
Alzheimer’s Disease: Challenges and a Therapeutic Opportunity to Treat It with a Neurotrophic Compound


Abstract
:1. Introduction
2. Ethiopathogenesis of AD
2.1. Amyloid β and Neurofibrillary Tangles
2.2. Relation between Key Player Proteins in AD Pathology and Plasticity
2.2.1. Presenilin 1 (PS1)
2.2.2. Tau
2.2.3. Amyloid-β
2.2.4. APOE
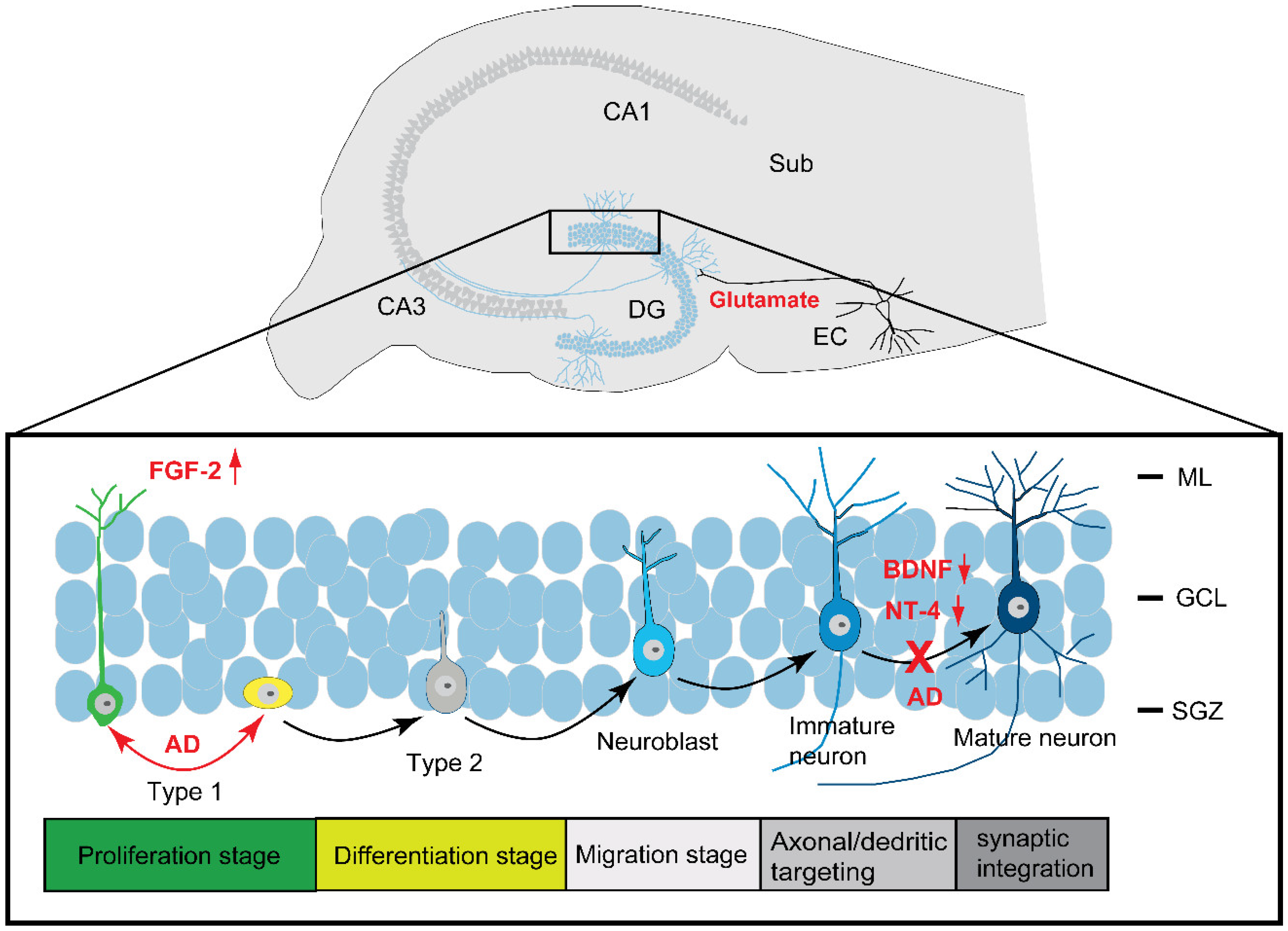
2.3. Neurogenesis in AD
2.3.1. Neurogenesis
2.3.2. Neurogenesis in AD
2.4. Neurodegeneration, Synaptic Deficit, and Synaptic Compensation
2.4.1. Neurodegeneration
2.4.2. Synaptic Deficit
2.4.3. Synaptic Compensation
2.5. Neuroinflammation
2.6. Cognitive Impairment
2.7. Growth Factors and Neurotrophins
3. Development of Drugs for AD
3.1. Shift from Large Molecules to Small Peptidergic Compounds
3.1.1. Peptides as Drugs
3.1.2. Neurotrophic Factor Peptide Mimetics as Potential Drugs for AD
3.1.3. Current Status of AD Approved and Developing Drugs in the Market
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Förstl, H.; Kurz, A. Clinical Features of Alzheimer’s Disease. Eur. Arch. Psychiatry Clin. Neurosci. 1999, 249, 288–290. [Google Scholar] [CrossRef]
- López, O.L.; DeKosky, S.T. Clinical Symptoms in Alzheimer’s Disease. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2008; Volume 89, pp. 207–216. ISBN 978-0-444-51898-9. [Google Scholar]
- Petersen, R.C.; Roberts, R.O.; Knopman, D.S.; Boeve, B.F.; Geda, Y.E.; Ivnik, R.J.; Smith, G.E.; Jack, C.R. Mild Cognitive Impairment: Ten Years Later. Arch. Neurol. 2009, 66, 1447–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKhann, G.; Knopman, D.S.; Chertkow, H.; Hymann, B.; Jack, C.R.; Kawas, C.; Klunk, W.; Koroshetz, W.; Manly, J.; Mayeux, R.; et al. The Diagnosis of Dementia Due to Alzheimer’s Disease: Recommendations from the National Institute on Aging- Alzheimer’s Association Workgroups on Diagnostic Guidelines for Alzheimer’s Disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Glenner, G.G.; Wong, C.W. Alzheimer’s Disease: Initial Report of the Purification and Characterization of a Novel Cerebrovascular Amyloid Protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Iqbal, K.; Zaidi, T.; Thompson, C.H.; Merz, P.A.; Wisniewski, H.M. Alzheimer Paired Helical Filaments: Bulk Isolation, Solubility, and Protein Composition. Acta Neuropathol. 1984, 62, 167–177. [Google Scholar] [CrossRef]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal Phosphorylation of the Microtubule-Associated Protein Tau (Tau) in Alzheimer Cytoskeletal Pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundke-Iqbal, I.; Iqbal, K.; Quinlan, M.; Tung, Y.C.; Zaidi, M.S.; Wisniewski, H.M. Microtubule-Associated Protein Tau. A Component of Alzheimer Paired Helical Filaments. J. Biol. Chem. 1986, 261, 6084–6089. [Google Scholar] [CrossRef]
- Babcock, K.R.; Page, J.S.; Fallon, J.R.; Webb, A.E. Adult Hippocampal Neurogenesis in Aging and Alzheimer’s Disease. Stem Cell Rep. 2021, 16, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer Disease-Associated Neurofibrillary Pathology Using Paraffin Sections and Immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Isla, T.; Price, J.L.; McKeel, D.W., Jr.; Morris, J.C.; Growdon, J.H.; Hyman, B.T. Profound Loss of Layer II Entorhinal Cortex Neurons Occurs in Very Mild Alzheimer’s Disease. J. Neurosci. 1996, 16, 4491–4500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomlinson, B.E.; Blessed, G.; Roth, M. Observations on the Brains of Demented Old People. J. Neurol. Sci. 1970, 11, 205–242. [Google Scholar] [CrossRef]
- Alafuzoff, I.; Iqbal, K.; Friden, H.; Adolfsson, R.; Winblad, B. Histopathological Criteria for Progressive Dementia Disorders: Clinical-Pathological Correlation and Classification by Multivariate Data Analysis. Acta Neuropathol. 1987, 74, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Hou, J.; Ping, J.; Cai, D. Advances in Developing Novel Therapeutic Strategies for Alzheimer’s Disease. Mol. Neurodegener. 2018, 13, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Yamamori, H.; Tatebayashi, Y.; Shafit-Zagardo, B.; Tanimukai, H.; Chen, S.; Iqbal, K.; Grundke-Iqbal, I. Failure of Neuronal Maturation in Alzheimer Disease Dentate Gyrus. J. Neuropathol. Exp. Neurol. 2008, 67, 78–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruts, M.; Hendriks, L.; Van Broeckhoven, C. The Presenilin Genes: A New Gene Family Involved in Alzheimer Disease Pathology. Hum. Mol. Genet. 1996, 5, 1449–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, R.F.; Hutton, M.; Fuldner, M.; Froelich, S.; Karran, E.; Talbot, C.; Crook, R.; Lendon, C.; Prihar, G.; He, C.; et al. The Structure of the Presenilin 1 (S182) Gene and Identification of Six Novel Mutations in Early Onset AD Families. Nat. Genet. 1995, 11, 219–222. [Google Scholar] [CrossRef]
- Strittmatter, W.J.; Roses, A.D. Apolipoprotein E and Alzheimer Disease. Proc. Natl. Acad. Sci. USA 1995, 92, 4725–4727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene Dose of Apolipoprotein E Type 4 Allele and the Risk of Alzheimer’s Disease in Late Onset Families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Disouky, A.; Lazarov, O. Adult Hippocampal Neurogenesis in Alzheimer’s Disease. In Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netherlands, 2021; Volume 177, pp. 137–156. ISBN 978-0-12-824143-1. [Google Scholar]
- Aggidis, A.; Chatterjee, S.; Townsend, D.; Fullwood, N.J.; Ortega, E.R.; Tarutani, A.; Hasegawa, M.; Lucas, H.; Mudher, A.; Allsop, D. Peptide-Based Inhibitors of Tau Aggregation as a Potential Therapeutic for Alzheimer’s Disease and Other Tauopathies. bioRxiv 2021. [Google Scholar] [CrossRef]
- Lazarov, O.; Marr, R.A. Neurogenesis and Alzheimer’s Disease: At the Crossroads. Exp. Neurol. 2010, 223, 267–281. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winner, B.; Kohl, Z.; Gage, F.H. Neurodegenerative Disease and Adult Neurogenesis: Neurodegenerative Disease and Adult Neurogenesis. Eur. J. Neurosci. 2011, 33, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Trojanowski, J.Q.; Lee, V.M.-Y. “Fatal Attractions” of Proteins: A Comprehensive Hypothetical Mechanism Underlying Alzheimer’s Disease and Other Neurodegenerative Disorders. Ann. N. Y. Acad. Sci. 2006, 924, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Cuello, A.C. Intracellular and Extracellular Aβ, a Tale of Two Neuropathologies. Brain Pathol. 2006, 15, 66–71. [Google Scholar] [CrossRef]
- Mayes, J.; Tinker-Mill, C.; Kolosov, O.; Zhang, H.; Tabner, B.J.; Allsop, D. β-Amyloid Fibrils in Alzheimer Disease Are Not Inert When Bound to Copper Ions but Can Degrade Hydrogen Peroxide and Generate Reactive Oxygen Species. J. Biol. Chem. 2014, 289, 12052–12062. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Braak, E. Neuropathological Stageing of Alzheimer-Related Changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, S.E.; Hyman, B.T.; Flory, J.; Damasio, A.R.; Hoesen, G.W.V.; Van Hoesen, G.W. The Topographical and Neuroanatomical Distribution of Neurofibrillary Tangles and Neuritic Plaques in the Cerebral Cortex of Patients with Alzheimer’s Disease. Cereb. Cortex 1991, 1, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Gibson, P.H.; Tomlinson, B.E. Numbers of Hirano Bodies in the Hippocampus of Normal and Demented People with Alzheimer’s Disease. J. Neurol. Sci. 1977, 33, 199–206. [Google Scholar] [CrossRef]
- Ingelsson, M.; Fukumoto, H.; Newell, K.L.; Growdon, J.H.; Hedley-Whyte, E.T.; Frosch, M.P.; Albert, M.S.; Hyman, B.T.; Irizarry, M.C. Early Abeta Accumulation and Progressive Synaptic Loss, Gliosis, and Tangle Formation in AD Brain. Neurology 2004, 62, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Isla, T.; Hollister, R.; West, H.; Mui, S.; Growdon, J.H.; Petersen, R.C.; Parisi, J.E.; Hyman, B.T. Neuronal Loss Correlates with but Exceeds Neurofibrillary Tangles in Alzheimer’s Disease. Ann. Neurol. 1997, 41, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Olichney, J.M.; Galasko, D.; Salmon, D.P.; Hofstetter, C.R.; Hansen, L.A.; Katzman, R.; Thal, L.J. Cognitive Decline Is Faster in Lewy Body Variant than in Alzheimer’s Disease. Neurology 1998, 51, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Arriagada, P.V.; Growdon, J.H.; Hedley-Whyte, E.T.; Hyman, B.T.; Hedleywhyte, E.T.; Hyman, B.T. Neurofibrillary Tangles but Not Senile Plaques Parallel Duration and Severity of Alzheimer’s Disease. Neurology 1992, 42, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Kaffy, J.; Brinet, D.; Soulier, J.-L.; Correia, I.; Tonali, N.; Fera, K.F.; Iacone, Y.; Hoffmann, A.R.F.; Khemtémourian, L.; Crousse, B.; et al. Designed Glycopeptidomimetics Disrupt Protein–Protein Interactions Mediating Amyloid β-Peptide Aggregation and Restore Neuroblastoma Cell Viability. J. Med. Chem. 2016, 59, 2025–2040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, S.I.A.; Linse, S.; Luheshi, L.M.; Hellstrand, E.; White, D.A.; Rajah, L.; Otzen, D.E.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Proliferation of Amyloid-Β42 Aggregates Occurs through a Secondary Nucleation Mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 9758–9763. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.; Allsop, D. Amyloid Deposition as the Central Event in the Aetiology of Alzheimer’s Disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Ballatore, C.; Lee, V.M.-Y.; Trojanowski, J.Q. Tau-Mediated Neurodegeneration in Alzheimer’s Disease and Related Disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar] [CrossRef]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM Structures of Tau Filaments from Alzheimer’s Disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Cowan, C.M.; Mudher, A. Are Tau Aggregates Toxic or Protective in Tauopathies? Front. Neurol. 2013, 4, 114. [Google Scholar] [CrossRef] [Green Version]
- Santa-Maria, I.; Varghese, M.; Ksiȩżak-Reding, H.; Dzhun, A.; Wang, J.; Pasinetti, G.M. Paired Helical Filaments from Alzheimer Disease Brain Induce Intracellular Accumulation of Tau Protein in Aggresomes. J. Biol. Chem. 2012, 287, 20522–20533. [Google Scholar] [CrossRef]
- Sung, P.-S.; Lin, P.-Y.; Liu, C.-H.; Su, H.-C.; Tsai, K.-J. Neuroinflammation and Neurogenesis in Alzheimer’s Disease and Potential Therapeutic Approaches. Int. J. Mol. Sci. 2020, 21, 701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacobini, E.; Gold, G. Alzheimer Disease Therapy–Moving from Amyloid-β to Tau. Nat. Rev. Neurol. 2013, 9, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Silver, M.H.; Newell, K.; Brady, C.; Hedley-White, E.T.; Perls, T.T. Distinguishing between Neurodegenerative Disease and Disease-Free Aging: Correlating Neuropsychological Evaluations and Neuropathological Studies in Centenarians. Psychosom. Med. 2002, 64, 493–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, M.S.; Kaye, J.A.; Ball, M.J. The Oregon Brain Aging Study: Neuropathology Accompanying Healthy Aging in the Oldest Old. Neurology 2000, 54, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Baazaoui, N. Effect of CNTF Derived Peptide, P021 on Cognition and Pathology in 3xTG-AD Mouse Model of Alzheimer’s Disease. Ph.D. Thesis, Graduate Center, City University of New York, New York, NY, USA, 2016. [Google Scholar]
- Gold, G.; Bouras, C.; Kövari, E.; Canuto, A.; Glaría, B.G.; Malky, A.; Hof, P.R.; Michel, J.P.; Giannakopoulos, P. Clinical Validity of Braak Neuropathological Staging in the Oldest-Old. Acta Neuropathol. 2000, 99, 583–584. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.P.; Snowdon, D.A.; Markesbery, W.R. Alzheimer’s Neurofibrillary Pathology and the Spectrum of Cognitive Function: Findings from the Nun Study. Ann. Neurol. 2002, 51, 567–577. [Google Scholar] [CrossRef]
- Berezovska, O.; Frosch, M.; McLean, P.; Knowles, R.; Koo, E.; Kang, D.; Shen, J.; Lu, F.M.; Lux, S.E.; Tonegawa, S.; et al. The Alzheimer-Related Gene Presenilin 1 Facilitates Notch 1 in Primary Mammalian Neurons. Mol. Brain Res. 1999, 69, 273–280. [Google Scholar] [CrossRef]
- Wen, P.H.; Friedrich, V.L.; Shioi, J.; Robakis, N.K.; Elder, G.A. Presenilin-1 Is Expressed in Neural Progenitor Cells in the Hippocampus of Adult Mice. Neurosci. Lett. 2002, 318, 53–56. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, H.; Ma, Y. Molecular Mechanisms of Altered Adult Hippocampal Neurogenesis in Alzheimer’s Disease. Mech. Ageing Dev. 2021, 195, 111452. [Google Scholar] [CrossRef]
- Lee, M.K.; Slunt, H.H.; Martin, L.J.; Thinakaran, G.; Kim, G.; Gandy, S.E.; Seeger, M.; Koo, E.; Price, D.L.; Sisodia, S.S. Expression of Presenilin 1 and 2 (PS1 and PS2) in Human and Murine Tissues. J. Neurosci. 1996, 16, 7513–7525. [Google Scholar] [CrossRef]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A Protein Factor Essential for Microtubule Assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bullmann, T.; de Silva, R.; Holzer, M.; Mori, H.; Arendt, T. Expression of Embryonic Tau Protein Isoforms Persist during Adult Neurogenesis in the Hippocampus. Hippocampus 2007, 17, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Fuster-Matanzo, A.; Llorens-Martín, M.; Jurado-Arjona, J.; Avila, J.; Hernández, F. Tau Protein and Adult Hippocampal Neurogenesis. Front. Neurosci. 2012, 6, 104. [Google Scholar] [CrossRef] [Green Version]
- Llorens-Martin, M.; Teixeira, C.M.; Fuster-Matanzo, A.; Jurado-Arjona, J.; Borrell, V.; Soriano, E.; Avila, J.; Hernández, F. Tau Isoform with Three Microtubule Binding Domains Is a Marker of New Axons Generated from the Subgranular Zone in the Hippocampal Dentate Gyrus: Implications for Alzheimer’s Disease. JAD 2012, 29, 921–930. [Google Scholar] [CrossRef] [Green Version]
- Komuro, Y.; Xu, G.; Bhaskar, K.; Lamb, B.T. Human Tau Expression Reduces Adult Neurogenesis in a Mouse Model of Tauopathy. Neurobiol. Aging 2015, 36, 2034–2042. [Google Scholar] [CrossRef] [Green Version]
- Pristerà, A.; Saraulli, D.; Farioli-Vecchioli, S.; Strimpakos, G.; Costanzi, M.; di Certo, M.G.; Cannas, S.; Ciotti, M.T.; Tirone, F.; Mattei, E.; et al. Impact of N-Tau on Adult Hippocampal Neurogenesis, Anxiety, and Memory. Neurobiol. Aging 2013, 34, 2551–2563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Criado-Marrero, M.; Sabbagh, J.J.; Jones, M.R.; Chaput, D.; Dickey, C.A.; Blair, L.J. Hippocampal Neurogenesis Is Enhanced in Adult Tau Deficient Mice. Cells 2020, 9, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dioli, C.; Patrício, P.; Trindade, R.; Pinto, L.G.; Silva, J.M.; Morais, M.; Ferreiro, E.; Borges, S.; Mateus-Pinheiro, A.; Rodrigues, A.J.; et al. Tau-Dependent Suppression of Adult Neurogenesis in the Stressed Hippocampus. Mol. Psychiatry 2017, 22, 1110–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollands, C.; Tobin, M.K.; Hsu, M.; Musaraca, K.; Yu, T.-S.; Mishra, R.; Kernie, S.G.; Lazarov, O. Depletion of Adult Neurogenesis Exacerbates Cognitive Deficits in Alzheimer’s Disease by Compromising Hippocampal Inhibition. Mol. Neurodegener. 2017, 12, 64. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Li, H.-L.; Tian, N.; Liu, F.; Wang, L.; Yin, Y.; Yue, L.; Ma, L.; Wan, Y.; Wang, J.-Z. Interneuron Accumulation of Phosphorylated Tau Impairs Adult Hippocampal Neurogenesis by Suppressing GABAergic Transmission. Cell Stem Cell 2020, 26, 331–345.e6. [Google Scholar] [CrossRef]
- Farioli-Vecchioli, S.; Ricci, V.; Middei, S. Adult Hippocampal Neurogenesis in Alzheimer’s Disease: An Overview of Human and Animal Studies with Implications for Therapeutic Perspectives Aimed at Memory Recovery. Neural Plast. 2022, 2022, 9959044. [Google Scholar] [CrossRef]
- Morales-Garcia, J.A.; Luna-Medina, R.; Alonso-Gil, S.; Sanz-Sancristobal, M.; Palomo, V.; Gil, C.; Santos, A.; Martinez, A.; Perez-Castillo, A. Glycogen Synthase Kinase 3 Inhibition Promotes Adult Hippocampal Neurogenesis in Vitro and in Vivo. ACS Chem. Neurosci. 2012, 3, 963–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maqbool, M.; Mobashir, M.; Hoda, N. Pivotal Role of Glycogen Synthase Kinase-3: A Therapeutic Target for Alzheimer’s Disease. Eur. J. Med. Chem. 2016, 107, 63–81. [Google Scholar] [CrossRef]
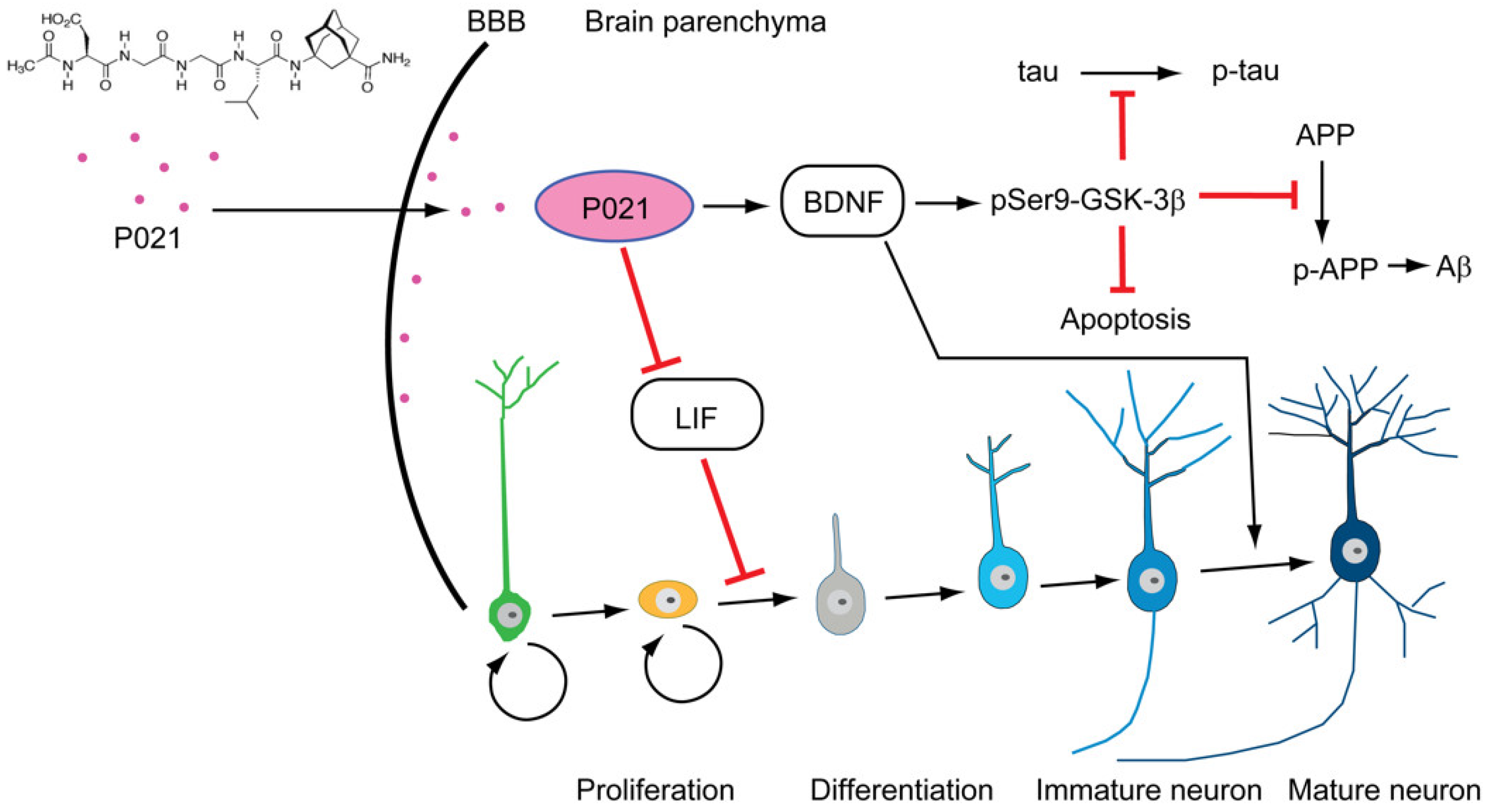
- Bolognin, S.; Buffelli, M.; Puoliväli, J.; Iqbal, K. Rescue of Cognitive-Aging by Administration of a Neurogenic and/or Neurotrophic Compound. Neurobiol. Aging 2014, 35, 2134–2146. [Google Scholar] [CrossRef] [PubMed]
- Kazim, S.F.; Blanchard, J.; Dai, C.L.; Tung, Y.C.; Laferla, F.M.; Iqbal, I.G.; Iqbal, K.; Faraz, S.; Blanchard, J.; Dai, C.L.; et al. Disease Modifying Effect of Chronic Oral Treatment with a Neurotrophic Peptidergic Compound in a Triple Transgenic Mouse Model of Alzheimer’ s Disease. Neurobiol. Dis. 2014, 71, 110–130. [Google Scholar] [CrossRef] [PubMed]
- Baazaoui, N.; Iqbal, K. Prevention of Dendritic and Synaptic Deficits and Cognitive Impairment with a Neurotrophic Compound. Alzheimer’s Res. Ther. 2017, 9, 45. [Google Scholar] [CrossRef]
- Baglietto-Vargas, D.; Sánchez-Mejias, E.; Navarro, V.; Jimenez, S.; Trujillo-Estrada, L.; Gómez-Arboledas, A.; Sánchez-Mico, M.; Sánchez-Varo, R.; Vizuete, M.; Dávila, J.C.; et al. Dual Roles of Aβ in Proliferative Processes in an Amyloidogenic Model of Alzheimer’s Disease. Sci. Rep. 2017, 7, 10085. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Halabisky, B.; Zhou, Y.; Palop, J.J.; Yu, G.; Mucke, L.; Gan, L. Imbalance between GABAergic and Glutamatergic Transmission Impairs Adult Neurogenesis in an Animal Model of Alzheimer’s Disease. Cell Stem Cell 2009, 5, 624–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armato, U.; Chakravarthy, B.; Pacchiana, R.; Whitfield, J.F. Alzheimer’s Disease: An Update of the Roles of Receptors, Astrocytes and Primary Cilia (Review). Int. J. Mol. Med. 2013, 31, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caviston, J.P.; Holzbaur, E.L.F. Huntingtin as an Essential Integrator of Intracellular Vesicular Trafficking. Trends Cell Biol. 2009, 19, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Simuni, T.; Sethi, K. Nonmotor Manifestations of Parkinson’s Disease. Ann. Neurol. 2008, 64 (Suppl. S2), S65–S80. [Google Scholar] [CrossRef] [PubMed]
- Suidan, G.L.; Ramaswamy, G. Targeting Apolipoprotein E for Alzheimer’s Disease: An Industry Perspective. Int. J. Mol. Sci. 2019, 20, 2161. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.-P.; Gilley, J.A.; Zhang, G.; Kernie, S.G. ApoE Is Required for Maintenance of the Dentate Gyrus Neural Progenitor Pool. Development 2011, 138, 4351–4362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahley, R.W.; Rall, S.C. Apolipoprotein E: Far More Than a Lipid Transport Protein. Annu. Rev. Genom. Hum. Genet. 2000, 1, 507–537. [Google Scholar] [CrossRef]
- Li, G.; Bien-Ly, N.; Andrews-Zwilling, Y.; Xu, Q.; Bernardo, A.; Ring, K.; Halabisky, B.; Deng, C.; Mahley, R.W.; Huang, Y. GABAergic Interneuron Dysfunction Impairs Hippocampal Neurogenesis in Adult Apolipoprotein E4 Knockin Mice. Cell Stem Cell 2009, 5, 634–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levi, O.; Michaelson, D.M. Environmental Enrichment Stimulates Neurogenesis in Apolipoprotein E3 and Neuronal Apoptosis in Apolipoprotein E4 Transgenic Mice. J. Neurochem. 2007, 100, 202–210. [Google Scholar] [CrossRef]
- Mu, Y.; Gage, F.H. Adult Hippocampal Neurogenesis and Its Role in Alzheimer’s Disease. Mol. Neurodegener. 2011, 6, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spalding, K.L.; Bergmann, O.; Alkass, K.; Bernard, S.; Salehpour, M.; Huttner, H.B.; Boström, E.; Westerlund, I.; Vial, C.; Buchholz, B.A.; et al. Dynamics of Hippocampal Neurogenesis in Adult Humans. Cell 2013, 153, 1219–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuster-Matanzo, A.; Llorens-Martín, M.; Hernández, F.; Avila, J. Role of Neuroinflammation in Adult Neurogenesis and Alzheimer Disease: Therapeutic Approaches. Mediat. Inflamm. 2013, 2013, 260925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Pareek, V.; Faiq, M.A.; Ghosh, S.K.; Kumari, C. Adult Neurogenesis in Humans: A Review of Basic Concepts, History, Current Research, and Clinical Implications. Innov. Clin. Neurosci. 2019, 16, 30–37. [Google Scholar] [PubMed]
- Schmidt-Hieber, C.; Jonas, P.; Bischofberger, J. Enhanced Synaptic Plasticity in Newly Generated Granule Cells of the Adult Hippocampus. Nature 2004, 429, 184–187. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Scott, B.W.; Wojtowicz, J.M. Heterogenous Properties of Dentate Granule Neurons in the Adult Rat. J. Neurobiol. 2000, 42, 248–257. [Google Scholar] [CrossRef]
- Yang, C.-H.; Di Antonio, A.; Kirschen, G.W.; Varma, P.; Hsieh, J.; Ge, S. Circuit Integration Initiation of New Hippocampal Neurons in the Adult Brain. Cell Rep. 2020, 30, 959–968.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elder, G.A.; De Gasperi, R.; Gama Sosa, M.A. Research Update: Neurogenesis in Adult Brain and Neuropsychiatric Disorders. Mt. Sinai J. Med. 2006, 73, 931–940. [Google Scholar] [PubMed]
- Bielefeld, P.; Durá, I.; Danielewicz, J.; Lucassen, P.J.; Baekelandt, V.; Abrous, D.N.; Encinas, J.M.; Fitzsimons, C.P. Insult-Induced Aberrant Hippocampal Neurogenesis: Functional Consequences and Possible Therapeutic Strategies. Behav. Brain Res. 2019, 372, 112032. [Google Scholar] [CrossRef] [PubMed]
- Sahay, A.; Scobie, K.N.; Hill, A.S.; O’Carroll, C.M.; Kheirbek, M.A.; Burghardt, N.S.; Fenton, A.A.; Dranovsky, A.; Hen, R. Increasing Adult Hippocampal Neurogenesis Is Sufficient to Improve Pattern Separation. Nature 2011, 472, 466–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruel-Jungerman, E.; Rampon, C.; Laroche, S. Adult Hippocampal Neurogenesis, Synaptic Plasticity and Memory: Facts and Hypotheses. Rev. Neurosci. 2007, 18, 93–114. [Google Scholar] [CrossRef] [PubMed]
- Goodman, T.; Trouche, S.; Massou, I.; Verret, L.; Zerwas, M.; Roullet, P.; Rampon, C. Young Hippocampal Neurons Are Critical for Recent and Remote Spatial Memory in Adult Mice. Neuroscience 2010, 171, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Akers, K.G.; Martinez-Canabal, A.; Restivo, L.; Yiu, A.P.; De Cristofaro, A.; Hsiang, H.-L.; Wheeler, A.L.; Guskjolen, A.; Niibori, Y.; Shoji, H.; et al. Hippocampal Neurogenesis Regulates Forgetting During Adulthood and Infancy. Science 2014, 344, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Nakashiba, T.; Cushman, J.D.; Pelkey, K.A.; Renaudineau, S.; Buhl, D.L.; McHugh, T.J.; Rodriguez Barrera, V.; Chittajallu, R.; Iwamoto, K.S.; McBain, C.J.; et al. Young Dentate Granule Cells Mediate Pattern Separation, Whereas Old Granule Cells Facilitate Pattern Completion. Cell 2012, 149, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Anacker, C.; Luna, V.M.; Stevens, G.S.; Millette, A.; Shores, R.; Jimenez, J.C.; Chen, B.; Hen, R. Hippocampal Neurogenesis Confers Stress Resilience by Inhibiting the Ventral Dentate Gyrus. Nature 2018, 559, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Deisseroth, K.; Singla, S.; Toda, H.; Monje, M.; Palmer, T.D.; Malenka, R.C. Excitation-Neurogenesis Coupling in Adult Neural Stem/Progenitor Cells. Neuron 2004, 42, 535–552. [Google Scholar] [CrossRef] [Green Version]
- Enwere, E. Aging Results in Reduced Epidermal Growth Factor Receptor Signaling, Diminished Olfactory Neurogenesis, and Deficits in Fine Olfactory Discrimination. J. Neurosci. 2004, 24, 8354–8365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aizawa, K.; Ageyama, N.; Terao, K.; Hisatsune, T. Primate-Specific Alterations in Neural Stem/Progenitor Cells in the Aged Hippocampus. Neurobiol. Aging 2011, 32, 140–150. [Google Scholar] [CrossRef]
- Leuner, B.; Kozorovitskiy, Y.; Gross, C.G.; Gould, E. Diminished Adult Neurogenesis in the Marmoset Brain Precedes Old Age. Proc. Natl. Acad. Sci. USA 2007, 104, 17169–17173. [Google Scholar] [CrossRef] [Green Version]
- Tobin, M.K.; Musaraca, K.; Disouky, A.; Shetti, A.; Bheri, A.; Honer, W.G.; Kim, N.; Dawe, R.J.; Bennett, D.A.; Arfanakis, K.; et al. Human Hippocampal Neurogenesis Persists in Aged Adults and Alzheimer’s Disease Patients. Cell Stem Cell 2019, 24, 974–982.e3. [Google Scholar] [CrossRef]
- Bergmann, O.; Spalding, K.L.; Frisén, J. Adult Neurogenesis in Humans. Cold Spring Harb. Perspect. Biol. 2015, 7, a018994. [Google Scholar] [CrossRef] [Green Version]
- Jin, K.; Peel, A.L.; Mao, X.O.; Xie, L.; Cottrell, B.A.; Henshall, D.C.; Greenberg, D.A. Increased Hippocampal Neurogenesis in Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2004, 101, 343–347. [Google Scholar] [CrossRef] [Green Version]
- Hock, C.; Heese, K.; Hulette, C.; Rosenberg, C.; Bryan, P.; Bank, B. Region-Specific Neurotrophin Imbalances in Alzheimer Disease. Arch. Neurol. 2000, 57, 846–851. [Google Scholar] [CrossRef] [Green Version]
- Stopa, E.G.; Gonzalez, A.M.; Chorsky, R.; Corona, R.J.; Alvarez, J.; Bird, E.D.; Baird, A. Basic Fibroblast Growth Factor in Alzheimer’s Disease. Biochem. Biophys. Res. Commun. 1990, 171, 690–696. [Google Scholar] [CrossRef]
- Chen, H.; Tung, Y.-C.C.; Li, B.; Iqbal, K.; Grundke-Iqbal, I. Trophic Factors Counteract Elevated FGF-2-Induced Inhibition of Adult Neurogenesis. Neurobiol. Aging 2007, 28, 1148–1162. [Google Scholar] [CrossRef] [PubMed]
- Crews, L.; Adame, A.; Patrick, C.; Delaney, A.; Pham, E.; Rockenstein, E.; Hansen, L.; Masliah, E. Increased BMP6 Levels in the Brains of Alzheimer’s Disease Patients and APP Transgenic Mice Are Accompanied by Impaired Neurogenesis. J. Neurosci. 2010, 30, 12252–12262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Jiménez, E.P.; Flor-García, M.; Terreros-Roncal, J.; Rábano, A.; Cafini, F.; Pallas-Bazarra, N.; Ávila, J.; Llorens-Martín, M. Adult Hippocampal Neurogenesis Is Abundant in Neurologically Healthy Subjects and Drops Sharply in Patients with Alzheimer’s Disease. Nat. Med. 2019, 25, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Fox, N.C.; Freeborough, P.A.; Rossor, M.N. Visualisation and Quantification of Rates of Atrophy in Alzheimer’s Disease. Lancet 1996, 348, 94–97. [Google Scholar] [CrossRef]
- Chan, D.; Fox, N.C.; Jenkins, R.; Scahill, R.I.; Crum, W.R.; Rossor, M.N. Rates of Global and Regional Cerebral Atrophy in AD and Frontotemporal Dementia. Neurology 2001, 57, 1756–1763. [Google Scholar] [CrossRef]
- Iqbal, K.; Grundke-Iqbal, I. Neurofibrillary Pathology Leads to Synaptic Loss and Not the Other Way around in Alzheimer Disease. J. Alzheimer’s Dis. JAD 2002, 4, 235–238. [Google Scholar] [CrossRef]
- Coleman, P.; Federoff, H.; Kurlan, R. A Focus on the Synapse for Neuroprotection in Alzheimer Disease and Other Dementias. Neurology 2004, 63, 1155–1162. [Google Scholar] [CrossRef]
- Kazim, S.F.; Blanchard, J.; Bianchi, R.; Iqbal, K. Early Neurotrophic Pharmacotherapy Rescues Developmental Delay and Alzheimer’s-like Memory Deficits in the Ts65Dn Mouse Model of Down Syndrome. Sci. Rep. 2017, 7, 45561. [Google Scholar] [CrossRef] [Green Version]
- Selkoe, D.J. Alzheimer’s Disease Is a Synaptic Failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [Green Version]
- DeKosky, S.T.; Scheff, S.W. Synapse Loss in Frontal Cortex Biopsies in Alzheimer’s Disease: Correlation with Cognitive Severity. Ann. Neurol. 1990, 27, 457–464. [Google Scholar] [CrossRef]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical Basis of Cognitive Alterations in Alzheimer’s Disease: Synapse Loss Is the Major Correlate of Cognitive Impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Scheff, S.W.; Price, D.A. Synaptic Pathology in Alzheimer’s Disease: A Review of Ultrastructural Studies. Neurobiol. Aging 2003, 24, 1029–1046. [Google Scholar] [CrossRef] [PubMed]
- Arendt, T. Synaptic Degeneration in Alzheimer’s Disease. Acta Neuropathol. 2009, 118, 167–179. [Google Scholar] [CrossRef]
- Knobloch, M.; Mansuy, I.M. Dendritic Spine Loss and Synaptic Alterations in Alzheimer’s Disease. Mol. Neurobiol. 2008, 37, 73–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, C.A.A.; Mann, D.M.A.M.; Sumpter, P.Q.Q.; Yates, P.O.O. A Quantitative Morphometric Analysis of the Neuronal and Synaptic Content of the Frontal and Temporal Cortex in Patients with Alzheimer’s Disease. J. Neurol. Sci. 1987, 78, 151–164. [Google Scholar] [CrossRef]
- Scheff, S.W.; Price, D. a Alzheimer’s Disease-Related Alterations in Synaptic Density: Neocortex and Hippocampus. J. Alzheimer’s Dis. JAD 2006, 9, 101–115. [Google Scholar] [CrossRef]
- Masliah, E.; Mallory, M.; Alford, M.; DeTeresa, R.; Hansen, L.A.; McKeel, D.W.; Morris, J.C. Altered Expression of Synaptic Proteins Occurs Early during Progression of Alzheimer’s Disease. Neurology 2001, 56, 127–129. [Google Scholar] [CrossRef] [Green Version]
- Coleman, P.D.; Yao, P.J. Synaptic Slaughter in Alzheimer’s Disease. Neurobiol. Aging 2003, 24, 1023–1027. [Google Scholar] [CrossRef]
- Baazaoui, N.; Iqbal, K. A Novel Therapeutic Approach to Treat Alzheimer’s Disease by Neurotrophic Support during the Period of Synaptic Compensation. J. Alzheimer’s Dis. 2018, 62, 1211–1218. [Google Scholar] [CrossRef] [Green Version]
- Kazim, S.F.; Iqbal, K. Neural Regeneration as a Disease-Modifying Therapeutic Strategy for Alzheimer’s Disease. In Neuroprotection in Alzheimer’s Disease; Elsevier: Amsterdam, The Netherlands, 2017; pp. 3–29. ISBN 978-0-12-803690-7. [Google Scholar]
- Leuba, G.; Savioz, A.; Vernay, A.; Carnal, B.; Kraftsik, R.; Tardif, E.; Riederer, I.; Riederer, B.M. Differential Changes in Synaptic Proteins in the Alzheimer Frontal Cortex with Marked Increase in PSD-95 Postsynaptic Protein. J. Alzheimer’s Dis. JAD 2008, 15, 139–151. [Google Scholar] [CrossRef]
- Leuba, G.; Walzer, C.; Vernay, A.; Carnal, B.; Kraftsik, R.; Piotton, F.; Marin, P.; Bouras, C.; Savioz, A. Postsynaptic Density Protein PSD-95 Expression in Alzheimer’s Disease and Okadaic Acid Induced Neuritic Retraction. Neurobiol. Dis. 2008, 30, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Bell, K.F.S.; Bennett, D.A.; Cuello, A.C. Paradoxical Upregulation of Glutamatergic Presynaptic Boutons during Mild Cognitive Impairment. J. Neurosci. 2007, 27, 10810–10817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperling, R. Functional MRI Studies of Associative Encoding in Normal Aging, Mild Cognitive Impairment, and Alzheimer’s Disease. Ann. N. Y. Acad. Sci. 2007, 1097, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Baazaoui, N.; Flory, M.; Iqbal, K. Synaptic Compensation as a Probable Cause of Prolonged Mild Cognitive Impairment in Alzheimer’s Disease: Implications from a Transgenic Mouse Model of the Disease. J. Alzheimer’s Dis. JAD 2017, 56, 1385–1401. [Google Scholar] [CrossRef]
- Chesnokova, V.; Pechnick, R.N.; Wawrowsky, K. Chronic Peripheral Inflammation, Hippocampal Neurogenesis, and Behavior. Brain Behav. Immun. 2016, 58, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Russo, I.; Barlati, S.; Bosetti, F. Effects of Neuroinflammation on the Regenerative Capacity of Brain Stem Cells: Neuroinflammation and Neurogenesis. J. Neurochem. 2011, 116, 947–956. [Google Scholar] [CrossRef] [Green Version]
- Martino, G.; Pluchino, S. Neural Stem Cells: Guardians of the Brain. Nat. Cell Biol. 2007, 9, 1031–1034. [Google Scholar] [CrossRef]
- Skaper, S.D.; Giusti, P.; Facci, L. Microglia and Mast Cells: Two Tracks on the Road to Neuroinflammation. FASEB J. 2012, 26, 3103–3117. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Brown, M.A. Innate Immunity in the Central Nervous System. J. Clin. Investig. 2012, 122, 1164–1171. [Google Scholar] [CrossRef]
- Sierra, A.; Encinas, J.M.; Deudero, J.J.P.; Chancey, J.H.; Enikolopov, G.; Overstreet-Wadiche, L.S.; Tsirka, S.E.; Maletic-Savatic, M. Microglia Shape Adult Hippocampal Neurogenesis through Apoptosis-Coupled Phagocytosis. Cell Stem Cell 2010, 7, 483–495. [Google Scholar] [CrossRef]
- Nikolakopoulou, A.M.; Dutta, R.; Chen, Z.; Miller, R.H.; Trapp, B.D. Activated Microglia Enhance Neurogenesis via Trypsinogen Secretion. Proc. Natl. Acad. Sci. USA 2013, 110, 8714–8719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, A.M.; Torres-Alemán, I. The Many Faces of Insulin-like Peptide Signalling in the Brain. Nat. Rev. Neurosci. 2012, 13, 225–239. [Google Scholar] [CrossRef]
- Mir, S.; Cai, W.; Carlson, S.W.; Saatman, K.E.; Andres, D.A. IGF-1 Mediated Neurogenesis Involves a Novel RIT1/Akt/Sox2 Cascade. Sci Rep. 2017, 7, 3283. [Google Scholar] [CrossRef] [PubMed]
- Sato, K. Effects of Microglia on Neurogenesis. Glia 2015, 63, 1394–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valero, J.; Bernardino, L.; Cardoso, F.L.; Silva, A.P.; Fontes-Ribeiro, C.; Ambrósio, A.F.; Malva, J.O. Impact of Neuroinflammation on Hippocampal Neurogenesis: Relevance to Aging and Alzheimer’s Disease. J. Alzheimer’s Dis. JAD 2017, 60, S161–S168. [Google Scholar] [CrossRef] [PubMed]
- Mosher, K.I.; Andres, R.H.; Fukuhara, T.; Bieri, G.; Hasegawa-Moriyama, M.; He, Y.; Guzman, R.; Wyss-Coray, T. Neural Progenitor Cells Regulate Microglia Functions and Activity. Nat. Neurosci. 2012, 15, 1485–1487. [Google Scholar] [CrossRef] [PubMed]
- Walton, N.M.; Sutter, B.M.; Laywell, E.D.; Levkoff, L.H.; Kearns, S.M.; Marshall, G.P.; Scheffler, B.; Steindler, D.A. Microglia Instruct Subventricular Zone Neurogenesis. Glia 2006, 54, 815–825. [Google Scholar] [CrossRef]
- Nakanishi, M.; Niidome, T.; Matsuda, S.; Akaike, A.; Kihara, T.; Sugimoto, H. Microglia-Derived Interleukin-6 and Leukaemia Inhibitory Factor Promote Astrocytic Differentiation of Neural Stem/Progenitor Cells: Microglia and NSPC Differentiation. Eur. J. Neurosci. 2007, 25, 649–658. [Google Scholar] [CrossRef]
- Valero, J.; Eiriz, M.F.; Santos, T.; Neiva, I.; Ferreira, R.; Malva, J.O. Microglia: The Bodyguard and the Hunter of the Adult Neurogenic Niche. In Advances in Stem Cell Research; Baharvand, H., Aghdami, N., Eds.; Humana Press: Totowa, NJ, USA, 2012; pp. 245–279. ISBN 978-1-61779-939-6. [Google Scholar]
- Gonzalez-Perez, O.; Gutierrez-Fernandez, F.; Lopez-Virgen, V.; Collas-Aguilar, J.; Quinones-Hinojosa, A.; Garcia-Verdugo, J.M. Immunological Regulation of Neurogenic Niches in the Adult Brain. Neuroscience 2012, 226, 270–281. [Google Scholar] [CrossRef] [Green Version]
- Behrens, M.M.; Ali, S.S.; Dugan, L.L. Interleukin-6 Mediates the Increase in NADPH-Oxidase in the Ketamine Model of Schizophrenia. J. Neurosci. 2008, 28, 13957–13966. [Google Scholar] [CrossRef] [Green Version]
- Samavati, L.; Lee, I.; Mathes, I.; Lottspeich, F.; Hüttemann, M. Tumor Necrosis Factor α Inhibits Oxidative Phosphorylation through Tyrosine Phosphorylation at Subunit I of Cytochrome c Oxidase. J. Biol. Chem. 2008, 283, 21134–21144. [Google Scholar] [CrossRef] [Green Version]
- Town, T.; Nikolic, V.; Tan, J. The Microglial “Activation” Continuum: From Innate to Adaptive Responses. J. Neuroinflamm. 2005, 2, 24. [Google Scholar] [CrossRef] [Green Version]
- Halliday, G.; Robinson, S.R.; Shepherd, C.; Kril, J. Alzheimer’s Disease and Inflammation: A Review of Cellular and Therapeutic Mechanisms. Clin. Exp. Pharm. Physiol. 2000, 27, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, G.J.; Drego, R.; Hakimian, E.; Masliah, E. Mechanisms of Cell Signaling and Inflammation in Alzheimer’s Disease. Curr. Drug Targets Inflamm. Allergy 2005, 4, 247–256. [Google Scholar] [CrossRef]
- Mega, M.S.; Cummings, J.L.; Fiorello, T.; Gornbein, J. The Spectrum of Behavioral Changes in Alzheimer’s Disease. Neurology 1996, 46, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Kessing, L.V. Depression and the Risk for Dementia. Curr. Opin. Psychiatry 2012, 25, 457–461. [Google Scholar] [CrossRef]
- Van Dam, D.; Vermeiren, Y.; Dekker, A.D.; Naudé, P.J.W.; De Deyn, P.P. Neuropsychiatric Disturbances in Alzheimer’s Disease: What Have We Learned from Neuropathological Studies? Curr. Alzheimer Res. CAR 2016, 13, 1145–1164. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, M.; Shao, H.; Zandi, P.; Lyketsos, C.G.; Welsh-Bohmer, K.A.; Norton, M.C.; Breitner, J.C.S.; Steffens, D.C.; Tschanz, J.T. Cache County Investigators Point and 5-Year Period Prevalence of Neuropsychiatric Symptoms in Dementia: The Cache County Study. Int. J. Geriat. Psychiatry 2008, 23, 170–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballard, C.; Corbett, A. Agitation and Aggression in People with Alzheimer’s Disease. Curr. Opin. Psychiatry 2013, 26, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Lyketsos, C.G. Mental and Behavioral Disturbances in Dementia: Findings From the Cache County Study on Memory in Aging. Am. J. Psychiatry 2000, 157, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Flory, M.; Soininen, H. Clinical Symptoms and Symptom Signatures of Alzheimer’s Disease Subgroups. J. Alzheimer’s Dis. JAD 2013, 37, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Coogan, A.N.; Schutová, B.; Husung, S.; Furczyk, K.; Baune, B.T.; Kropp, P.; Häßler, F.; Thome, J. The Circadian System in Alzheimer’s Disease: Disturbances, Mechanisms, and Opportunities. Biol. Psychiatry 2013, 74, 333–339. [Google Scholar] [CrossRef]
- Wittko, I.M.; Schanzer, A.; Kuzmichev, A.; Schneider, F.T.; Shibuya, M.; Raab, S.; Plate, K.H. VEGFR-1 Regulates Adult Olfactory Bulb Neurogenesis and Migration of Neural Progenitors in the Rostral Migratory Stream In Vivo. J. Neurosci. 2009, 29, 8704–8714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leventhal, C.; Rafii, S.; Rafii, D.; Shahar, A.; Goldman, S.A. Endothelial Trophic Support of Neuronal Production and Recruitment from the Adult Mammalian Subependyma. Mol. Cell. Neurosci. 1999, 13, 450–464. [Google Scholar] [CrossRef]
- Yuan, H.; Chen, R.; Wu, L.; Chen, Q.; Hu, A.; Zhang, T.; Wang, Z.; Zhu, X. The Regulatory Mechanism of Neurogenesis by IGF-1 in Adult Mice. Mol. Neurobiol. 2015, 51, 512–522. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, G.; Wu, Y.; Sha, H.; Zhang, P.; Jia, J. BDNF Promotes EGF-Induced Proliferation and Migration of Human Fetal Neural Stem/Progenitor Cells via the PI3K/Akt Pathway. Molecules 2011, 16, 10146–10156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarov, O.; Mattson, M.P.; Peterson, D.A.; Pimplikar, S.W.; van Praag, H. When Neurogenesis Encounters Aging and Disease. Trends Neurosci. 2010, 33, 569–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colangelo, A.M.; Follesa, P.; Mocchetti, I. Differential Induction of Nerve Growth Factor and Basic Fibroblast Growth Factor MRNA in Neonatal and Aged Rat Brain. Mol. Brain Res. 1998, 53, 218–225. [Google Scholar] [CrossRef]
- Colangelo, A.; Cirillo, G.; Alberghina, L.; Papa, M.; Westerhoff, H. Neural Plasticity and Adult Neurogenesis: The Deep Biology Perspective. Neural Regen Res. 2019, 14, 201. [Google Scholar] [CrossRef]
- Chao, M.V. Neurotrophins and Their Receptors: A Convergence Point for Many Signalling Pathways. Nat. Rev. Neurosci. 2003, 4, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Emsley, J. Endogenous and Exogenous Ciliary Neurotrophic Factor Enhances Forebrain Neurogenesis in Adult Mice. Exp. Neurol. 2003, 183, 298–310. [Google Scholar] [CrossRef]
- Blanchard, J.; Chohan, M.O.; Li, B.; Liu, F.; Iqbal, K.; Grundke-iqbal, I. Beneficial Effect of a CNTF Tetrapeptide on Adult Hippocampal Neurogenesis, Neuronal Plasticity, and Spatial Memory in Mice. J. Alzheimer’s Dis. JAD 2010, 21, 1185–1195. [Google Scholar] [CrossRef] [PubMed]
- Hagg, T. Molecular Regulation of Adult CNS Neurogenesis: An Integrated View. Trends Neurosci. 2005, 28, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Stevens, C.F.; Gage, F.H. Astroglia Induce Neurogenesis from Adult Neural Stem Cells. Nature 2002, 417, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Chojnacki, A.; Shimazaki, T.; Gregg, C.; Weinmaster, G.; Weiss, S. Glycoprotein 130 Signaling Regulates Notch1 Expression and Activation in the Self-Renewal of Mammalian Forebrain Neural Stem Cells. J. Neurosci. 2003, 23, 1730–1741. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; He, Z.; Ruan, J.; Ma, Z.; Liu, Y.; Gong, C.; Iqbal, K.; Sun, S.; Chen, H. Role of Ciliary Neurotrophic Factor in the Proliferation and Differentiation of Neural Stem Cells. J. Alzheimer’s Dis. JAD 2013, 37, 587–592. [Google Scholar] [CrossRef]
- Yang, P.; Arnold, S.A.; Habas, A.; Hetman, M.; Hagg, T. Ciliary Neurotrophic Factor Mediates Dopamine D2 Receptor-Induced CNS Neurogenesis in Adult Mice. J. Neurosci. 2008, 28, 2231–2241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, S.; Aldrich, T.H.; Stahl, N.; Pan, L.; Taga, T.; Kishimoto, T.; Ip, N.Y.; Yancopoulos, G.D. LIFR Beta and Gp130 as Heterodimerizing Signal Transducers of the Tripartite CNTF Receptor. Science 1993, 260, 1805–1808. [Google Scholar] [CrossRef]
- Kazim, S.F.; Iqbal, K. Neurotrophic Factor Small-Molecule Mimetics Mediated Neuroregeneration and Synaptic Repair: Emerging Therapeutic Modality for Alzheimer’s Disease. Mol. Neurodegener. 2016, 11, 50. [Google Scholar] [CrossRef] [Green Version]
- Lu, B.; Nagappan, G.; Guan, X.; Nathan, P.J.; Wren, P. BDNF-Based Synaptic Repair as a Disease-Modifying Strategy for Neurodegenerative Diseases. Nat. Rev. Neurosci. 2013, 14, 401–416. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Cao, L.; Wang, L.M.; Guo, C.; Ye, J.L.; Chai, Y.F.; Yan, Z.Y. Development of Neurotrophic Molecules for Treatment of Neurodegeneration. Curr. Protein Pept. Sci. 2001, 2, 261–276. [Google Scholar] [CrossRef]
- Longo, F.; Yang, T.; Knowles, J.; Xie, Y.; Moore, L.; Massa, S. Small Molecule Neurotrophin Receptor Ligands: Novel Strategies for Targeting Alzheimers Disease Mechanisms. Curr. Alzheimer Res. CAR 2007, 4, 503–506. [Google Scholar] [CrossRef]
- Massa, S.M.; Yang, T.; Xie, Y.; Shi, J.; Bilgen, M.; Joyce, J.N.; Nehama, D.; Rajadas, J.; Longo, F.M. Small Molecule BDNF Mimetics Activate TrkB Signaling and Prevent Neuronal Degeneration in Rodents. J. Clin. Investig. 2010, 120, 1774–1785. [Google Scholar] [CrossRef] [Green Version]
- Longo, F.M.; Massa, S.M. Neurotrophin-Based Strategies for Neuroprotection. J. Alzheimer’s Dis. JAD 2004, 6, S13–S17. [Google Scholar] [CrossRef]
- Lau, J.L.; Dunn, M.K. Therapeutic Peptides: Historical Perspectives, Current Development Trends, and Future Directions. Bioorganic Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef]
- Yadav, A.; Pandey, D.; Ashraf, G.M. Rachana Peptide Based Therapy for Neurological Disorders. Curr. Protein Pept. Sci. 2021, 22, 656–665. [Google Scholar] [CrossRef]
- Mota, I.F.L.; de Lima, L.S.; De M. Santana, B.; de A. M. Gobbo, G.; Bicca, J.V.M.L.; Azevedo, J.R.M.; Veras, L.G.; de A. A. Taveira, R.; Pinheiro, G.B.; Mortari, M.R. Alzheimer’s Disease: Innovative Therapeutic Approaches Based on Peptides and Nanoparticles. Neuroscientist 2021, 107385842110164. [Google Scholar] [CrossRef]
- Banks, W.A. Developing Drugs That Can Cross the Blood-Brain Barrier: Applications to Alzheimer’s Disease. BMC Neurosci. 2008, 9, S2. [Google Scholar] [CrossRef] [Green Version]
- Lien, S.; Lowman, H.B. Therapeutic Peptides. Trends Biotechnol. 2003, 21, 556–562. [Google Scholar] [CrossRef]
- Jennings, D.; Huntwork-Rodriguez, S.; Henry, A.G.; Sasaki, J.C.; Meisner, R.; Diaz, D.; Solanoy, H.; Wang, X.; Negrou, E.; Bondar, V.V.; et al. Preclinical and Clinical Evaluation of the LRRK2 Inhibitor DNL201 for Parkinson’s Disease. Sci. Transl. Med. 2022, 14, eabj2658. [Google Scholar] [CrossRef]
- Panza, F.; Solfrizzi, V.; Seripa, D.; Imbimbo, B.P.; Lozupone, M.; Santamato, A.; Zecca, C.; Barulli, M.R.; Bellomo, A.; Pilotto, A.; et al. Tau-Centric Targets and Drugs in Clinical Development for the Treatment of Alzheimer’s Disease. BioMed Res. Int. 2016, 2016, 3245935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saragovi, H.U.; Gehring, K. Development of Pharmacological Agents for Targeting Neurotrophins and Their Receptors. Trends Pharmacol. Sci. 2000, 21, 93–98. [Google Scholar] [CrossRef]
- Kelleher-Andersson, J. Neurogenesis as a Potential Therapeutic Strategy for Neurodegenerative Disorders. J. Alzheimer’s Dis. 2004, 6, S19–S25. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Kazim, S.F.; Bolognin, S.; Blanchard, J. Shifting Balance from Neurodegeneration to Regeneration of the Brain: A Novel Therapeutic Approach to Alzheimer’s Disease and Related Neurodegenerative Conditions. Neural Regen. Res. 2014, 9, 1518–1519. [Google Scholar] [CrossRef] [PubMed]
- Chohan, M.O.; Li, B.; Blanchard, J.; Tung, Y.-C.; Heaney, A.T.; Rabe, A.; Iqbal, K.; Grundke-Iqbal, I. Enhancement of Dentate Gyrus Neurogenesis, Dendritic and Synaptic Plasticity and Memory by a Neurotrophic Peptide. Neurobiol. Aging 2011, 32, 1420–1434. [Google Scholar] [CrossRef]
- Li, B.; Wanka, L.; Blanchard, J.; Liu, F.; Chohan, M.O.; Iqbal, K.; Grundke-Iqbal, I. Neurotrophic Peptides Incorporating Adamantane Improve Learning and Memory, Promote Neurogenesis and Synaptic Plasticity in Mice. FEBS Lett. 2010, 584, 3359–3365. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Liu, Y.; Dai, C.-L.; Baazaoui, N.; Tung, Y.C.; Liu, F.; Iqbal, K. Neurotrophic Treatment Initiated During Early Postnatal Development Prevents the Alzheimer-Like Behavior and Synaptic Dysfunction. J. Alzheimer’s Dis. JAD 2021, 82, 631–646. [Google Scholar] [CrossRef]
- Blanchard, J.; Bolognin, S.; Chohan, M.O.; Rabe, A.; Iqbal, K.; Grundke-Iqbal, I. Rescue of Synaptic Failure and Alleviation of Learning and Memory Impairments in a Trisomic Mouse Model of down Syndrome. J. Neuropathol. Exp. Neurol. 2011, 70, 1070–1079. [Google Scholar] [CrossRef] [Green Version]
- Phiel, C.J.; Wilson, C.A.; Lee, V.M.-Y.; Klein, P.S. GSK-3alpha Regulates Production of Alzheimer’s Disease Amyloid-Beta Peptides. Nature 2003, 423, 435–439. [Google Scholar] [CrossRef]
- Serenó, L.; Coma, M.; Rodríguez, M.; Sánchez-Ferrer, P.; Sánchez, M.B.; Gich, I.; Agulló, J.M.; Pérez, M.; Avila, J.; Guardia-Laguarta, C.; et al. A Novel GSK-3β Inhibitor Reduces Alzheimer’s Pathology and Rescues Neuronal Loss in Vivo. Neurobiol. Dis. 2009, 35, 359–367. [Google Scholar] [CrossRef]
- Beurel, E.; Michalek, S.M.; Jope, R.S. Innate and Adaptive Immune Responses Regulated by Glycogen Synthase Kinase-3 (GSK3). Trends Immunol. 2010, 31, 24–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, W.; Wang, Y.; Liu, Y.; Dai, C.-L.; Tung, Y.-C.; Liu, F.; Iqbal, K. Prenatal to Early Postnatal Neurotrophic Treatment Prevents Alzheimer-like Behavior and Pathology in Mice. Alzheimer’s Res. 2020, 12, 102. [Google Scholar] [CrossRef] [PubMed]
- Kazim, S.F.; Blanchard, J.; Dai, C.; Tung, Y.; Grundke-Iqbal, I.; Iqbal, K. O3–13–02: Chronic Treatment with a CNTF-derived Peptide Reverses Dendritic and Synaptic Plasticity Deficits, Cognitive Impairment and Tau Pathology in an Alzheimer’s Disease Mouse Model. Alzheimer’s Dement. 2013, 9, 548. [Google Scholar] [CrossRef]
- Blanchard, J.; Wanka, L.; Tung, Y.-C.C.; Cárdenas-Aguayo, M.D.C.; Laferla, F.M.; Iqbal, K.; Grundke-Iqbal, I. Pharmacologic Reversal of Neurogenic and Neuroplastic Abnormalities and Cognitive Impairments without Affecting Aβ and Tau Pathologies in 3xTg-AD Mice. Acta Neuropathol. 2010, 120, 605–621. [Google Scholar] [CrossRef] [PubMed]
- Bolognin, S.; Blanchard, J.; Wang, X.; Basurto-Islas, G.; Tung, Y.C.; Kohlbrenner, E.; Grundke-Iqbal, I.; Iqbal, K. An Experimental Rat Model of Sporadic Alzheimer’s Disease and Rescue of Cognitive Impairment with a Neurotrophic Peptide. Acta Neuropathol. 2012, 123, 133–151. [Google Scholar] [CrossRef] [Green Version]
- Rockenstein, E.; Ubhi, K.; Doppler, E.; Novak, P.; Moessler, H.; Li, B.; Blanchard, J.; Grundke-Iqbal, I.; Iqbal, K.; Mante, M.; et al. Regional Comparison of the Neurogenic Effects of CNTF-Derived Peptides and Cerebrolysin in AβPP Transgenic Mice. J. Alzheimer’s Dis. JAD 2011, 27, 743–752. [Google Scholar] [CrossRef] [Green Version]
- Kazim, S.F.; Cardenas-Aguayo, M.D.C.; Arif, M.; Blanchard, J.; Fayyaz, F.; Grundke-Iqbal, I.; Iqbal, K. Sera from Children with Autism Induce Autistic Features Which Can Be Rescued with a CNTF Small Peptide Mimetic in Rats. PLoS ONE 2015, 10, e0118627. [Google Scholar] [CrossRef]
- Khatoon, S.; Chalbot, S.; Bolognin, S.; Puoliväli, J.; Iqbal, K. Elevated Tau Level in Aged Rat Cerebrospinal Fluid Reduced by Treatment with a Neurotrophic Compound. J. Alzheimer’s Dis. JAD 2015, 47, 557–564. [Google Scholar] [CrossRef]
- Baazaoui, N.; Iqbal, K. Prevention of Amyloid-β and Tau Pathologies, Associated Neurodegeneration, and Cognitive Deficit by Early Treatment with a Neurotrophic Compound. J. Alzheimer’s Dis. JAD 2017, 58, 215–230. [Google Scholar] [CrossRef]
- Liu, Y.; Wei, W.; Baazaoui, N.; Liu, F.; Iqbal, K. Inhibition of AMD-Like Pathology with a Neurotrophic Compound in Aged Rats and 3xTg-AD Mice. Front. Aging Neurosci. 2019, 11, 309. [Google Scholar] [CrossRef]
- Fish, P.V.; Steadman, D.; Bayle, E.D.; Whiting, P. New Approaches for the Treatment of Alzheimer’s Disease. Bioorganic Med. Chem. Lett. 2019, 29, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Lee, G.; Nahed, P.; Kambar, M.E.Z.N.; Zhong, K.; Fonseca, J.; Taghva, K. Alzheimer’s Disease Drug Development Pipeline: 2022. AD Transl. Res. Clin. Interv. 2022, 8, e12295. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J. New Approaches to Symptomatic Treatments for Alzheimer’s Disease. Mol. Neurodegener. 2021, 16, 2. [Google Scholar] [CrossRef] [PubMed]
- Rabinovici, G.D. Controversy and Progress in Alzheimer’s Disease—FDA Approval of Aducanumab. N. Engl. J. Med. 2021, 385, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Mintun, M.A.; Lo, A.C.; Duggan Evans, C.; Wessels, A.M.; Ardayfio, P.A.; Andersen, S.W.; Shcherbinin, S.; Sparks, J.; Sims, J.R.; Brys, M.; et al. Donanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2021, 384, 1691–1704. [Google Scholar] [CrossRef] [PubMed]
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.Y.K.; Lannfelt, L.; Bradley, H.; Rabe, M.; Koyama, A.; et al. A Randomized, Double-Blind, Phase 2b Proof-of-Concept Clinical Trial in Early Alzheimer’s Disease with Lecanemab, an Anti-Aβ Protofibril Antibody. Alzheimer’s Res. Ther. 2021, 13, 80. [Google Scholar] [CrossRef]
- Tible, O.P.; Riese, F.; Savaskan, E.; von Gunten, A. Best Practice in the Management of Behavioural and Psychological Symptoms of Dementia. Adv. Neurol. Disord. 2017, 10, 297–309. [Google Scholar] [CrossRef]
- Leonpacher, A.K.; Peters, M.E.; Drye, L.T.; Makino, K.M.; Newell, J.A.; Devanand, D.P.; Frangakis, C.; Munro, C.A.; Mintzer, J.E.; Pollock, B.G.; et al. Effects of Citalopram on Neuropsychiatric Symptoms in Alzheimer’s Dementia: Evidence from the CitAD Study. Am. J Psychiatry 2016, 173, 473–480. [Google Scholar] [CrossRef] [Green Version]
- Porsteinsson, A.P.; Drye, L.T.; Pollock, B.G.; Devanand, D.P.; Frangakis, C.; Ismail, Z.; Marano, C.; Meinert, C.L.; Mintzer, J.E.; Munro, C.A.; et al. Effect of Citalopram on Agitation in Alzheimer Disease: The CitAD Randomized Clinical Trial. JAMA 2014, 311, 682–691. [Google Scholar] [CrossRef]
- Faculty of Psychiatry of Old Age and Committee for Therapeutic Interventions and Evidence-Based Practice. Professional Practice Guideline 10 Antipsychotic Medications as a Treatment of Behavioural and Psychological Symptoms of Dementia; Royal Australian and New Zealand College of Psychiatrists: Melbourne, Australia, 2016. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
| Neuropathological Features | Composition | References |
|---|---|---|
| Amyloid plaques | Composed of Aβ 40 and Aβ 42 fragments that result from the sequential cleavage of AβPP by the enzymes β-secretase and γ-secretase in neurons. There are two types of amyloid plaques: diffuse and dense core based on their morphology and their positive or negative staining with Thioflavin-S or Congo Red. | [28,29,30,31] |
| Dense core plaques | Dense core amyloid plaques stain with thioflavin-S and Congo Red and they are typically surrounded by dystrophic neuritis, reactive astrocytes and activated microglial cells, and are associated with synaptic loss. | [28,29,30,31] |
| Diffuse plaques | They are amorphous plaques with an undefined contour and amorphous amyloid deposits which are negatively stained with Congo red and thioflavin S. | [29] |
| Amyloid β | Amyloid β: a 40 or 42 amino acid peptide derived from amyloid precursor protein (APP) after its sequential cleavage by β- and γ-secretases. | [29] |
| Cerebral amyloid angiopathy (CAA) | It is the consequence of the deposition of Aβ in the vessel walls. The major constituent of CAA is the soluble form of Aβ (Aβ40) | [29] |
| Neurofibrillary tangles (NFTs) | They are primarily made up of paired helical filaments (PHFs) that are fibrils of 10 nm in diameter that form pairs with a helical tridimensional conformation at a regular periodicity of ~65 nm They are caused by the aggregation of the hyperphosphorylated tau in neurons of the misfolded tau that become extraneuronal (“ghost” tangles) when tangle-bearing neurons die. | [28,30] |
| Neuropil threads | They are axonal and dendritic segments formed by the aggregated and hyperphosphorylated tau that are usually associated with the NFT in AD. | [29] |
| Granuovacuolar degeneration (GVD) and Hirano bodies | GVD mainly formed by large double-membrane bodies with an unknown origin and significance. Usually detected in the cytoplasm of hippocampal pyramidal neurons of AD patients. | [31] |
| Glial responses (Neuroinflammation) | A significant positive correlation was reported between both astrocytosis and microgliosis and NFT burden but not between both reactive glial cell types and amyloid burden, which suggests that neuroinflammation is tightly linked to neurofibrillary degeneration. | [29,32] |
| Neuronal and synaptic loss | Neuronal loss is the major cause of cortical atrophy. Synaptic Loss contributes along with neuronal loss to cortical atrophy. | [11,29,33] |
| α-synuclein positive Lewy bodies | AD patients that present α-synuclein positive Lewy bodies exhibit acceleration in the disease process and a more aggressive and rapid cognitive decline compared to pure AD patients. | [34] |
| Cognitive decline | Episodic memory is the first area affected in the AD process, followed by impairment in the executive functions, apraxia, visuospatial navigation deficits, visuo-perceptive deficits, and semantic memory, which consequently results the full-blown dementia syndrome. | [29,35] |
| Psychiatric Symptoms | Prevalence | References |
|---|---|---|
| Depression | Its prevalence is around 20–50% in AD patients. | [151,152] |
| Apathy | Its prevalence could reach up to 80% Is the most common and persistent neuropsychological feature in AD | [152] |
| Agitation, irritability and aggression | Its prevalence is between 48% and 80% with symptoms that persistfor months and happen across all AD stages. | [153,154] |
| Anxiety and phobia | Prevalence was reported to range from 7.9% to 29.8% | [152,155] |
| Psychotic symptoms (delusions and hallucinations) | The prevalence is between 30–50% in AD. Hallucination was found to diagnose AD with 14% sensitivity and 99% specificity. | [152,156] |
| Sleep disorders | Common behavioral disturbances in AD. Prevalence between 25% to 50% of patients ~75% of patients sleep for extended periods during the day. | [152,157] |
| Hypokinesia | Could diagnose AD with 30% sensitivity and 99% specificity | [156] |
| Paranoia | Could diagnose AD with 15% sensitivity and 99% specificity | [156] |
| Rigidity | Could diagnose AD with 16% sensitivity and 100% specificity | [156] |
| Tremors | Could diagnose AD with 16% sensitivity and 96% specificity | [156] |
| Drug | Effects | References |
|---|---|---|
| Four cholinesterase inhibitors: Donepezil (Aricept™), rivastigmine (Exelon™), and galantamine (Razadyne™). Tacrine: No longer available on the market | Targets cholinergic innervations in the nucleus basalis. | [207] |
| One NMDA receptor antagonist: Memantine (Namenda™). | N-methyl-d-aspartate receptor antagonist (NMDA) that blocks glutamate from binding to its receptors. This prevents excessive excitotoxicity and neuronal cell death, which is thought to contribute to the pathogenesis of AD. | [207,208] |
| GV-971 (Oligomannate™), an oligosaccharide | Reduction of systemic inflammation and neuroinflammation - Approved in China | [207] |
| Aducanumab | First disease-modifying therapy (DMT). Became available on the market in 2021 for MCI due to AD and mild AD dementia An anti-amyloid monoclonal antibody Accelerated regulatory mechanism based on demonstration of amyloid plaque lowering | [207,209] |
| Donanemab and lecanemab | Monoclonal antibodies Under review by the US Food and Drug Administration (FDA). | [210,211] |
| Phase 3 clinical trials 31 agents | 21 DMTs (5 biologic and 16 small molecules) | [207] |
| Phase 2 clinical trials 82 agents | 71 DMTs (26 biologics and 45 small molecules). | [207] |
| Phase 1 clinical trials 30 agents | 27 DMTs (9 biologics and 18 small molecules) | [207] |
| Antipsychotic drugs | ||
| Acetylcholinesterase inhibitors | May improve apathy, delusions and hallucinations, and less commonly improve aggression, depression, disinhibited behaviors, irritability or nocturnal disruption in patients with mild to moderate dementia | [212] |
| Selective serotonin reuptake inhibitors (SSRIs) | Effective in the management of depression and anxiety in people with dementia that cannot be treated by non-pharmacological interventions alone. For AD patients, citalopram was reported to decrease agitation and to likely improve other symptoms such as delusions, suggesting that it may have antipsychotic effect | [213,214] |
| Antipsychotics Risperidone, Quetiapine, Olanzapine | Have only a modest effect in managing the psychological symptoms that accompany AD and other neurodegenerative diseases The level of effectiveness of these drugs varies between patients. | [215] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baazaoui, N.; Iqbal, K. Alzheimer’s Disease: Challenges and a Therapeutic Opportunity to Treat It with a Neurotrophic Compound. Biomolecules 2022, 12, 1409. https://doi.org/10.3390/biom12101409
Baazaoui N, Iqbal K. Alzheimer’s Disease: Challenges and a Therapeutic Opportunity to Treat It with a Neurotrophic Compound. Biomolecules. 2022; 12(10):1409. https://doi.org/10.3390/biom12101409
Chicago/Turabian StyleBaazaoui, Narjes, and Khalid Iqbal. 2022. "Alzheimer’s Disease: Challenges and a Therapeutic Opportunity to Treat It with a Neurotrophic Compound" Biomolecules 12, no. 10: 1409. https://doi.org/10.3390/biom12101409
APA StyleBaazaoui, N., & Iqbal, K. (2022). Alzheimer’s Disease: Challenges and a Therapeutic Opportunity to Treat It with a Neurotrophic Compound. Biomolecules, 12(10), 1409. https://doi.org/10.3390/biom12101409