombin, Trypsin |
| SerpinB11 | Epipin | - | - |
| SerpinB12 | Yukopin | + | Trypsin, Plasmin |
| SerpinB13 | Headpin, Hurpin | + | Cathepsin K, L, V |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kryvalap, Y.; Czyzyk, J. The Role of Proteases and Serpin Protease Inhibitors in β-Cell Biology and Diabetes. Biomolecules 2022, 12, 67. https://doi.org/10.3390/biom12010067
Kryvalap Y, Czyzyk J. The Role of Proteases and Serpin Protease Inhibitors in β-Cell Biology and Diabetes. Biomolecules. 2022; 12(1):67. https://doi.org/10.3390/biom12010067
Chicago/Turabian StyleKryvalap, Yury, and Jan Czyzyk. 2022. "The Role of Proteases and Serpin Protease Inhibitors in β-Cell Biology and Diabetes" Biomolecules 12, no. 1: 67. https://doi.org/10.3390/biom12010067