
From Small Peptides to Large Proteins against Alzheimer’sDisease


 ,
,  ,
,  , ,
, ,  , ,
, ,  , and
, and
Abstract
:1. Introduction
2. The Hallmark Lesions of AD: β-Amyloid and Tau Proteins
2.1. Amyloid β-Peptide (Abeta)
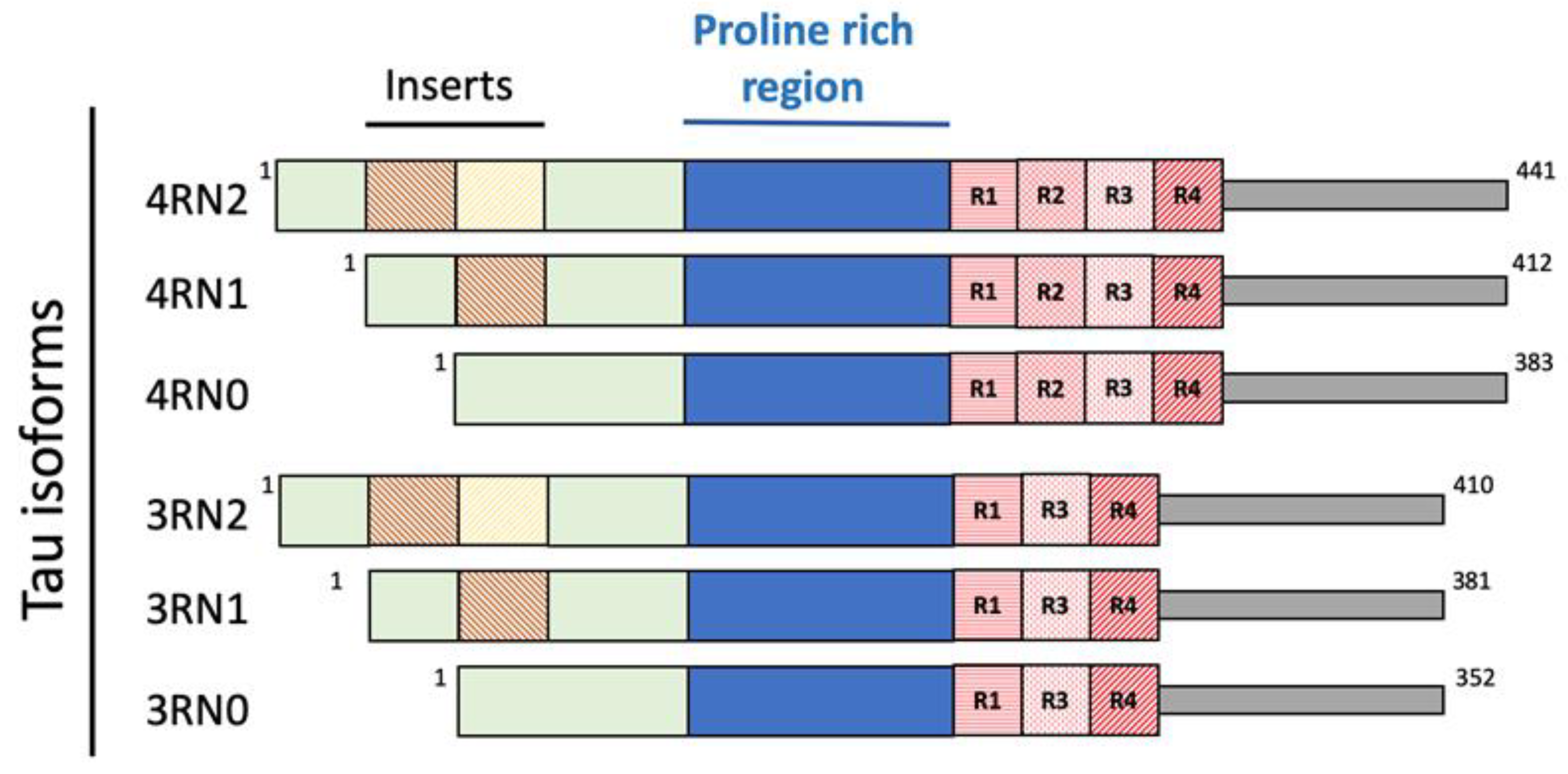
2.2. Tau Protein
2.3. Proteins and Metal Ions in AD
2.4. Peptide-Based Scaffolds to Target Cu Ions as Therapeutics
3. Oxidative Stress and Its Involvement in AD Onset
4. The Antioxidant Properties of Egg-Derived Peptides
5. Cholinesterase and BACE Inhibitory Activity of Egg-Derived Peptides
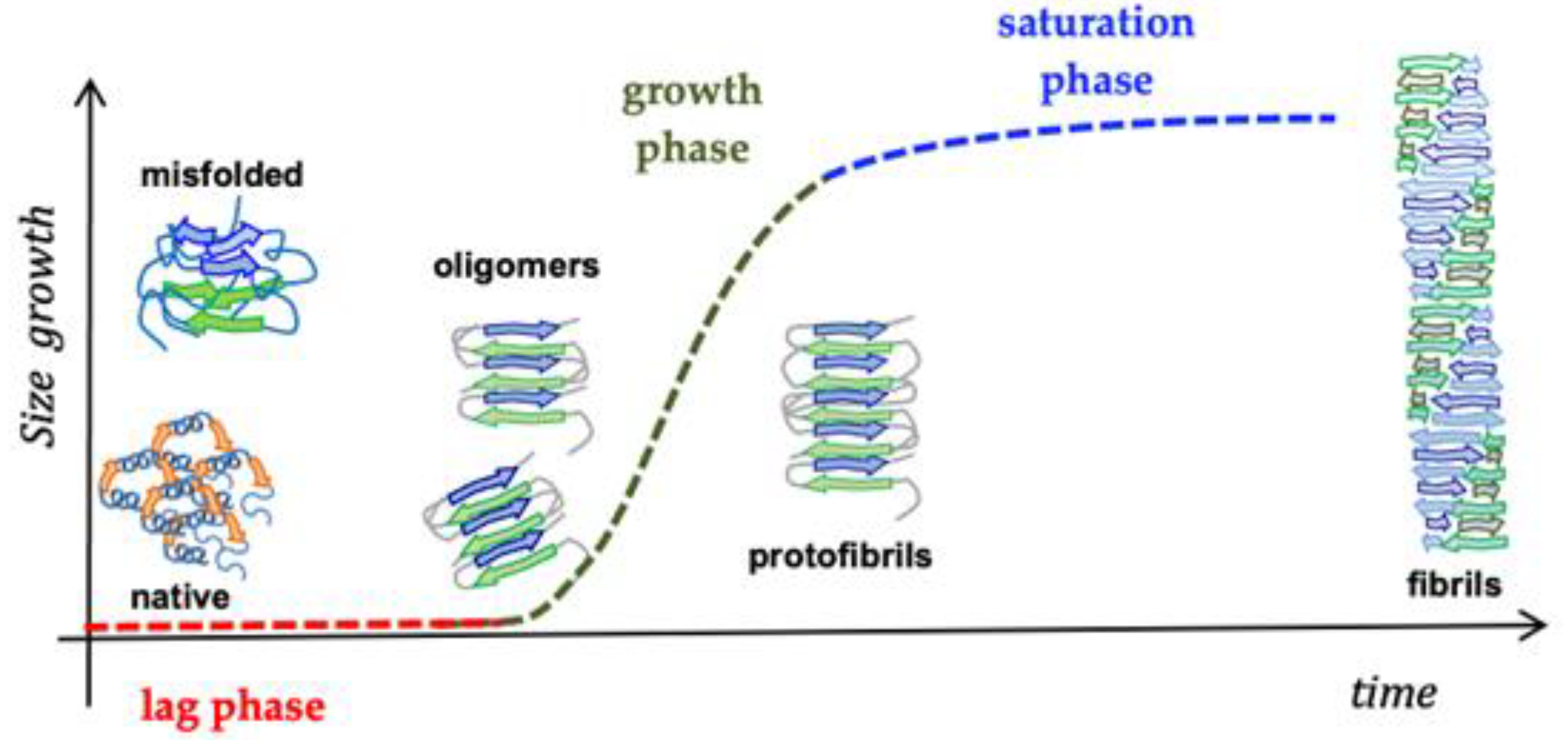
6. Beta-Sheet Breaker (BSB) Peptides as Abeta Aggregation-Inhibitor
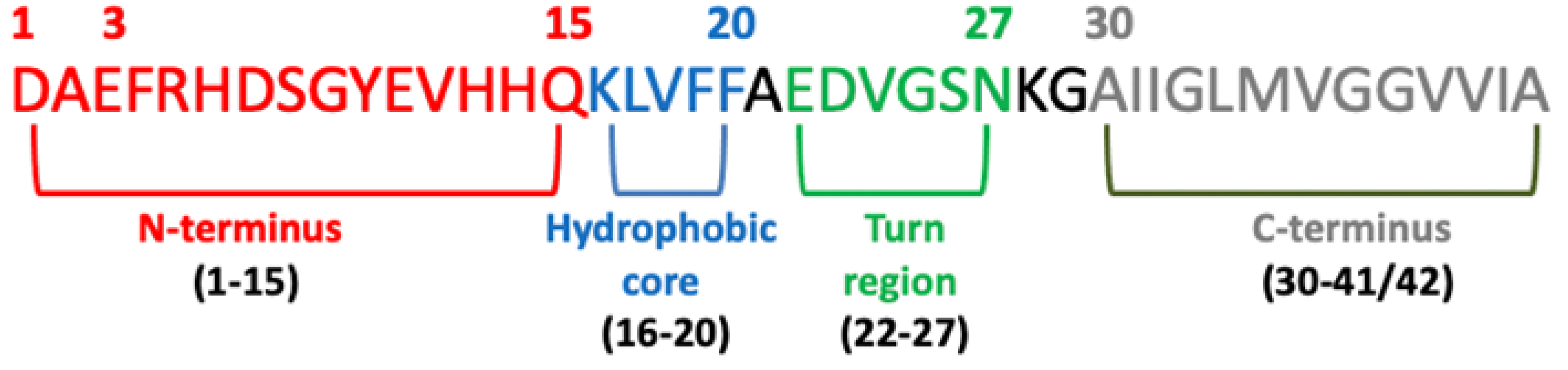
6.1. N-Terminus Sequence
6.2. Hydrophobic Core
6.3. C-Terminus (31–40/42)
7. The Blood–Brain Barrier (BBB) and AD
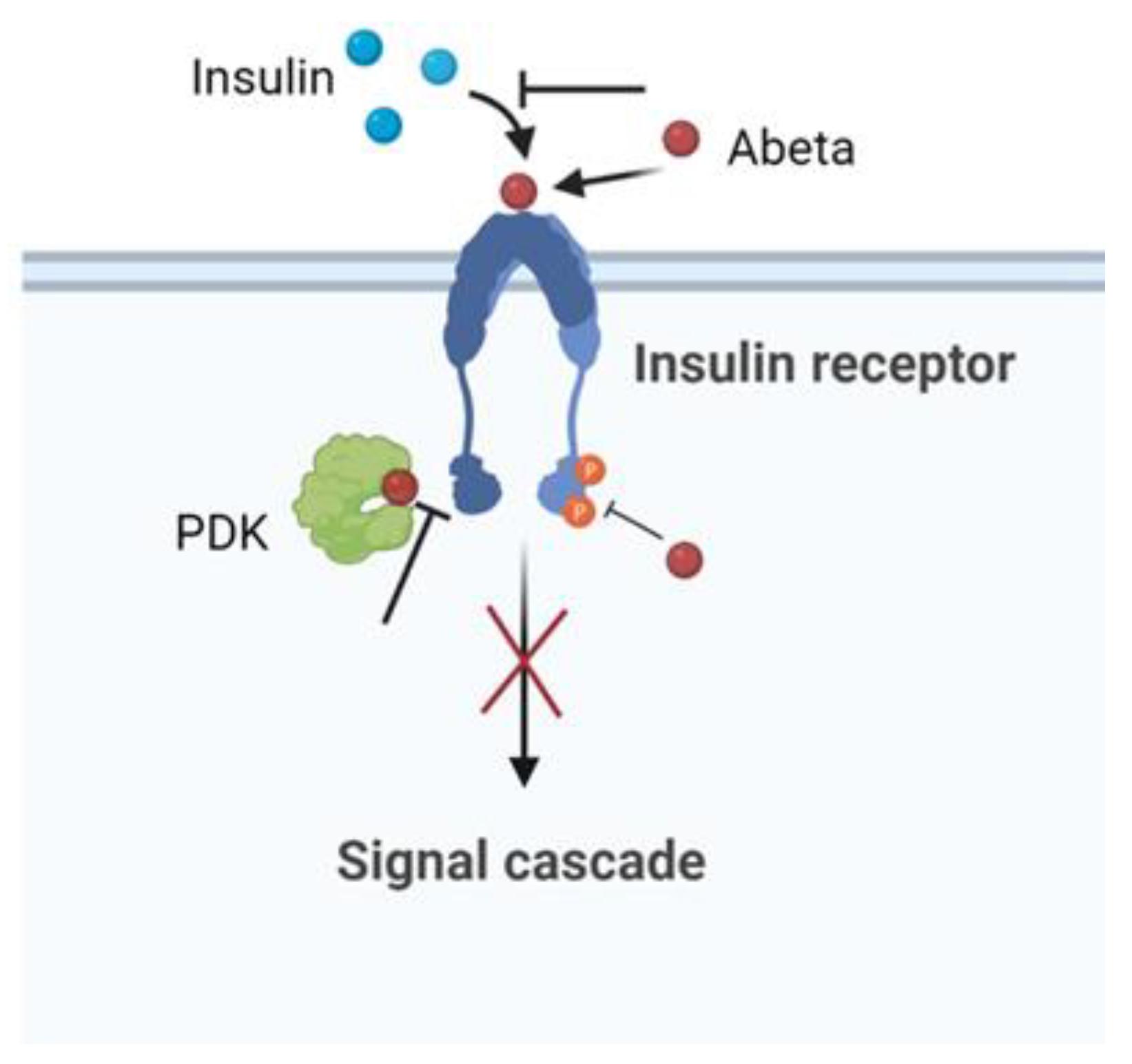
8. The Insulin Effect against AD
9. Large-Size Proteins and AD: The Case of the Heat Shock Proteins (HSPs)
9.1. HSPs and Abeta
9.2. HSPs and Tau Protein
10. Current Treatment of AD
11. Summary
12. Conclusions
Author Contributions
Funding
Institutional Review Board statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Picone, P.; Di Carlo, M.; Nuzzo, D. Obesity and Alzheimer’s disease: Molecular bases. Eur. J. Neurosci. 2020, 52, 3944–3950. [Google Scholar] [CrossRef] [PubMed]
- Raudino, F. Non-cognitive symptoms and related conditions in the Alzheimer’s Disease: A literature review. Neurol. Sci. 2013, 34, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Jessen, F.; Amariglio, R.E.; van Boxtel, M.; Breteler, M.; Ceccaldi, M.; Chételat, G.; Dubois, B.; Dufouil, C.; Ellis, K.A.; van der Flier, W.M.; et al. A conceptual framework for research on subjective cognitive decline in preclinical Alzheimer’s disease. Alzheimers Dement. 2014, 10, 844–852. [Google Scholar] [CrossRef] [PubMed]
- Mariani, E.; Monastero, R.; Mecocci, P. Mild cognitive impairment: A systematic review. J. Alzheimers Dis. 2007, 12, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Feldman, H.H.; Frisoni, G.B.; Hampel, H.; Jagust, W.J.; Johnson, K.A.; Knopman, D.S.; et al. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016, 87, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Delmotte, K.; Schaeverbeke, J.; Poesen, K.; Vandenberghe, R. Prognostic value of amyloid/tau/neurodegeneration (ATN) classification based on diagnostic cerebrospinal fluid samples for Alzheimer’s disease. Alzheimers Res. Ther. 2021, 13, 84. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol. Aging 1995, 16, 271–284. [Google Scholar] [CrossRef]
- Di Carlo, M.; Giacomazza, D.; San Biagio, P.L. Alzheimer’s Disease: Biological aspects, therapeutic perspectives and diagnostic tools. J. Phys. Condens. Matter 2012, 24, 244102. [Google Scholar] [CrossRef] [PubMed]
- Chow, N.; Korenberg, J.R.; Chen, X.-N.; Neve, R.L. APP-BP1, a novel protein that binds to the carboxyl-terminal region of the Amyloid Precursor Protein. J. Biol. Chem. 1996, 271, 11339–11346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilquet, V.; De Strooper, B. Amyloid-beta precursor protein processing in neurodegeneration. Curr. Opin. Neurobiol. 2004, 14, 582–588. [Google Scholar] [CrossRef]
- Dawkins, E.; Small, D.H. Insights into the physiological function of the β-amyloid precursor protein: Beyond Alzheimer’s disease. J. Neurochem. 2014, 129, 756–769. [Google Scholar] [CrossRef]
- Morley, J.E.; Farr, S.A.; Nguyen, A.D.; Xu, F. What is the physiological function of amyloid-beta protein? J. Nutr. Health Aging 2019, 23, 225–226. [Google Scholar] [CrossRef] [PubMed]
- Flood, J.F.; Morley, J.E.; Roberts, E. Amnestic effects in mice of four synthetic peptides homologous to amyloid beta protein from patients with Alzheimer disease. Proc. Natl. Acad. Sci. USA 1991, 88, 3363–3366. [Google Scholar] [CrossRef]
- Pearson, H.A.; Peers, C. Physiological roles for amyloid β peptide. J. Physiol. 2006, 575, 5–10. [Google Scholar] [CrossRef]
- Flammang, B.; Paradossi-Piquard, R.; Sevalle, J.; Bebayle, D.; Dabert-Gay, A.-S.; Thevenet, A.; Lauritzen, I.; Checler, F. Evidence that the amyloid-β protein precursor intracellular domain, AICD, derives from β-secretase-generated C-terminal fragment. J. Alzheimers Dis. 2012, 30, 145–153. [Google Scholar] [CrossRef]
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2012, 2, a006270. [Google Scholar] [CrossRef]
- Becker-Pauly, C.; Pietrzik, C.U. The metalloprotease meprin β is an alternative β-secretase of APP. Front. Mol. Neurosci. 2017, 9, 159. [Google Scholar] [CrossRef]
- Tanabe, C.; Hotoda, N.; Sasagawa, N.; Sehara-Fujisawa, A.; Maruyama, K.; Ishiura, S. ADAM19 is tightly associated with constitutive Alzheimer’s disease APP alpha-secretase in A172 cells. Biochem. Biophys. Res. Commun. 2006, 352, 111–117. [Google Scholar] [CrossRef]
- Postina, R.; Schroeder, A.; Dewachter, I.; Bohl, J.; Schmitt, U.; Kojro, E.; Prinzen, C.; Endres, K.; Hiemke, C.; Blessing, M.; et al. A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J. Clin. Investig. 2004, 113, 1456–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haass, C. Take five–BACE and the gamma-secretase quartet conduct Alzheimer’s amyloid beta-peptide generation. EMBO J. 2004, 23, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Moussa-Pacha, N.M.; Abdin, S.M.; Omar, H.A.; Alniss, H.; Al-Tel, T.H. BACE1 inhibitors: Current status and future directions in treating Alzheimer’s disease. Med. Res. Rev. 2020, 40, 339–384. [Google Scholar] [CrossRef]
- Coimbra, J.R.M.; Marques, D.F.F.; Baptista, S.J.; Pereira, C.M.F.; Moreira, P.I.; Dinis, T.C.P.; Santos, A.E.; Salvador, J.A.R. Highlights in BACE1 inhibitors for Alzheimer’s disease treatment. Front. Chem. 2018, 6, 178. [Google Scholar] [CrossRef] [PubMed]
- Hansson, C.A.; Frykman, S.; Farmery, M.R.; Tjenberg, L.O.; Nilsberth, C.; Pursglove, S.E.; Ito, A.; Winblad, B.; Cowburn, R.F.; Thyberg, J. Nicastrin, presenilin, APH-1, and PEN-2 form active gamma-secretase complexes in mitochondria. J. Biol. Chem. 2004, 279, 51654–51660. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgings, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Wingo, T.S.; Lah, J.J.; Levey, A.I.; Cutler, D.J. Autosomal recessive causes likely in early-onset Alzheimer disease. Arch. Neurol. 2012, 69, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Buell, A.K. The growth of amyloid fibrils: Rates and mechanisms. Biochem. J. 2019, 476, 2677–2703. [Google Scholar] [CrossRef] [PubMed]
- Deleanu, M.; Hernandez, J.-F.; Cippelletti, L.; Biron, J.P.; Rossi, E.; Taverna, M.; Cottet, H.; Chamieh, J. Unraveling the speciation of b-amyloid peptides during the aggregation process by Taylor dispersion analysis. Anal. Chem. 2021, 93, 6523–6533. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.; Wernstedt, C.; Wilander, E.; Hayden, D.W.; O’Brian, T.D.; Johnson, K.H. Amyloid fibrils in human insulinoma and islets of Langherans of the diabetic cat are derived from a neuropeptide-like protein also present in normal islet cells. Proc. Natl. Acad. Sci. USA 1987, 84, 3881–3885. [Google Scholar] [CrossRef]
- Akter, R.; Cao, P.; Noor, H.; Ridgway, Z.; Tu, L.-T.; Wang, H.; Wong, A.G.; Zhang, X.; Abedini, A.; Schmidt, A.M.; et al. Islet amyloid polypeptide: Structure, function, and pathology. J. Diab. Res. 2016, 2798269. [Google Scholar] [CrossRef] [Green Version]
- Seeliger, J.; Weise, K.; Opitz, N.; Winter, R. The Effect of Aβ on IAPP Aggregation in the Presence of an Isolated β-Cell Membrane. J. Mol. Biol. 2012, 421, 348–363. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.A.; Moore, K.B.E.; Saraswati, A.P.; Fortin, J.S. Recent advances in the discovery of the therapeutics to curtail islet amyloid polypeptide aggregation in Type 2 Diabetes treatment. Advanced Biology 2022, 2022, 2101301. [Google Scholar] [CrossRef] [PubMed]
- García-Viñuales, S.; Ilie, I.M.; Santoro, A.M.; Romanucci, V.; Zarrelli, A.; Di Fabio, G.; Caflisch, A.; Milardi, D. Silybins inhibit human IAPP amyloid growth and toxicity through stereospecific interactions. Biochim. Biophys. Acta-Proteins Proteom. 2021, 1870, 140772. [Google Scholar] [CrossRef] [PubMed]
- Bedrood, S.; Li, Y.; Isas, J.M.; Hegde, B.G.; Baxa, U.; Haworth, I.S.; Langen, R. Fibril structure of human islet amyloid polypeptide. J. Biol. Chem. 2012, 287, 5235–5241. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, P.; Solomon, T.; Malajczuk, C.J.; Mancera, R.L.; Howard, M.; Arrigan, D.W.M.; Newsholme, P.; Martins, R.N. Role of the cell membrane interface in modulating production and uptake of Alzheimer’s beta amyloid protein. Biochim. Biophys. Acta-Biomembr. 2018, 1860, 1639–1651. [Google Scholar] [CrossRef]
- Bharadwaj, P.; Solomon, T.; Sahoo, B.R.; Ignasiak, K.; Gaskin, S.; Rowles, J.; Verdile, G.; Howard, M.J.; Bond, C.S.; Ramamoorthy, A.; et al. Amylin and beta amyloid proteins interact to form amorphous heterocomplexes with enhanced toxicity in neuronal cells. Sci. Rep. 2020, 10, 10356. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Hu, R.; Ren, B.; Chen, H.; Jiang, B.; Ma, J.; Zheng, J. Molecular understanding of Aβ-hIAPP cross-seeding assemblies on lipid membranes. ACS Chem. Neurosci. 2017, 8, 524–537. [Google Scholar] [CrossRef] [PubMed]
- Simic, G.; Babic Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milosevic, N.; Bazadona, D.; Buée, L.; da Silva, R.R.; Di Giovanni, G.; et al. Tau protein hyperphoshorilation and aggregation in Alzheimer’s Disease and other tauopathies, and possible neuroprotective strategies. Biomolecules 2016, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Verwilst, P.; Kim, H.S.; Kim, S.; Kang, C.; Kim, J.S. Shedding light on tau protein aggregation: The progress in developing highly selective fluorophores. Chem. Soc. Rev. 2018, 47, 2249–2265. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, F.; Avila, J. Tauopathies. Cell. Mol. Life Sci. 2007, 64, 2219–2233. [Google Scholar] [CrossRef]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Kopke, E.; Tung, Y.C.; Shaikh, S.; Alonso, A.C.; Iqbal, K.; Grundke-Iqbal, I. Microtubule-associated protein tau: Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J. Biol. Chem. 1993, 268, 24374–24384. [Google Scholar] [CrossRef]
- Madeiros, R.; Baglietto-Vargas, D.; LaFerla, F.M. The role of Tau in Alzheimer’s Disease and related disorders. CNS Neurosci. Ther. 2011, 17, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M. Structure of NTF: Biochemical approach. Adv. Exp. Med. Biol. 2019, 1184, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, S.; Eftekharzadeh, B.; Tepper, K.; Zoltowska, K.M.; Bennett, R.E.; Dujardin, S.; Laskowski, P.R.; MacKenzie, D.; Kamath, T.; Commins, C.; et al. Tau protein liquid-liquid phase separation can initiate Tau aggregation. EMBO J. 2018, 37, e98049. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Vigers, M.; McCarty, J.; Rauch, J.N.; Fredrickson, G.H.; Wilson, M.Z.; Shea, J.-E.; Han, S.; Kosik, K.S. The prolin-rich domain promotes Tau liquid-liquid phase separation in cells. J. Cell Biol. 2020, 219, e202006054. [Google Scholar] [CrossRef]
- Min, S.W.; Cho, S.-H.; Zhou, Y.; Schroeder, S.; Haroutunian, V.; Seeley, W.W.; Huang, E.J.; Shen, Y.; Masliah, E.; Mukherjee, C.; et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 2010, 67, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Cook, C.; Carlomagno, Y.; Gendron, T.F.; Dunmore, J.; Scheffel, K.; Stetler, C.; Davis, M.; Dickson, D.; Jarpe, M.; DeTure, M.; et al. Acetylation of the KXGS motifs in tau is a critical determinant in modulation of tau aggregation and clearance. Human. Mol. Genet. 2014, 23, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef]
- Liu, Y.; Nguyen, M.; Robert, A.; Meunier, B. Metal Ions in Alzheimer’s Disease: A Key Role or Not? ACC Chem. Res. 2019, 52, 2026–2035. [Google Scholar] [CrossRef] [PubMed]
- Cristovao, J.S.; Santos, R.; Gomes, C.M. Metals and neuronal metal bindind proteins implicated in Alzheimer’s disease. Oxidative Med. Cell. Long. 2016, 2016, 9812178. [Google Scholar] [CrossRef]
- Cecarini, V.; Gee, J.; Fioretti, E.; Amici, M.; Angeletti, M.; Eleuteri, A.M.; Keller, J.N. Protein oxidation and cellular homeostasis: Enphasis on metabolism. Biochim. Biophys. Acta 2007, 1773, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Shichiri, M. The role of lipid peroxidation in neurological disorders. J. Clin. Biochem. Nutr. 2014, 54, 151–160. [Google Scholar] [CrossRef]
- Chen, W.-T.; Liao, Y.-H.; Yu, H.-M.; Cheng, I.H.; Chen, Y.-R. Distinct effects of Zn2+, Cu2+, Fe3+, and Al3+ on amyloid-β stability, oligomerization, and aggregation: Amyloid-β destabilization promotes annular protofibril formation. J. Biol. Chem. 2011, 286, 9646–9656. [Google Scholar] [CrossRef] [PubMed]
- Baum, L.; Chan, I.H.S.; Cheung, S.K.-K.; Goggins, W.B.; Mok, V.; Lam, L.; Leung, V.; Hui, E.; Ng, C.; Woo, J.; et al. Serum zinc is decreased in Alzheimer’s disease and serum arsenic correlates positively with cognitive ability. BioMetals 2010, 23, 173–179. [Google Scholar] [CrossRef]
- Bishop, G.M.; Robinson, S.R.; Liu, Q.; Perry, G.; Atwood, C.S.; Smith, M.A. Iron: A pathological mediator of Alzheimer disease? Dev. Neurosci. 2002, 24, 184–187. [Google Scholar] [CrossRef]
- Tougu, V.; Karafin, A.; Zovo, K.; Chung, R.S.; Howells, C.; West, A.K.; Palumaa, P. Zn(II)- and Cu(II)-induced non-fibrillar aggregates of amyloid-β (1–42) peptide are transformed to amyloid fibrils, both spontaneously and under the influence of metal chelators. J. Neurochem. 2009, 110, 1784–1795. [Google Scholar] [CrossRef] [PubMed]
- Sarell, C.J.; Wilkinson, S.R.; Viles, J.H. Substoichiometric levels of Cu2+ ions accelerate the kinetics of fiber formation and promote cell toxicity of amyloid-β from Alzheimer disease. J. Biol. Chem. 2010, 285, 41533–41540. [Google Scholar] [CrossRef] [PubMed]
- Wild, K.; August, A.; Pietrzik, C.U.; Kins, S. Structure and synaptic function of metal binding to amyloid precursor protein and its proteolytic fragments. Front. Mol. Neurosci. 2017, 10, 21. [Google Scholar] [CrossRef]
- Caballero, A.B.; Terol-Ordaz, L.; Espargaró, A.; Vázquez, G.; Nicolás, E.; Sabaté, R.; Gamez, P. Histidine-Rich Oligopeptides to Lessen Copper-Mediated Amyloid-β Toxicity. Chemistry 2016, 22, 7268–7280. [Google Scholar] [CrossRef] [PubMed]
- Esmieu, C.; Ferrand, G.; Borghesani, V.; Hureau, C. Impact of N-Truncated Aβ Peptides on Cu- and Cu(Aβ)-Generated ROS: Cu I Matters! Chemistry 2021, 27, 1777–1786. [Google Scholar] [CrossRef] [PubMed]
- Koh, J.-Y.; Lee, S.-J. Metallothionein-3 as a multifunctional player in the control of cellular processes and diseases. Mol. Brain 2020, 13, 116. [Google Scholar] [CrossRef] [PubMed]
- Atrián-Blasco, E.; Santoro, A.; Pountney, D.L.; Meloni, G.; Hureau, C.; Faller, P. Chemistry of mammalian metallothioneins and their interaction with amyloidogenic peptides and proteins. Chem. Soc. Rev. 2017, 46, 7683–7693. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Xu, Q.; Cheng, H.; Tan, X. The efficacy and pharmacological mechanism of Zn7MT3 to protect against Alzheimer’s disease. Sci. Rep. 2017, 7, 13763. [Google Scholar] [CrossRef]
- Cristovao, J.S.; Gomes, C.M. S100 proteins in Alzheimer’s disease. Front. Neurosci. 2019, 13, 463. [Google Scholar] [CrossRef] [PubMed]
- McAllister, B.B.; Dyck, R.H. Zinc transporter 3 (ZnT3) and vescicular zinc in central nervous system function. Neurosci. Biobehav. Rev. 2017, 80, 329–350. [Google Scholar] [CrossRef] [PubMed]
- Adlard, P.A.; Parncutt, J.M.; Finkelstein, D.I.; Bush, A.I. Cognitive loss in zinc transporter-3 knock-out mice: A phenocopy for the synaptic and memory deficits of Alzheimer’s disease? J. Neurosci. 2010, 30, 1631–1636. [Google Scholar] [CrossRef]
- Mrak, R.E.; Griffin, W.S.T. The role of activated astrocytes and of the neurotrophic cytokine S100B in the pathogenesis of Alzheimer’s disease. Neurobiol. Aging 2001, 22, 915–922. [Google Scholar] [CrossRef]
- Yamamoto, A.; Shin, R.-W.; Hasegawa, K.; Naiki, H.; Sato, H.; Yoshimasu, F.; Kitamoto, T. Iron (III) induces aggregation of hyperphosphorylated tau and its reduction to iron (II) reverses the aggregation: Implications in the formation of neurofibrillary tangles of Alzheimer’s disease. J. Neurochem. 2002, 82, 1137–1147. [Google Scholar] [CrossRef]
- Savelieff, M.G.; Nam, G.; Kang, J.; Lee, H.J.; Lee, M.; Lim, M.H. Development of Multifunctional Molecules as Potential Therapeutic Candidates for Alzheimer’s Disease, Parkinson’s Disease, and Amyotrophic Lateral Sclerosis in the Last Decade. Chem. Rev. 2019, 119, 1221–1322. [Google Scholar] [CrossRef]
- Trapani, G.; Satriano, C.; La Mendola, D. Peptides and their Metal Complexes in Neurodegenerative Diseases: From Structural Studies to Nanomedicine Prospects. Curr. Med. Chem. 2018, 25, 715–747. [Google Scholar] [CrossRef] [PubMed]
- Maiti, B.K.; Govil, N.; Kundu, T.; Moura, J.J.G. Designed Metal-ATCUN Derivatives: Redox- and Non-redox-Based Applications Relevant for Chemistry, Biology, and Medicine. iScience 2020, 23, 101792. [Google Scholar] [CrossRef] [PubMed]
- Folk, D.S.; Franz, K.J. A prochelator activated by beta-secretase inhibits Abeta aggregation and suppresses copper-induced reactive oxygen species formation. J. Am. Chem. Soc. 2010, 132, 4994–4995. [Google Scholar] [CrossRef]
- Hu, X.; Zhang, Q.; Wang, W.; Yuan, Z.; Zhu, X.; Chen, B.; Chen, X. Tripeptide GGH as the Inhibitor of Copper-Amyloid-β-Mediated Redox Reaction and Toxicity. ACS Chem. Neurosci. 2016, 7, 1255–1263. [Google Scholar] [CrossRef]
- Sharma, L.K.; Lu, J.; Bai, Y. Mitochondrial respiratory complex I: Structure, function and implication in human diseases. Curr. Med. Chem. 2009, 16, 1266–1277. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human diseases. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Rada, B.; Leto, T.L. Oxidative innate immune defenses by Nox/Duox family NADPH oxidases. Contrib. Microbiol. 2008, 15, 164–187. [Google Scholar] [CrossRef]
- Sinenko, S.A.; Starkova, T.Y.; Kurzmin, A.A.; Tomilin, A.N. Physiological signaling functions of reactive oxygen species in stem cells: From flies to man. Front. Cell Dev. Biol. 2021, 9, 714370. [Google Scholar] [CrossRef] [PubMed]
- Di Carlo, M.; Giacomazza, D.; Picone, P.; Nuzzo, D.; San Biagio, P.L. Are oxidative stress and mitochondrial dysfunction the key players in the neurodegenerative diseases? Free Ras. Res. 2012, 46, 1327–1338. [Google Scholar] [CrossRef] [PubMed]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [Green Version]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative stress in neurodegenerative diseases: From a mitochondrial point of view. Oxidative Med. Cell. Long. 2019, 2019, 2105607. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, F.; Ada, R.H.I.; Bansal, R.; Broersen, K. Review of oxidative stress products and related genes in early Alzheimer’s disease. J. Alz. Dis. 2021, 83, 977–1001. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Merkesbery, W.R. Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer’s disease. Nucleic Acid Res. 2007, 35, 7497–7504. [Google Scholar] [CrossRef]
- Collin, F.; Cheignon, C.; Hureau, C. Oxidative stress as a biomarker for Alzheimer’s disease. Biomark. Med. 2018, 12, 201–203. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta 2014, 1842, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Ionescu-Tucker, A.; Cotman, C.W. Emerging roles of oxidative stress in brain aging and Alzheimer’s disease. Neurobiol. Aging 2021, 107, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Tonnies, E.; Trushina, E. Oxidative stress, synaptic dysfunction, and Alzheimer’s disease. J. Alz. Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [PubMed]
- Funke, A.; van Groen, T.; Kadish, I.; Bartnik, D.; Nagel-Steger, L.; Brener, O.; Sehl, T.; Batra-Safferling, R.; Moriscot, C.; Schoehn, G.; et al. Oral treatment with the d-enantiomeric peptide D3 improves the pathology and behavior of Alzheimer’s Disease transgenic mice. ACS Chem. Neurosci. 2010, 1, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Benedé, S.; Molina, E. Chicken egg proteins and derived peptides with antioxidant properties. Foods 2020, 9, 735. [Google Scholar] [CrossRef] [PubMed]
- Abeyrathne, E.D.N.S.; Huang, X.; Ahn, D.U. Antioxidant, angiotensin-converting enzyme inhibitory activity and other functional properties of egg white proteins and their derived peptides—A review. Poult. Sci. 2018, 97, 1462–1468. [Google Scholar] [CrossRef] [PubMed]
- Nimalaratne, C.; Wu, J. Hen egg as an antioxidant food commodity: A review. Nutrients 2015, 7, 8274–8293. [Google Scholar] [CrossRef] [PubMed]
- Nimalaratne, C.; Bandara, N.; Wu, J. Purification and characterization of antioxidant peptides from enzymatically hydrolyzed chicken egg white. Food Chem. 2015, 188, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Jin, Y.; Lin, S.; Jones, G.S.; Chen, F. Purification and identification of novel antioxidant peptides from egg white protein and their antioxidant activities. Food Chem. 2015, 175, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Davalos, A.; Miguel, M.; Bartolomé, B.; López-Fandiño, R. Antioxidant activity of peptides derived from egg white proteins by enzymatic hydrolysis. J. Food Prot. 2004, 67, 1939–1944. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Shen, S.; Nimalaratne, C.; Li, C.; Majumder, K.; Wu, J. Effects of addition of egg ovotransferrin-derived peptides on the oxygen radical absorbance capacity of different teas. Food Chem. 2012, 135, 1600–1607. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Katayama, S.; Mine, Y. Antioxidant activity of tryptic digests of hen egg yolk phosvitin: Antioxidant activity of phosvitin peptides. J. Sci. Food Agric. 2007, 87, 2604–2608. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Dong, W.; Wu, S.; Shen, J.; Zhao, W.; Ding, L.; Liu, J.; Zheng, F. Identification of ovalbumin-derived peptides as multi-target inhibitors of AChE, BChE, and BACE1. J. Sci. Food Agric. 2020, 100, 2648–2655. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Wu, S.; Zhao, W.; Ding, L.; Fan, Y.; Shiuan, D.; Liu, J.; Chen, F. Anti-Alzheimers activity and molecular mechanism of albumin-derived peptides against AChE and BChE. Food Funct. 2018, 9, 1173–1178. [Google Scholar] [CrossRef]
- Nuzzo, D.; Frinchi, F.; Giardina, C.; Scordino, M.; Zuccarini, M.; De Simone, C.; Di Carlo, M.; Belluardo, N.; Mudò, G.; Di Liberto, V. Neuroprotective and Antioxidant Role of Oxotremorine-M, a Non-selective Muscarinic Acetylcholine Receptors Agonist, in a Cellular Model of Alzheimer Disease. Cell Mol. Neurobiol. 2022, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Wu, S.; Zhao, W.; Ding, L.; Shiuan, D.; Zheng, F.; Li, J.; Liu, J. Biological evaluation and interaction mechanism of beta-site APP cleaving enzyme 1 inhibitory pentapeptide from egg albumin. Food Sci. Hum. Wellness 2020, 9, 162–167. [Google Scholar] [CrossRef]
- Mason, J.M.; Kokkoni, N.; Stott, K.; Doig, A.J. Design strategies for anti-amyloid agents. Curr. Opin. Struct. Biol. 2003, 13, 526–532. [Google Scholar] [CrossRef]
- Goyal, D.; Shuaib, S.; Mann, S.; Goyal, B. Rationally designed peptides and peptidomimetics as inhibitors of amyloid-β (Ab) aggregation: Potential therapeutics of Alzheimer’s Disease. ACS Comb. Sci. 2017, 19, 55–80. [Google Scholar] [CrossRef]
- Belluti, F.; Rampa, A.; Gobbi, S.; Bisi, A. Small-molecule inhibitors/modulators of amyloid-β peptide aggregation and toxicity for the treatment of Alzheimer’s disease: A patent review (2010–2012). Expert Opin. Ther. Pat. 2013, 23, 581–596. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Kindy, M.S.; Baumann, M.; Frangione, B. Inhibition of Alzheimer’s amyloidosis by peptides that prevent β-sheet conformation. Biochem. Biophys. Res. Comm. 1996, 116, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Yee, A.; Brewer, H.B., Jr.; Das, S.; Potter, H. Amyloid-associated proteins alpha 1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer beta-protein into filaments. Nature 1994, 372, 92–94. [Google Scholar] [CrossRef]
- Boyett, K.W.; DiCarlo, G.; Jantzen, P.T.; Jackson, J.; O’Leary, C.; Wilcock, D.; Morgan, D.; Gordon, M.N. Increased fibrillar beta-amyloid in response to human C1q injections into hippocampus and cortex of APP + PS1 transgenic mice. Neurochem. Res. 2003, 28, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, H.; Liu, L.; Murray, I.V.J.; Axelsen, P.H. A mechanistic link between oxidative stress and membrane mediated amyloidogenesis revealed by infrared spectroscopy. Biochim. Biophys. Acta-Biomembr. 2007, 1768, 1913–1922. [Google Scholar] [CrossRef] [PubMed]
- Stellato, F.; Fusco, Z.; Chiaraluce, R.; Consalvi, V.; Dinarelli, S.; Placidi, E.; Petrosino, M.; Rossi, G.C.; Minicozzi, V.; Morante, S. The effect of β-sheet breaker peptides on metal associated Amyloid-β peptide aggregation process. Biophys. Chem. 2017, 229, 110–114. [Google Scholar] [CrossRef]
- Snow, A.D.; Wight, T.N. Proteoglycans in the pathogenesis of Alzheimer’s disease and other amyloidosis. Neurobiol. Aging 1989, 10, 481–497. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Selkoe, D.J. Abeta oligomers—A decade of discovery. J. Neurochem. 2007, 101, 1172–1184. [Google Scholar] [CrossRef]
- Gardberg, A.S.; Dice, L.T.; Ou, S.; Rich, R.L.; Helmbrecht, E.; Ko, J.; Wetzel, R.; Myszka, D.G.; Patterson, P.H.; Dealwis, C. Molecular basis for passive immunotherapy of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 15659–15664. [Google Scholar] [CrossRef]
- Wasmer, C.; Lange, A.; Van Melckebeke, H.; Siemer, A.B.; Riek, R.; Meier, B.H. Amyloid fibrils of the HET-s (218–289) prion form a beta solenoid with a triangular hydrophobic core. Science 2008, 319, 1523–1526. [Google Scholar] [CrossRef] [PubMed]
- Maji, S.K.; Ogorzalek Loo, R.R.; Inayathullah, M.; Spring, S.M.; Vollers, S.S.; Condron, M.M.; Bitan, G.; Loo, J.A.; Teplow, D.B. Amino acid position-specific contributions to amyloid beta-protein oligomerization. J. Biol. Chem. 2009, 284, 23580–23591. [Google Scholar] [CrossRef] [PubMed]
- Fradinger, E.A.; Monien, B.H.; Urbanc, B.; Lomakin, A.; Tan, M.; Li, H.; Spring, S.M.; Condron, M.M.; Cruz, L.; Xie, L.C.; et al. C-terminal peptides coassemble into Abeta42 oligomers and protect neurons against Abeta42-induced neurotoxicity. Proc. Natl. Acad. Sci. USA 2008, 105, 14175–14180. [Google Scholar] [CrossRef]
- Lee, J.; Kwon, I.; Jang, S.S.; Cho, A.E. Investigation of the effect of erythrosine B on amyloid beta peptide using molecular modeling. J. Mol. Model. 2016, 22, 92. [Google Scholar] [CrossRef]
- Greco, V.; Naletova, I.; Ahmed, I.M.M.; Vaccaro, S.; Messina, L.; La Mendola, D.; Bellia, F.; Sciuto, S.; Satriano, C.; Rizzarelli, E. Hyaluronan-carnosine conjugates inhibit Aβ aggregation and toxicity. Sci. Rep. 2020, 10, 15998. [Google Scholar] [CrossRef] [PubMed]
- Wen, G.; Chen, D.; Qin, W.; Zhou, B.; Wang, Y.; Liu, Z.; Du, J.; Zhou, Q.; Quan, J.; Bu, X. Stabilizing amyloid-β peptide by the N-terminus capture is capable of preventing and eliminating amyloid-β oligomers. Chem. Commun. 2017, 53, 7673–7676. [Google Scholar] [CrossRef] [PubMed]
- Bieler, S.; Soto, C. Beta-sheet breakers for Alzheimer’s disease therapy. Curr. Drug Targets 2004, 5, 553–558. [Google Scholar] [CrossRef]
- Soto, C.; Sigurdsson, E.M.; Morelli, L.; Kumar, R.A.; Castano, E.M.; Frangione, B. Beta-sheet breaker peptides inhibit fibrillogenesis ia rat brain model of amyloidosis: Implications for Alzheimer’s therapy. Nat. Med. 1998, 4, 822–826. [Google Scholar] [CrossRef]
- Jani, V.; Sonavane, U.; Joshi, R. Destabilization potential of beta sheet breaker peptides on Abeta fibril structure: An insight from molecular dynamics simulation study. RSC Adv. 2021, 11, 23557–23573. [Google Scholar] [CrossRef] [PubMed]
- Jamula, A.; Ludwiczak, J.; Stepkowski, D. β-sheet breakers with consecutive phenylalanines: Insights into mechanism of dissolution of β-amyloid fibrils. Proteins 2021, 89, 762–780. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Rahimi, F.; Bitan, G. Modulation of amyloid β-protein (Aβ) assembly by homologous C-terminal fragments as a strategy for inhibiting Aβ toxicity. ACS Chem. Neurosci. 2016, 7, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Villabona-Rueda, A.; Erice, C.; Pardo, C.A.; Stins, M.F. The evolving concept pf the blood brain barrier (BBB): From a single static barrier to a heterogeneous and dynamic relay center. Front. Cell. Neurosci. 2019, 13, 405. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The Blood-Brain Barrier. Cold Spring Harbor. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed]
- Schofield, C.L.; Rodrigo-Navarro, A.; Dalby, M.J.; Van Agtmael, T.; Salmeron-Sanchez, M. Biochemical- and biophysical-induced barrigenesis in the blood-brain barrier: A review of barrigenic factor for use in vitro models. Adv. NanoBiomed. Res. 2021, 1, 2000068. [Google Scholar] [CrossRef]
- Wang, D.; Chen, F.; Han, Z.; Yin, Z.; Ge, X.; Lei, P. Relationship between amyloid-β deposition and blood-brain barrier dysfunction in Alheimer’s disease. Front. Cell. Neurosci. 2021, 15, 695479. [Google Scholar] [CrossRef]
- Kleinridders, A.; Ferris, H.A.; Cai, W.; Kahn, C.R. Insulin action in brain regulates systemic metabolism and brain function. Diabetes 2014, 63, 2232–2243. [Google Scholar] [CrossRef]
- Csajbok, E.A.; Tamas, G. Cerebral cortex: A target and source of insulin? Diabetologia 2016, 59, 1609–1615. [Google Scholar] [CrossRef]
- Margolis, R.U.; Altszuler, N. Insulin in the cerebrospinal fluid. Nature 1967, 215, 1375–1376. [Google Scholar] [CrossRef]
- Woods, S.C.; Porte, D., Jr. Relationship between plasma and cerebrospinal fluid insulin levels of dogs. Am. J. Physiol 1977, 233, E331–E334. [Google Scholar] [CrossRef]
- Blazquez, E.; Velazquez, E.; Hurtado-Carneiro, V.; Ruiz-Albusac, J.M. Insulin in the brain: Its pathophysiological implications for states related with central insulin resistance, Type 2 Diabetes and Alzheimer’s disease. Front. Endocrinol. 2014, 5, 161. [Google Scholar] [CrossRef]
- Konishi, M.; Sakaguchi, M.; Lockhart, S.M.; Cai, W.; Li, M.E.; Homan, E.P.; Rask-Madsen, C.; Kahn, C.R. Endothelial insulin receptors differentially control insulin signaling kinetics in peripheral tissues and brain of mice. Proc. Natl. Acad. Sci. USA 2017, 114, E8478–E8487. [Google Scholar] [CrossRef]
- Sartorius, T.; Peter, A.; Heni, M.; Maetzeler, W.; Fritsche, A.; Haring, H.-U.; Hennige, A.M. The brain response to peripheral insulin declines with age: A contribution of the blood—brain barrier? PLoS ONE 2015, 10, e0126804. [Google Scholar] [CrossRef] [PubMed]
- Nuzzo, D.; Picone, P.; Baldassano, S.; Caruana, L.; Messina, E.; Marino Gammazza, A.; Cappello, F.; Mulè, F.; Di Carlo, M. Insulin resistance as common molecular denominator linking obesity to Alzheimer’s disease. Curr. Alzheimer Res. 2015, 12, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Kellar, D.; Craft, S. Brain insulin resistance in Alzheimer’s disease and related disorders: Mechanisms and therapeutic approaches. Lancet Neurol. 2020, 19, 758–766. [Google Scholar] [CrossRef]
- Accardi, G.; Caruso, C.; Colonna-Romano, G.; Camarda, C.; Monastero, R.; Candore, G. Can Alzheimer disease be a form of type 3 diabetes? Rejuvenation Res. 2012, 15, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Willette, A.A.; Johnson, S.C.; Birdsill, A.C.; Sager, M.A.; Christian, B.; Baker, L.D.; Craft, S.; Oh, J.; Statz, E.; Hermann, B.P.; et al. Insulin resistance predicts brain amyloid deposition in late middle-aged adults. Alzheimers Dement. 2015, 11, 504–510.e1. [Google Scholar] [CrossRef] [PubMed]
- Ekblad, L.L.; Johansson, J.; Helin, S.; Viitanen, M.; Laine, H.; Puukka, P.; Jula, A.; Rinne, J.O. Midlife insulin resistance, APOE genotype, and late-life brain amyloid accumulation. Neurology 2018, 90, e1150–e1157. [Google Scholar] [CrossRef] [PubMed]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—Is this type 3 diabetes? J. Alzheimers Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.-Q.; De Felice, F.G.; Fernandez, S.; Chen, H.; Lambert, M.P.; Quon, M.J.; Krafft, G.A.; Klein, W.L. Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J. 2008, 22, 246–260. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-K.; Kumar, P.; Fu, Q.; Rosen, K.M.; Querfurth, H.W. The insulin/AKT signaling pathway is targeted by intracellular β-amyloid. Mol. Biol. Cell 2009, 20, 1533–1544. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.-Q.; Townsend, M. Insulin resistance and amyloidogenesis as common molecular foundation for type 2 diabetes and Alzheimer’s disease. Biochim. Biophys. Acta-Mol. Basis Disease 2009, 1792, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Long, K.; Williams, T.L.; Urbanc, B. Insulin inhibits Aβ42 aggregation and prevents Aβ42-induced membrane disruption. Biochemistry 2019, 58, 4519–4529. [Google Scholar] [CrossRef] [PubMed]
- Imamura, T.; Yanagihara, Y.T.; Ohyagi, Y.; Nakamura, N.; Iinuma, K.M.; Yamasaki, R.; Asai, H.; Maeda, M.; Murakami, K.; Irie, K.; et al. Insulin deficiency promotes formation of toxic amyloid-β42 conformer co-aggregating with hyper-phosphorylated tau oligomer in an Alzheimer’s disease model. Neurobiol. Dis. 2020, 137, 104739. [Google Scholar] [CrossRef]
- Picone, P.; Giacomazza, D.; Vetri, V.; Carrotta, R.; Militello, V.; San Biagio, P.L.; Di Carlo, M. Insulin-activated Akt rescues Aβ oxidative stress-induced cell death by orchestrating molecular trafficking. Aging Cell 2011, 10, 832–843. [Google Scholar] [CrossRef] [PubMed]
- Galizzi, G.; Di Carlo, M. Insulin and its key role for mitochondrial function/dysfunction and quality control: A shared link between dysmetabolism and neurodegeneration. Biology 2022, 11, 943. [Google Scholar] [CrossRef] [PubMed]
- Picone, P.; Ditta, L.A.; Sabatino, M.A.; Militello, V.; San Biagio, P.L.; Di Giacinto, M.L.; Cristaldi, L.; Nuzzo, D.; Dispenza, C.; Giacomazza, D.; et al. Ionizing radiation-engineered nanogels as insulin nanocarriers for the development of a new strategy for the treatment of Alzheimer’s disease. Biomaterials 2016, 80, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Picone, P.; Sabatino, M.A.; Ditta, L.A.; Amato, A.; San Biagio, P.L.; Mulè, F.; Giacomazza, D.; Dispenza, C.; Di Carlo, M. Nose-to-brain delivery of insulin enhanced by a nanogel carrier. J. Control. Release 2018, 270, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Di Carlo, M.; Picone, P.; Carrotta, R.; Giacomazza, D.; San Biagio, P.L. Insulin promotes survival of amyloid-beta oligomers neuroblastoma damaged cells via caspase 9 inhibition and Hsp70 upregulation. J. Biomed. Biotechnol. 2010, 2010, 147835. [Google Scholar] [CrossRef]
- Mao, Y.-F.; Guo, Z.; Zheng, T.; Jiang, Y.; Yan, Y.; Yin, X.; Chen, Y.; Zhang, B. Intranasal insulin alleviates cognitive deficits and amyloid pathology in young adult APPswe/PS1dE9 mice. Aging Cell 2016, 15, 893–902. [Google Scholar] [CrossRef]
- Pandini, G.; Pace, V.; Copani, A.; Squatrito, S.; Milardi, D.; Vigneri, R. Insulin has multiple antiamyloidogenic effects on human neuronal cells. Endocrinology 2013, 154, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Benedict, C.; Hallschmid, M.; Hatke, A.; Schultes, B.; Fehm, H.L.; Born, J.; Kern, W. Intranasal insulin improves memory in humans. Psyconeuroendocrinology 2009, 29, 1326–1334. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Asthana, S.; Newcomer, J.W.; Wilkinson, C.W.; Tio Matos, I.; Baker, L.D.; Cherrier, M.; Lofgreen, C.; Latendresse, S.; Petrova, A.; et al. Enhancement of memory in Alzheimer disease with insulin and somatostatin, but not glucose. Arch. Gen. Psychiatry 1999, 56, 1135–1140. [Google Scholar] [CrossRef] [PubMed]
- Claxton, A.; Baker, L.D.; Hanson, A.; Trittschuh, E.H.; Cholerton, B.; Morgan, A.; Callaghan, M.; Arbuckle, M.; Behl, C.; Craft, S. Long-acting intranasal insulin Detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer’s disease dementia. J. Alzheimers Dis. 2015, 44, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Nuzzo, D.; Picone, P. Multiple Sclerosis: Focus on extracellular and artificial vesicles, nanoparticles as potential therapeutic approaches. Int. J. Mol. Sci. 2021, 22, 8866. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Sharma, A.R.; Lee, S.; Bhattacharya, M.; Nam, J.; Chakraborty, C. Advances in nanocarriers enabled brain targeted drug delivery across blood brain barrier. Int. J. Pharm. 2019, 559, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Picone, P.; Palumbo, F.S.; Federico, S.; Pitarresi, G.; Adamo, G.; Bongiovanni, A.; Chaves, A.; Cancemi, P.; Muccilli, V.; Giglio, V.; et al. Nano-structured myelin: New nanovesicles for targeted delivery to white matter and microglia, from brain-to-brain. Mater. Today Bio. 2021, 12, 100146. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Peng, Z.; Seven, E.S.; Leblanc, R.M. Crossing the blood-brain barrier with nanoparticles. J. Contr. Release 2018, 28, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Dhuria, S.V.; Hanson, L.R.; Frey, W.R., 2nd. Intranasal delivery to the central nervous system: Mechanisms and experimental considerations. J. Pharm. Sci. 2010, 99, 1654–1673. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, J.J.; Thorne, R.G. Intranasal delivery of biologics to the central nervous system. Adv. Drug Deliv. Rev. 2012, 64, 614–628. [Google Scholar] [CrossRef]
- Yang, P.; Liu, H.J.; Cheng, S.M.; Wang, Z.L.; Cheng, X.; Yu, H.X.; Liu, X.F. Direct transport of VEGF from the nasal cavity to brain. Neurosci. Lett. 2009, 449, 108–111. [Google Scholar] [CrossRef]
- Jogani, V.; Jinturkar, K.; Vyas, T.; Misra, A. Recent patents review on intranasal administration for CNS drug delivery. Recent Patents Drug Deliv. Formul. 2008, 2, 25–40. [Google Scholar] [CrossRef]
- Kamei, N.; Takeda-Morishita, M. Brain delivery of insulin boosted by intranasal coadministration with cell-penetrating peptides. J. Control. Release 2015, 197, 105–110. [Google Scholar] [CrossRef]
- Illum, L.; Davis, S.S. Absorption enhancers for nasal drug delivery. Clin. Pharmacokinet. 2003, 42, 1107–1128. [Google Scholar] [CrossRef]
- McMartin, C.; Hutchinson, L.E.; Hyde, R.; Peters, G.E. Analysis of structural requirements for the absorption of drugs and macromolecules from the nasal cavity. J. Pharm. Sci. 1987, 76, 535–540. [Google Scholar] [CrossRef]
- Hendrick, J.P.; Hartl, F.U. Molecular chaperone functions of Heat-Shock Proteins. Ann. Rev. Biochem. 1993, 62, 349–384. [Google Scholar] [CrossRef]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In Vivo aspects of protein folding and quality control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. Recent advances in understanding catalysis of protein folding by molecular chaperones. FEBS Lett. 2020, 594, 2770–2781. [Google Scholar] [CrossRef] [PubMed]
- Fink, A.L. Chaperone-mediated protein folding. Physiol. Rev. 1993, 79, 425–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saibil, H. Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 2013, 14, 630–642. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.Y.; Lee, S.; Cyr, D.M. Mechanisms for regulating Hsp70 function by Hsp40. Cell Stress Chaperon 2003, 8, 309–316. [Google Scholar] [CrossRef]
- Frank, E.; Madsen, O.; van Rheede, T.; Ricard, J.; Huynen, M.A.; de Jong, W.W. Evolutionary diversity of vertebrate small heat shock proteins. J. Mol. Evol. 2004, 59, 792–805. [Google Scholar] [CrossRef] [PubMed]
- Stengel, F.; Baldwin, A.J.; Painter, A.J.; Benesh, J.L.P. Quaternary dynamics and plasticity underlie small heat shock protein chaperone function. Proc. Natl. Acad. Sci. USA 2010, 107, 2007–2012. [Google Scholar] [CrossRef]
- Young, J.C.; Moarefi, I.; Hartl, F.U. Hsp90: A specialized but essential protein-folding tool. J. Cell Biol. 2001, 154, 267–273. [Google Scholar] [CrossRef]
- Bukau, B.; Horwich, A.L. The Hsp70 and Hsp60 chaperones machines. Cell 1998, 92, 351–366. [Google Scholar] [CrossRef]
- Mogk, A.; Bukau, B.; Kampinga, H.H. Cellular handling of protein aggregates by disaggregation machines. Mol. Cell. 2018, 69, 214–226. [Google Scholar] [CrossRef]
- Muchowski, P.J.; Wacker, J.L. Modulation of neurodegeneration by molecular chaperones. Nat. Rev. Neurosci. 2005, 6, 11–22. [Google Scholar] [CrossRef]
- Molecular mechanisms used by chaperones to reduce the toxicity of aberrant protein oligomers. Proc. Natl. Acad. Sci. USA 2012, 109, 12479–12484. [CrossRef]
- Chaari, A. Molecular chaperones biochemistry and role in neurodegenerative diseases. Int. J. Biol. Macromol. 2019, 131, 396–411. [Google Scholar] [CrossRef]
- Tittelmeier, J.; Nachman, E.; Nussbaum-Krammer, C. Molecular chaperones: A double-edged sword in neurodegenerative diseases. Front. Aging Neurosci. 2020, 12, 581374. [Google Scholar] [CrossRef] [PubMed]
- Bahr, T.; Katuri, J.; Liang, T.; Bai, Y. Mitochondrial chaperones in human health and disease. Free Rad. Biomed. 2021, 179, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Nachman, E.; Wentink, A.S.; Madiona, K.; Bousset, L.; Katsinelos, T.; Allison, K.; Kampinga, H.; McEwan, W.A.; Jahn, T.R.; Melki, R.; et al. Disassembly of Tau fibrils by the human Hsp70 disaggregation machinery generates small seeding competent species. J. Biol. Chem. 2020, 295, 9676–9690. [Google Scholar] [CrossRef]
- Ring, J.; Tadic, J.; Ristic, S.; Poglisch, M.; Bergmann, M.; Radic, N.; Mossmann, D.; Liang, Y.T.; Maglione, M.; Jerkovic, A.; et al. The Hsp40 chaperone Ydj1 drives amiloyd beta 42 toxicity. EMBO Mol. Med. 2022, 14, e13952. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Michaels, T.C.T.; Linse, S.; Mansson, C.; Emanuelsson, C.; Presto, J.; Johansson, J.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Kinetic analysis reveals the diversity of microscopic mechanisms through which molecular chaperones suppress amyloid formation. Nat. Commun. 2016, 7, 10948. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmus, M.M.M.; de Waal, R.M.W.; Verbeek, M.M. Heat Shock proteins and amateur chaperones in amyloid accumulation and clearance in Alzheimer’s Disease. Mol. Neurobiol. 2007, 35, 203–216. [Google Scholar] [CrossRef]
- Bobkova, N.V.; Evgen’ev, M.; Garbuz, D.G.; Kulikov, A.M.; Morozov, A.; Samokhin, A.; Velmeshev, D.; Medvinskaya, N.; Nesterova, I.; Pollock, A.; et al. Exogenous Hsp70 delays senescence and improves cognitive function in aging mice. PNAS 2015, 112, 16006–16011. [Google Scholar] [CrossRef]
- Webster, J.M.; Darling, A.L.; Uversky, V.N.; Blair, L.J. Small heat shock proteins, big impact on protein aggregation in neurodegenerative disease. Front. Pharmacol. 2019, 10, 1047. [Google Scholar] [CrossRef]
- Vendredy, L.; Adrianssens, E.; Timmerman, V. Small heat shock proteins in neurodegenerative diseases. Cell Stress Chaperon 2020, 25, 679–699. [Google Scholar] [CrossRef]
- Mangione, M.R.; Vilasi, S.; Marino, C.; Librizzi, F.; Canale, C.; Spigolon, D.; Bucchieri, F.; Fucarino, A.; Passantino, R.; Cappello, F.; et al. Hsp60, amateur chaperon in amyloid-beta fibrillogenesis. Biochim. Biophys. Acta 2016, 1860, 2474–2483. [Google Scholar] [CrossRef]
- Marino, C.; Krishnan, B.; Cappello, F.; Taglialatela, G. Hsp60 protects against Amyloid β oligomers synaptic toxicity via modification of toxic oligomer conformation. ACS Chem. Neurosci. 2019, 10, 2858–2867. [Google Scholar] [CrossRef]
- Lu, R.C.; Tan, M.S.; Wang, H.; Xie, A.M.; Yu, J.T.; Tan, L. Heat shock protein 70 in Alzheimer’s disease. Biomed. Res. Int. 2014, 2014, 435203. [Google Scholar] [CrossRef]
- Evans, C.G.; Wisén, S.; Gestwicki, J.E. Heat shock proteins 70 and 90 inhibit early stages of amyloid β-(1.42) aggregation in vitro. J. Mol. Biol. 2006, 44, 33182–33191. [Google Scholar] [CrossRef]
- Franco, A.; Velasco-Carneros, L.; Alvarez, N.; Orozco, N.; Moro, F.; Prado, A.; Muga, A. Unzipping the secrets of amyloid disassembly by the human disaggregase. Cells 2021, 10, 2745. [Google Scholar] [CrossRef] [PubMed]
- Harari, A.; Zoltsman, G.; Levin, T.; Rosenzweig, R. Hsp104 N-terminal domain interaction with substrates plays a regulatory role in protein disaggregation. FEBS J. 2022, 289, 5359–5377. [Google Scholar] [CrossRef] [PubMed]
- Low, K.J.Y.; Venkatraman, A.; Metha, J.S.; Pervushin, K. Molecular mechanism of amyloid disaggregation. J. Adv. Res. 2022, 36, 113–132. [Google Scholar] [CrossRef] [PubMed]
- Yakubu, U.M.; Morano, K.A. Suppression of aggregate and amyloid formation by novel intrinsically disordered region in metazoan Hsp110 chaperones. J. Biol. Chem. 2019, 296, 100567. [Google Scholar] [CrossRef] [PubMed]
- Baughman, H.E.R.; Clouser, A.F.; Klevit, R.E.; Nath, A. HspB1 and Hsc70 chaperones engage distinct tau species and have different inhibitory effects on amyloid formation. J. Biol. Chem. 2018, 293, 2687–2700. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.L.; Dahrendorff, J.; Creodore, S.G.; Dickey, C.A.; Blair, L.J.; Uversky, V.N. Small heat shock protein 22 kDa can modulate the aggregation and liquid-liquid phase separation behavior of tau. Protein Sci. 2021, 7, 1350–1359. [Google Scholar] [CrossRef] [PubMed]
- Irvin, R.; Faust, O.; Petrovic, I.; Grayer Wolf, S.; Hofmann, H.H.; Rosenzweig, R. Hsp40s play complementary roles in the prevention of tau amyloid formation. eLife 2021, 10, e69601. [Google Scholar] [CrossRef]
- Kundel, F.; De, S.; Flagmeier, P.; Horrocks, M.H.; Kjaergaard, M.; Shammas, S.L.; Jackson, S.E.; Dobson, C.M.; Klenerman, D. Hsp70 inhibits the nucleation and elongation of tau and sequesters tau aggregates with high affinity. ACS Chem. Biol. 2018, 13, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Radli, M.; Rüdiger, S.G.D. Dancing with the diva: Hsp90-client interactions. J. Mol. Biol. 2018, 430, 3029–3040. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, S.; Zhang, L.; Lu, J.; Zhao, C.; Luo, F.; Li, D.; Li, X.; Liu, C. Heat shock protein 104 (Hsp104) chaperones soluble Tau via a mechanism distinct from its disaggregase activity. J. Biol. Chem. 2019, 294, 4956–4965. [Google Scholar] [CrossRef] [PubMed]
- Peak, S.L.; Gracia, L.; Lora, G.; Jinwal, K. Hsp90-interacting co-chaperones and their family proteins in Tau regulation: Introducing a novel role for Cdc37L1. Neuroscience 2021, 453, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.; Maeda, S.; Vossel, K.; Mucke, L. The many faces of tau. Neuron 2011, 70, 410–426. [Google Scholar] [CrossRef]
- Ryder, B.D.; Wydorski, P.M.; Hou, Z.; Joachimiak, L.A. Chaperoning shape-shifting tau in disease. Trends Biochim. Sci. 2022, 47, 301–312. [Google Scholar] [CrossRef]
- Patterson, K.R.; Ward, S.M.; Combs, B.; Voss, K.; Kanaan, N.M.; Morfini, G.; Brady, S.T.; Gamblin, T.C.; Binder, L.I. Heat shock protein 70 prevents both tau aggregation and the inhibitory effects of preexisting tau aggregates on fast axonal transport. Biochemistry 2011, 50, 10300–10310. [Google Scholar] [CrossRef]
- Weickert, S.; Wawrzyniuk, M.; John, L.H.; Rudiger, S.G.D.; Drescher, M. The mechanism of Hsp90-induced oligomerizaton of Tau. Sci. Adv. 2020, 6, eaax6999. [Google Scholar] [CrossRef] [PubMed]
- Tortosa, E.; Santa-Maria, I.; Moreno, F.; Lim, F.; Perez, M.; Avila, J. Binding of Hsp90 to tau promotes a conformational change and aggregation of tau protein. J. Alz. Dis. 2009, 17, 319–325. [Google Scholar] [CrossRef]
- Blair, L.J.; Nordhues, B.A.; Hill, S.E.; Scaglione, K.M.; O’Leary, J.C.; Fontaine, S.N.; Breydo, L.; Zhang, B.; Li, P.; Wang, L.; et al. Accelerated neurodegeneration through chaperone-mediated oligomerization of tau. J. Clin. Investig. 2013, 123, 4158–4169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shelton, L.B.; Baker, J.D.; Zheng, D.; Sullivan, L.E.; Solanki, P.K.; Webster, J.M.; Sun, Z.; Sabbagh, J.J.; Nordhues, B.A.; Koren, J.; et al. Hsp90 activator Aha1 drives production of pathological tau aggregates. Proc. Natl. Acad. Sci. USA 2017, 114, 9707–9712. [Google Scholar] [CrossRef] [PubMed]
- Mangialasche, F.; Solomon, A.; Winblad, B.; Mecocci, P.; Kivipelto, M. Alzheimer’s disease: Clinical trials and drug development. Lancet Neurol. 2010, 9, 702–716. [Google Scholar] [CrossRef]
- Arvanitakis, Z.; Shah, R.C.; Bennett, D.A. Diagnosis and management of dementia: Review. JAMA 2019, 322, 1589–1599. [Google Scholar] [CrossRef]
- Vaz, M.; Silva, V.; Monteiro, C.; Silvestre, S. Role of aducanumab in the treatment of Alzheimer’s Disease: Challenges and opportunities. Clin. Interv. Aging 2022, 17, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Lythgoe, M.P.; Jenei, K.; Prasad, V. Regulatory decisions diverge over aducanumab for Alzheimer’s disease. BMJ 2022, 376, e069780. [Google Scholar] [CrossRef] [PubMed]
- Hallschmid, M. Intranasal insulin for Alzheimer’s disease. CNS Drug 2021, 35, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Handattu, S.P.; Garber, D.W.; Monroe, C.E.; vanGroen, T.; Kadish, I.; Nayyar, G.; Cao, D.; Palgunachari, M.N.; Li, L.; Anantharamaiah, G.M. Oral apolipoprotein a-i mimetic peptide improves cognitive function and reduces amyloid burden in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2009, 34, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Citron, M. Alzheimer’s disease: Strategies for disease modification. Nat. Rev. Drug Discov. 2010, 9, 387–398. [Google Scholar] [CrossRef]
- van Bokhoven, P.; de Wilde, A.; Vermunt, L.; Leferink, P.S.; Heetveld, S.; Cummings, J.; Scheltens, P.; Vijverberg, E.G.B. The Alzheimer’s disease drug development landscape. Alzheimers Res. Ther. 2021, 13, 186. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide/Protein | MW (Da) | Biological Effect/Target |
|---|---|---|
| Egg-derived peptides | 25–250 | Antioxidant effect [89,90,97] Cholinesterase and BACE inhibitory activity [98,99,100,101] |
| BSB peptides | 25–250 | Destabilizing effect against Abeta fibrillation process [116,117,118,123] Anti-inflammatory effect [119] |
| DF-4 peptide | 2300 | Reduction of the microglia and astrocyte activation [217] Decrease of Abeta deposition [217] Increase the cognitive function [217] |
| Insulin | 5800 | Akt insulin survival pathway activation [142,146] Destabilizing effect against Abeta fibrillation process [144] Inhibition of the intrinsic apoptotic pathway and mitochondrial protection [150] Antioxidant effect [146] |
| MT3 | 6000 | Maintenance of metal ion brain homeostasis [62] |
| ZnT3 | 65,000 | Maintenance of Zn2+ brain homeostasis [66] |
| S100 family | 10,000–12,000 | Promotion of neuronal and neurite growth [68] |
| Copper- chelating peptides | 250–1000 | Ability in chelating Cu2+ ions, reducing ROS formation and amyloid toxicity [60,73] |
| HSPs | 15,000–100,000 | Chaperon activity [171] Reduction of the neurotoxicity of misfolded protein aggregates [168,171] Inhibition of Abeta aggregation [192] Solubilization of protein aggregates [170,171] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Picone, P.; Sanfilippo, T.; Vasto, S.; Baldassano, S.; Guggino, R.; Nuzzo, D.; Bulone, D.; San Biagio, P.L.; Muscolino, E.; Monastero, R.; et al. From Small Peptides to Large Proteins against Alzheimer’sDisease. Biomolecules 2022, 12, 1344. https://doi.org/10.3390/biom12101344
Picone P, Sanfilippo T, Vasto S, Baldassano S, Guggino R, Nuzzo D, Bulone D, San Biagio PL, Muscolino E, Monastero R, et al. From Small Peptides to Large Proteins against Alzheimer’sDisease. Biomolecules. 2022; 12(10):1344. https://doi.org/10.3390/biom12101344
Chicago/Turabian StylePicone, Pasquale, Tiziana Sanfilippo, Sonya Vasto, Sara Baldassano, Rossella Guggino, Domenico Nuzzo, Donatella Bulone, Pier Luigi San Biagio, Emanuela Muscolino, Roberto Monastero, and et al. 2022. "From Small Peptides to Large Proteins against Alzheimer’sDisease" Biomolecules 12, no. 10: 1344. https://doi.org/10.3390/biom12101344
APA StylePicone, P., Sanfilippo, T., Vasto, S., Baldassano, S., Guggino, R., Nuzzo, D., Bulone, D., San Biagio, P. L., Muscolino, E., Monastero, R., Dispenza, C., & Giacomazza, D. (2022). From Small Peptides to Large Proteins against Alzheimer’sDisease. Biomolecules, 12(10), 1344. https://doi.org/10.3390/biom12101344