
Calcium Signaling Pathway Is Involved in the Shedding of ACE2 Catalytic Ectodomain: New Insights for Clinical and Therapeutic Applications of ACE2 for COVID-19

 ,
, {kind=link}
Abstract
1. Introduction
2. ACE2 Molecular Structure
3. The ACE2 Catalytic Ectodomain Undergoes Shedding
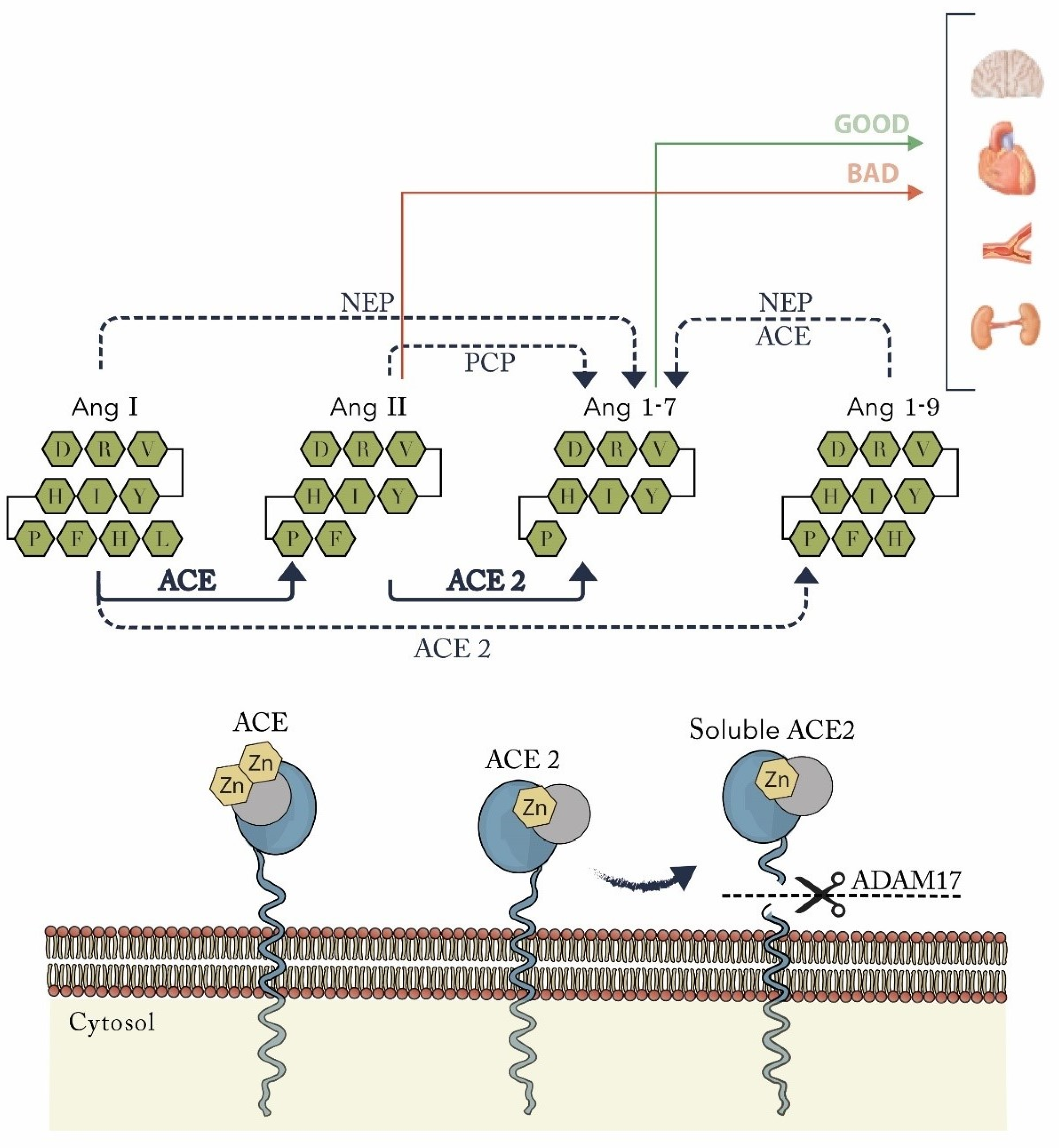
4. ACE2 and Its Unique Cleavage of Key Vasoactive Peptides: The Essential Role for the Renin-Angiotensin System
5. The Soluble ACE2 Plasma Activity and Its Association with Cardiac Remodeling, Endothelial Dysfunction and Prognosis in Heart Failure and Cardiovascular Disease
6. COVID-19 and Its Association with Thromboembolic Events
7. ACE2 Plays an Important Role in Thrombogenic and Inflammatory Activity
8. The Calcium Signaling Pathway Is Involved in ACE2 Release: The Role of Vitamin D in COVID-19?
9. ACE2 Shedding and ACE2 Cell Expression as Prognostic Markers in COVID-19
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AC | anticoagulation |
| ACE | angiotensin-converting enzyme |
| ACE2 | angiotensin-converting enzyme-2 |
| ACEIs | angiotensin-converting enzyme inhibitors |
| ACTIV-4 | anti-thrombotic for adults hospitalized with COVID-19 |
| ADAM17 | a disintegrin and metalloproteinase domain-containing protein 17 |
| AF | atrial fibrillation |
| AI | anemia of inflammation |
| ARDS | acute respiratory distress syndrome |
| AT | antithrombin |
| ATTACC | antithrombotic therapy to ameliorate complications of COVID-19 |
| AT1 | angiotensin I |
| AT1-5 | angiotensin 1-5 |
| AT1-7 | angiotensin 1-7 |
| AT1-9 | angiotensin 1-9 |
| AT2 | angiotensin II |
| AT1R | angiotensin I receptor |
| BBM | duodenal brush border membranes |
| CAD | coronary artery disease |
| CaM | calmodulin |
| CI | confidence interval |
| CRP | c-reactive protein |
| DM | diabetes |
| DIC | disseminated intravascular coagulation |
| DOACs | direct oral anticoagulants |
| DVT | deep vein thrombosis |
| ED | emergency department |
| EDTA | ethylenediaminetetraacetic acid |
| Fc | fragment crystallizable region |
| GPVI | platelet membrane glycoprotein |
| HF | heart failure |
| HR | hazard ratio |
| hrsACE2 | human recombinant soluble ACE2 |
| ICU | intensive care unit |
| IFN-α | interferon-alpha |
| IL | interleukin |
| LA | left atrial |
| LDL | low-density lipoprotein |
| LMWH | low molecular weight heparin |
| LVEF | left ventricular ejection fraction |
| MACE | major adverse cardiovascular events |
| NOS | nitric oxide synthase |
| NRP1 | neuropilin-1 |
| NYHA | New York Heart Association |
| OR | odds ratio |
| PE | pulmonary embolism |
| PT | prothrombin time |
| PURE | Prospective Urban Rural Epidemiology |
| RBD | receptor binding domain |
| REMAP-CAP | randomized, embedded, multifactorial adaptive platform trial for community-acquired pneumonia |
| SARS-CoV-1 | severe acute respiratory syndrome coronavirus |
| SARS-CoV-2 | severe acute respiratory syndrome coronavirus 2 |
| SARSr-CoVs | severe acute respiratory syndrome-related coronaviruses |
| SEMA3A | semaphorin 3A |
| TMPRSS2 | transmembrane serine protease 2 |
| TNF-α | tumor necrosis factor-alpha |
| UFH | unfractionated heparin |
| VTE | venous thromboembolism |
References
- Zhu, N.; Zhang, D.; Wenling, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from patients with Pneumonia in China. N. Engl. J. Med. 2019, 382, 727–733. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCov and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar]
- Lu, G.; Liu, D. SARS-like Virus in the Middle East: A Truly Bat-Related Coronavirus Causing Human Diseases. Protein Cell 2020, 3, 803–805. [Google Scholar] [CrossRef] [PubMed][Green Version]
- World Health Organization. Rolling Updates on Coronavirus Disease (COVID-19). 2020. Available online: https://www.who.int/emergencies/diseases/ (accessed on 28 February 2021).
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Li, F. Receptor recognition mechanisms of coronaviruses: A decade of structural studies. J. Virol. 2015, 89, 1954–1964. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.; et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk factors associated with acude respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef]
- Yin, S.; Huang, M.; Li, D.; Tang, N. Difference of coagulation features between severe pneumonia induced by SARS-CoV2 and non-SARS-CoV2. J. Thromb. Thrombolysis 2021, 51, 1107–1110. [Google Scholar] [CrossRef]
- Tan, L.; Wang, Q.; Zhang, D.; Ding, J.; Huang, Q.; Tang, Y.Q.; Wang, Q.; Miao, H. Lymphopenia predicts disease severity of COVID-19: A descriptive and predictive study. Signal Transduct. Target. Ther. 2020, 5, 33. [Google Scholar] [CrossRef] [PubMed]
- Gandini, O.; Criniti, A.; Ballesio, L.; Giglio, S.; Galardo, G.; Gianni, W.; Santoro, L.; Angeloni, A.; Lubrano, C. Serum Ferritin is an independent risk factor for Acute Respiratory Distress Syndrome in COVID-19. J. Infect. 2020, 81, 979–997. [Google Scholar] [CrossRef]
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A human homolog of angiotensin-converting enzyme: Cloning and function- al expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243. [Google Scholar] [CrossRef]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-convert- ing enzyme-related carboxypeptidase (ACE2) converts angiotensin I to an- giotensin 1-9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef]
- Towler, P.; Staker, B.; Prasad, S.G.; Menon, S.; Tang, J.; Parsons, T.; Ryan, D.; Fisher, M.; Williams, D.; Dales, N.A.; et al. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J. Biol. Chem. 2004, 279, 17996–18007. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Travanty, E.A.; Oko, L.; Edeen, K.; Berglund, A.; Wang, J.; Ito, Y.; Holmes, K.V.; Mason, R.J. Innate Immune Response of Human Alveolar Type II Cells Infected with Severe Acute Respiratory Syndrome–Coronavirus. Am. J. Respir. Cell Mol. Biol. 2013, 48, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.P.; Look, D.C.; Hickey, M.; Shi, L.; Pewe, L.; Netland, J.; Farzan, M.; Wohlford-Lenane, C.; Perlman, S.; McCray, P.B., Jr. Infection of human airway epithelia by SARS coronavirus is associated with ACE2 expression and localization. Adv. Exp. Med. Biol. 2006, 581, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Iwata, M.; Silva Enciso, J.E.; Greenberg, B.H. Selective and specific regulation of ectodomain shedding of angiotensin-converting enzyme 2 by tumor necrosis factor alpha-converting enzyme. Am. J. Physiol. Cell Physiol. 2009, 297, C1318–C1329. [Google Scholar] [CrossRef]
- Vickers, C.; Hales, P.; Kaushik, V.; Dick, L.; Gavin, J.; Tang, J.; Godbout, K.; Parsons, T.; Baronas, E.; Hsieh, F.; et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J. Biol. Chem. 2002, 277, 14838–14843. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Perlot, T.; Rehman, A.; Trichereau, J.; Ishiguro, H.; Paolino, M.; Sigl, V.; Hanada, T.; Hanada, R.; Lipinski, S.; et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 2012, 487, 477–481. [Google Scholar] [CrossRef]
- Klempin, F.; Mosienko, V.; Matthes, S.; Villela, D.C.; Todiras, M.; Penninger, J.M.; Bader, M.; Santos, R.A.S.; Alenina, N. Depletion of angiotensin-converting enzyme 2 reduces brain serotonin and impairs the running-induced neurogenic response. Cell Mol. Life Sci. 2018, 75, 3625–3634. [Google Scholar] [CrossRef]
- Basu, R.; Poglitsch, M.; Yogasundaram, H.; Thomas, J.; Rowe BHOudit, G.Y. Roles of angiotensin peptides and recombinant human ACE2 in heart failure. J. Am. Coll. Cardiol. 2017, 69, 805–819. [Google Scholar] [CrossRef]
- Wang, K.; Basu, R.; Poglitsch, M.; Bakal, J.A.; Oudit, G.Y. Elevated Angiotensin 1-7/Angiotensin II Ratio Predicts Favorable Outcomes in Patients With Heart Failure. Circ. Heart Fail. 2020, 13, e006939. [Google Scholar] [CrossRef] [PubMed]
- Haschke, M.; Schuster, M.; Poglitsch, M.; Loibner, H.; Salzberg, M.; Bruggisser, M.; Penninger, J.; Krahenbuhl, S. Pharmacokinetics and pharmacodynamics of recombinant human angiotensin-converting enzyme 2 in healthy human subjects. Clin. Pharmacokinet. 2013, 52, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Kokkonen, J.O.; Lindstedt, K.A.; Kovanen, P.T. Role for chymase in heart failure: Angiotensin II-dependent or -independent mechanisms? Circulation 2003, 107, 2522–2524. [Google Scholar] [CrossRef][Green Version]
- Epelman, S.; Tang, W.H.; Chen, S.Y.; Lente, F.V.; Francis, G.S.; Sen, S. Detection of soluble angiotensin-converting enzyme 2 in heart failure: Insights into the endogenous counter-regulatory pathway of the renin-angiotensin-aldosterone system. J. Am. Coll. Cardiol. 2008, 52, 750–754. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.B.; Zhong, J.C.; Grant, M.B.; Oudit, G.Y. Role of the ACE2/Angio 1-7 axis of the Renin-Angiotensin System in Heart Failure. Circ. Res. 2016, 118, 1313–1326. [Google Scholar] [CrossRef] [PubMed]
- Epelman, S.; Shrestha, K.; Troughton, R.W.; Francis, G.S.; Sen, S.; Klein, A.L.; Tang, W.H.W. Soluble Angiotensin-Converting Enzyme 2 in Human Heart Failure: Relation With Myocardial Function and Clinical Outcomes. J. Card. Fail. 2009, 15, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Schuster, A.; Borowski, A.G.; Thakur, A.; Li, L.; Tang, W.H.W. Soluble angiotensin converting enzyme 2 levels in chronic heart failure is associated with decreased exercise capacity and increased oxidative stress-mediated endotelial dysfunction. Transl. Res. 2019, 212, 8088. [Google Scholar] [CrossRef]
- Soro-Paavonen, A.; Gordin, D.; Forsblom, C.; Rosengard-Barlund, M.; Waden, J.; Thorn, L.; Sandholm, N.; Thormas, M.C.; Groop, P. Circulating ACE2 activity is increased in patients with type 1 diabetes and vascular complications. J. Hypertens. 2012, 30, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Pérez, J.T.; Riera, M.; Bosch, X.; Caralt, T.M.; Perea, R.J.; Pascual, J.; Soler, M.J. Role of circulating angiotensin coverting enzyme 2 in left ventricular remodeling following myocardial infarction: A prospective controlled study. PLoS ONE 2013, 8, e61695. [Google Scholar] [CrossRef]
- Walters, T.E.; Kalman, J.M.; Patel, S.K.; Mearns, M.; Velkoska, E.; Burrell, L.M. Angiotensin converting enzyme 2 activity and human atrial fibrillation: Increased plasma angiotensin converting enzyme 2 activity is associated with atrial fibrillation and more advanced left atrial structural remodelling. Europace 2017, 19, 1280–1287. [Google Scholar] [CrossRef]
- Ramchand, J.; Patel, S.K.; Kearney, L.G.; Matalanis, G.; Farouque, O.; Srivastava, P.M.; Burrell, L.M. Plasma ACE2 activity predicts mortality in aortic stenosis and is associated with severe myocardial fibrosis. JACC Cardiovasc Imaging 2020, 13, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Narula, S.; Yusuf, S.; Chong, M.; Ramasundarahettige, C.; Rangarajan, S.; Bangdiwala, S.I.; van Eikels, M.; Leineweber, K.; Wu, A.; Pigeyre, M.; et al. Plasma ACE2 and risk of death or cardiometabolic diseases: A case-cohort analysis. Lancet 2020, 396, 968–976. [Google Scholar] [CrossRef]
- Plotkin, A.; Weaver, F.A.; Abou-Zamzam, A.; Malas, M.B.; Lee, J.T.; Han, S.M.; Ding, L.; Magee, G.A. Thromboembolism risk of COVID-19 is high and associated with a higher risk of mortality: A systematic review and meta-analysis. EClinicalMedicine 2020, 29, 100639. [Google Scholar] [CrossRef]
- Pellegrini, J.A.S.; Rech, T.H.; Schwarz, P.; de Oliveira, A.C.T.; Vieceli, T.; Moraes, R.B.; Sekine, L.; Viana, M.V. Incidence of venous thromboembolism among patients with severe COVID-19 requiring mechanical ventilation compared to other causes of respiratory failure: A prospective cohort study. J. Thromb Thrombolysis 2021, 52, 482–492. [Google Scholar] [CrossRef]
- Miró, Ò.; Jiménez, S.; Mebazaa, A.; Freund, Y.; Burillo-Putze, G.; Martín, A.; Martín-Sánchez, F.J.; García-Lamberechts, E.J.; Alquézar-Arbé, A.; Jacob, J.; et al. Pulmonary embolism in patients with COVID-19: Incidence, risk factors, clinical characteristics, and outcome. Eur. Heart J. 2021, 42, 3127–3142. [Google Scholar] [CrossRef] [PubMed]
- Iba, T.; Levy, J.H.; Warkentin, T.E.; Thachil, J.; van der Poll, T.; Levi, M. Scientific and Standardization Committee on DIC, and the Scientific and Standardization Committee on Perioperative and Critical Care of the International Society on Thrombosis and Haemostasis. Diagnosis and management of sepsis-induced coagulopathy and disseminated intravascular coagulation. J. Thromb. Haemost. 2019, 17, 1989–1994. [Google Scholar] [CrossRef]
- McBane, R.D., 2nd; Torres Roldan, V.D.; Niven, A.S.; Pruthi, R.K.; Franco, P.M.; Linderbaum, J.A.; Casanegra, A.I.; Oyen, L.J.; Houghton, D.E.; Marshall, A.L.; et al. Anticoagulation in COVID-19: A Systematic Review, Meta-analysis, and Rapid Guidance From Mayo Clinic. Mayo Clin. Proc. 2020, 95, 2467–2486. [Google Scholar] [CrossRef] [PubMed]
- Nadkarni, G.N.; Lala, A.; Bagiella, E.; Chang, H.L.; Moreno, P.R.; Pujadas, E.; Arvind, V.; Bose, S.; Charney, A.W.; Chen, M.D.; et al. Anticoagulation, Bleeding, Mortality, and Pathology in Hospitalized Patients With COVID-19. J. Am. Coll. Cardiol. 2020, 76, 1815–1826. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.S.; Connors, J.M. Anticoagulation in COVID-19: Reaction to the ACTION trial. Lancet 2021, 397, 2226–2228. [Google Scholar] [CrossRef]
- Fraga-Silva, R.A.; Sorg, B.; Wankhede, M.; Dedeugd, C.; Jun, J.Y.; Baker, M.B.; Li, Y.; Castellano, R.K.; Katovich, M.J.; Raizada, M.K.; et al. ACE2 Activation promotes antithrombotic activity. Mol. Med. 2010, 16, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Fraga-Silva, R.A.; Pinheiro, S.V.; Goncalves, A.C.; Alenina, N.; Bader, M.; Santos, R.A. The antithrombotic effect of angiotensin-(1-7) involves mas-mediated NO release from platelets. Mol. Med. 2008, 14, 28–35. [Google Scholar] [CrossRef]
- Senchenkova, E.Y.; Russell, J.; Almeida-Paula, L.D.; Harding, J.W.; Granger, D.N. Angiotensin II-mediated microvascular thrombosis. Hypertension 2010, 56, 1089–1095. [Google Scholar] [CrossRef]
- Sampaio, W.O.; Souza dos Santos, R.A.; Faria-Silva, R.; da Mata Machado, L.T.; Schiffrin, E.L.; Touyz, R.M. Angiotensin-(1-7) through receptor mas mediates endothelial nitric oxide synthase activation via Akt-dependent pathways. Hypertension 2007, 49, 185–192. [Google Scholar] [CrossRef]
- Liu, P.P.; Blet, A.; Smyth, D.; Li, H. The Science Underlying COVID-19: Implications for the Cardiovascular System. Circulation 2020, 142, 68–78. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef]
- Khan, A.; Beenthin, C.; Zeno, B.; Albertson, T.E.; Boyd, J.; Christie, J.D.; Hall, R.; Poirier, G.; Ronco, J.J.; Tidswell, M.; et al. A pilot clinical trial of recombinant human angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Crit. Care 2017, 21, 234. [Google Scholar] [CrossRef]
- Verdecchia, P.; Cavallini, C.; Spanevello, A.; Angeli, F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 2020, 76, 14–20. [Google Scholar] [CrossRef]
- Grasselli, G.; Zangrillo, A.; Zanella, A.; Antonelli, M.; Cabrini, L.; Castelli, A.; Cereda, D.; Coluccello, A.; Foti, G.; Fumagalli, R.; et al. COVID-19 Lombardy ICU Network. Baseline Characteristics and Outcomes of 1591 Patients Infected With SARS-CoV-2 Admitted to ICUs of the Lombardy Region, Italy. JAMA 2020, 323, 1574–1581. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Codo, A.C.; Davanzo, G.G.; Monteiro, L.B.; de Souza, G.F.; Muraro, S.P.; Virgilio-da-Silva, J.V.; Prodonoff, J.S.; Carregari, V.C.; de Biagi Junior, C.A.O.; Crunfli, F.; et al. Elevated Glucose Levels Favor SARS-CoV-2 Infection and Monocyte Response through a HIF-1α/Glycolysis-Dependent Axis. Cell Metab. 2020, 32, 437–446.e5. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Zhang, J.; Schiavon, C.R.; He, M.; Chen, L.; Shen, H.; Zhang, Y.; Yin, Q.; Cho, Y.; Andrade, L.; et al. SARS-CoV-2 Spike Protein Impairs Endothelial Function via Downregulation of ACE 2. Circ. Res. 2021, 128, 1323–1326. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, M.; Saito, J.; Zhao, H.; Sakamoto, A.; Hirota, K.; Ma, D. Inflammation Triggered by SARS-CoV-2 and ACE2 Augment Drives Multiple Organ Failure of Severe COVID-19: Molecular Mechanisms and Implications. Inflammation 2021, 44, 13–34. [Google Scholar] [CrossRef]
- Leisman, D.E.; Ronner, L.; Pinotti, R.; Taylor, M.D.; Sinha, P.; Calfee, C.S.; Hirayama, A.V.; Mastroianni, F.; Turtle, C.J.; Harhay, M.O.; et al. Cytokine elevation in severe and critical COVID-19: A rapid systematic review, meta-analysis, and comparison with other inflammatory syndromes. Lancet Respir. Med. 2020, 8, 1233–1244. [Google Scholar] [CrossRef]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef]
- Lambert, D.W.; Clarke, N.E.; Hooper, N.M.; Turner, A.J. Calmodulin interacts with angiotensin-converting enzyme-2 (ACE2) and inhibits shedding of its ectodomain. FEBS Lett. 2008, 582, 385–390. [Google Scholar] [CrossRef]
- Kahn, J.; Walcheck, B.; Migaki, G.I.; Jutila, M.A.; Kishimoto, T.K. Calmodulin regulates L-selectin adhesion molecule expression and function through a protease-dependent mechanism. Cell 1998, 92, 809–818. [Google Scholar] [CrossRef]
- Andrews, R.K.; Suzuki-Inoue, K.; Shen, Y.; Tulasne, D.; Watson, S.P.; Berndt, M.C. Interaction of calmodulin with the cytoplasmic domain of platelet glycoprotein VI. Blood 2002, 99, 4219–4221. [Google Scholar] [CrossRef]
- Lai, Z.W.; Lew, R.A.; Yarski, M.A.; Mu, F.T.; Andrews, R.K.; Smith, A.I. The identification of a calmodulin-binding domain within the cytoplasmic tail of angiotensin-converting enzyme-2. Endocrinology 2009, 150, 2376–2381. [Google Scholar] [CrossRef]
- Bártová, E.; Legartová, S.; Krejčí, J.; Arcidiacono, O.A. Cell differentiation and aging accompanied by depletion of the ACE2 protein. Aging 2020, 12, 22495–22508. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Abrams, C.; Wang, L.; Gizzi, A.; He, L.; Lin, R.; Chen, Y.; Loll, P.J.; Pascal, J.M.; Zhang, J.F. Structural basis for calmodulin as a dynamic calcium sensor. Structure 2012, 20, 911–923. [Google Scholar] [CrossRef] [PubMed]
- Bikle, D.D.; Munson, S. 1,25-Dihydroxyvitamin D increases calmodulin binding to specific proteins in the chick duodenal brush border membrane. J. Clin. Investig. 1985, 76, 2312–2316. [Google Scholar] [CrossRef] [PubMed]
- Ellison, T.I.; Dowd, D.R.; MacDonald, P.N. Calmodulin-dependent kinase IV stimulates vitamin D receptor-mediated transcription. Mol. Endocrinol. 2005, 19, 2309–2319. [Google Scholar] [CrossRef] [PubMed]
- Robertshaw, H.J.; Brennan, F.M. Release of tumour necrosis factor alpha (TNFalpha) by TNFalpha cleaving enzyme (TACE) in response to septic stimuli in vitro. Br. J. Anaesth. 2005, 94, 222–228. [Google Scholar] [CrossRef]
- Jia, H.P.; Look, D.C.; Tan, P.; Shi, L.; Hickey, M.; Gakhar, L.; Chappell, M.C.; Wohlford-Lenane, C.; McCray, P.B., Jr. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L84–L96. [Google Scholar] [CrossRef]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Hurtado Del Pozo, C.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913.e7. [Google Scholar] [CrossRef]
- Lei, C.; Qian, K.; Li, T.; Zhang, S.; Fu, W.; Ding, M.; Hu, S. Neutralization of SARS-CoV-2 Spike Pseudotyped Virus by Recombinant ACE2-Ig. Nat. Commun. 2020, 11, 2070. [Google Scholar] [CrossRef]
- Tada, T.; Fan, C.; Chen, J.S.; Kaur, R.; Stapleford, K.A.; Gristick, H.; Dcosta, B.M.; Wilen, C.B.; Nimigean, C.M.; Landau, N.R. An ACE2 Microbody Containing a Single Immunoglobulin Fc Domain Is a Potent Inhibitor of SARS-CoV-2. Cell Rep. 2020, 33, 108528. [Google Scholar] [CrossRef]
- Meltzer, D.O.; Best, T.J.; Zhang, H.; Vokes, T.; Arora, V.; Solway, J. Association of Vitamin D Status and Other Clinical Characteristics With COVID-19 Test Results. JAMA Netw Open 2020, 3, e2019722. [Google Scholar] [CrossRef]
- Jain, A.; Chaurasia, R.; Sengar, N.S.; Singh, M.; Mahor, S.; Narain, S. Analysis of vitamin D level among asymptomatic and critically ill COVID-19 patients and its correlation with inflammatory markers. Sci. Rep. 2020, 10, 20191. [Google Scholar] [CrossRef] [PubMed]
- Bennouar, S.; Bachir Cherif, A.; Temmar, M.; Fauvel, J.P.; Bouafia, M.T.; Abdi, S. Vitamin D Deficiency and Low Serum Calcium as Predictors of Poor Prognosis in Patients with Severe COVID-19. J. Am. Coll. Nutr. 2021, 40, 104–110. [Google Scholar] [CrossRef]
- Rosenthal, N.; Cao, Z.; Gundrum, J.; Sianis, J.; Safo, S. Risk Factors Associated With In-Hospital Mortality in a US National Sample of Patients With COVID-19. JAMA Netw Open 2020, 3, e2029058. [Google Scholar] [CrossRef]
- Entrenas Castillo, M.; Entrenas Costa, L.M.; Vaquero Barrios, J.M.; Alcalá Díaz, J.F.; López Miranda, J.; Bouillon, R.; Quesada Gomez, J.M. Effect of calcifediol treatment and best available therapy versus best available therapy on intensive care unit admission and mortality among patients hospitalized for COVID-19: A pilot randomized clinical study. J. Steroid Biochem. Mol. Biol. 2020, 203, 105751. [Google Scholar] [CrossRef]
- Annweiler, G.; Corvaisier, M.; Gautier, J.; Dubée, V.; Legrand, E.; Sacco, G.; Annweiler, C. Vitamin D Supplementation Associated to Better Survival in Hospitalized Frail Elderly COVID-19 Patients: The GERIA-COVID Quasi-Experimental Study. Nutrients 2020, 12, 3377. [Google Scholar] [CrossRef]
- Mercante, G.; Ferreli, F.; De Virgilio, A.; Gaino, F.; Di Bari, M.; Colombo, G.; Russo, E.; Costantino, A.; Pirola, F.; Cugini, G.; et al. Prevalence of Taste and Smell Dysfunction in Coronavirus Disease 2019. JAMA Otolaryngol. Head Neck Surg. 2020, 146, 723–728. [Google Scholar] [CrossRef]
- Boscolo-Rizzo, P.; Borsetto, D.; Fabbris, C.; Spinato, G.; Frezza, D.; Menegaldo, A.; Mularoni, F.; Gaudioso, P.; Cazzador, D.; Marciani, S.; et al. Evolution of Altered Sense of Smell or Taste in Patients With Mildly Symptomatic COVID-19. JAMA Otolaryngol. Head Neck Surg. 2020, 146, 729–732. [Google Scholar] [CrossRef]
- Sisó-Almirall, A.; Kostov, B.; Mas-Heredia, M.; Vilanova-Rotllan, S.; Sequeira-Aymar, E.; Sans-Corrales, M.; Sant-Arderiu, E.; Cayuelas-Redondo, L.; Martínez-Pérez, A.; García-Plana, N.; et al. Prognostic factors in Spanish COVID-19 patients: A case series from Barcelona. PLoS ONE 2020, 15, e0237960. [Google Scholar] [CrossRef]
- Talavera, B.; García-Azorín, D.; Martínez-Pías, E.; Trigo, J.; Hernández-Pérez, I.; Valle-Peñacoba, G.; Simón-Campo, P.; de Lera, M.; Chavarría-Miranda, A.; López-Sanz, C.; et al. Anosmia is associated with lower in-hospital mortality in COVID-19. J. Neurol. Sci. 2020, 419, 117163. [Google Scholar] [CrossRef]
- Trigo, J.; García-Azorín, D.; Planchuelo-Gómez, Á.; Martínez-Pías, E.; Talavera, B.; Hernández-Pérez, I.; Valle-Peñacoba, G.; Simón-Campo, P.; de Lera, M.; Chavarría-Miranda, A.; et al. Factors associated with the presence of headache in hospitalized COVID-19 patients and impact on prognosis: A retrospective cohort study. J. Headache Pain 2020, 21, 94. [Google Scholar] [CrossRef]
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.E.; Williamson, M.K.; Antón-Plágaro, C.; Shoemark, D.K.; Simón-Gracia, L.; Bauer, M.; Hollandi, R.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 2020, 370, 861–865. [Google Scholar] [CrossRef]
- Kielian, M. Enhancing host cell infection by SARS-CoV-2. Science 2020, 370, 765–766. [Google Scholar] [CrossRef]
- Kolodkin, A.L.; Levengood, D.V.; Rowe, E.G.; Tai, Y.T.; Giger, R.J.; Ginty, D.D. Neuropilin is a semaphorin III receptor. Cell 1997, 90, 753–762. [Google Scholar] [CrossRef]
- Plein, A.; Fantin, A.; Ruhrberg, C. Neuropilin regulation of angiogenesis, arteriogenesis, and vascular permeability. Microcirculation 2014, 21, 315–323. [Google Scholar] [CrossRef]
- Gray, M.J.; Wey, J.S.; Belcheva, A.; McCarty, M.F.; Trevino, J.G.; Evans, D.B.; Ellis, L.M.; Gallick, G.E. Neuropilin-1 suppresses tumorigenic properties in a human pancreatic adenocarcinoma cell line lacking neuropilin-1 coreceptors. Cancer Res. 2005, 65, 3664–3670. [Google Scholar] [CrossRef]
- Liang, W.; Guan, W.; Chen, R.; Wang, W.; Li, J.; Xu, K.; Li, C.; Ai, Q.; Lu, W.; Liang, H.; et al. Cancer patients in SARS-CoV-2 infection: A nationwide analysis in China. Lancet Oncol. 2020, 21, 335–337. [Google Scholar] [CrossRef]
- Onder, G.; Rezza, G.; Brusaferro, S. Case-Fatality Rate and Characteristics of Patients Dying in Relation to COVID-19 in Italy. JAMA 2020, 323, 1775–1776. [Google Scholar] [CrossRef]
- Miyashita, H.; Mikami, T.; Chopra, N.; Yamada, T.; Chernyavsky, S.; Rizk, D.; Cruz, C. Do patients with cancer have a poorer prognosis of COVID-19? An experience in New York City. Ann. Oncol. 2020, 31, 1088–1089. [Google Scholar] [CrossRef]
- Aydillo, T.; Gonzalez-Reiche, A.S.; Aslam, S.; van de Guchte, A.; Khan, Z.; Obla, A.; Dutta, J.; van Bakel, H.; Aberg, J.; García-Sastre, A.; et al. Shedding of Viable SARS-CoV-2 after Immunosuppressive Therapy for Cancer. N. Engl. J. Med. 2020, 383, 2586–2588. [Google Scholar] [CrossRef]
- Crackower, M.A.; Sarao, R.; Oudit, G.Y.; Yagil, C.; Kozieradzki, I.; Scanga, S.E.; Oliveira-dos-Santos, A.J.; da Costa, J.; Zhang, L.; Pei, Y.; et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002, 417, 822–828. [Google Scholar] [CrossRef]
- Hanchate, N.K.; Giacobini, P.; Lhuillier, P.; Parkash, J.; Espy, C.; Fouveaut, C.; Leroy, C.; Baron, S.; Campagne, C.; Vanacker, C.; et al. SEMA3A, a gene involved in axonal pathfinding, is mutated in patients with Kallmann syndrome. PLoS Genet. 2012, 8, e1002896. [Google Scholar] [CrossRef]
- Laurendon, T.; Radulesco, T.; Mugnier, J.; Gérault, M.; Chagnaud, C.; El Ahmadi, A.A.; Varoquaux, A. Bilateral transient olfactory bulb edema during COVID-19-related anosmia. Neurology 2020, 95, 224–225. [Google Scholar] [CrossRef]
- Li, C.W.; Syue, L.S.; Tsai, Y.S.; Li, M.C.; Lo, C.L.; Tsai, C.S.; Chen, P.L.; Ko, W.C.; Lee, N.Y. Anosmia and olfactory tract neuropathy in a case of COVID-19. J. Microbiol. Immunol. Infect. 2021, 54, 93–96. [Google Scholar] [CrossRef]
- Aragão, M.; Leal, M.; Filho, O.C.; Fonseca, T.; Valença, M. Anosmia in COVID-19 Associated with Injury to the Olfactory Bulbs Evident on MRI. AJNR Am. J. Neuroradiol. 2020, 41, 1703–1706. [Google Scholar] [CrossRef]
- Ramani, A.; Müller, L.; Ostermann, P.N.; Gabriel, E.; Abida-Islam, P.; Müller-Schiffmann, A.; Mariappan, A.; Goureau, O.; Gruell, H.; Walker, A.; et al. SARS-CoV-2 targets neurons of 3D human brain organoids. EMBO J. 2020, 39, e106230. [Google Scholar] [CrossRef]
- Daneshgaran, G.; Dubin, D.P.; Gould, D.J. Cutaneous Manifestations of COVID-19: An Evidence-Based Review. Am. J. Clin. Dermatol. 2020, 21, 627–639. [Google Scholar] [CrossRef]
- Hubiche, T.; Le Duff, F.; Chiaverini, C.; Giordanengo, V.; Passeron, T. Negative SARS-CoV-2 PCR in patients with chilblain-like lesions. Lancet Infect. Dis. 2021, 21, 315–316. [Google Scholar] [CrossRef]
- Hubiche, T.; Cardot-Leccia, N.; Le Duff, F.; Seitz-Polski, B.; Giordana, P.; Chiaverini, C.; Giordanengo, V.; Gonfrier, G.; Raimondi, V.; Bausset, O.; et al. Clinical, Laboratory, and Interferon-Alpha Response Characteristics of Patients With Chilblain-like Lesions During the COVID-19 Pandemic. JAMA Dermatol. 2021, 157, 202–206. [Google Scholar] [CrossRef]
- Patel, V.B.; Parajuli, N.; Oudit, G.Y. Role of angiotensin-converting enzyme 2 (ACE2) in diabetic cardiovascular complications. Clin. Sci. 2014, 126, 471–482. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Escobar, A.; Vera-Vera, S.; Jurado-Román, A.; Jiménez-Valero, S.; Galeote, G.; Moreno, R. Calcium Signaling Pathway Is Involved in the Shedding of ACE2 Catalytic Ectodomain: New Insights for Clinical and Therapeutic Applications of ACE2 for COVID-19. Biomolecules 2022, 12, 76. https://doi.org/10.3390/biom12010076
García-Escobar A, Vera-Vera S, Jurado-Román A, Jiménez-Valero S, Galeote G, Moreno R. Calcium Signaling Pathway Is Involved in the Shedding of ACE2 Catalytic Ectodomain: New Insights for Clinical and Therapeutic Applications of ACE2 for COVID-19. Biomolecules. 2022; 12(1):76. https://doi.org/10.3390/biom12010076
Chicago/Turabian StyleGarcía-Escobar, Artemio, Silvio Vera-Vera, Alfonso Jurado-Román, Santiago Jiménez-Valero, Guillermo Galeote, and Raúl Moreno. 2022. "Calcium Signaling Pathway Is Involved in the Shedding of ACE2 Catalytic Ectodomain: New Insights for Clinical and Therapeutic Applications of ACE2 for COVID-19" Biomolecules 12, no. 1: 76. https://doi.org/10.3390/biom12010076
APA StyleGarcía-Escobar, A., Vera-Vera, S., Jurado-Román, A., Jiménez-Valero, S., Galeote, G., & Moreno, R. (2022). Calcium Signaling Pathway Is Involved in the Shedding of ACE2 Catalytic Ectodomain: New Insights for Clinical and Therapeutic Applications of ACE2 for COVID-19. Biomolecules, 12(1), 76. https://doi.org/10.3390/biom12010076