

Lung Organoids in Smoking Research: Current Advances and Future Promises

Abstract
1. Introduction
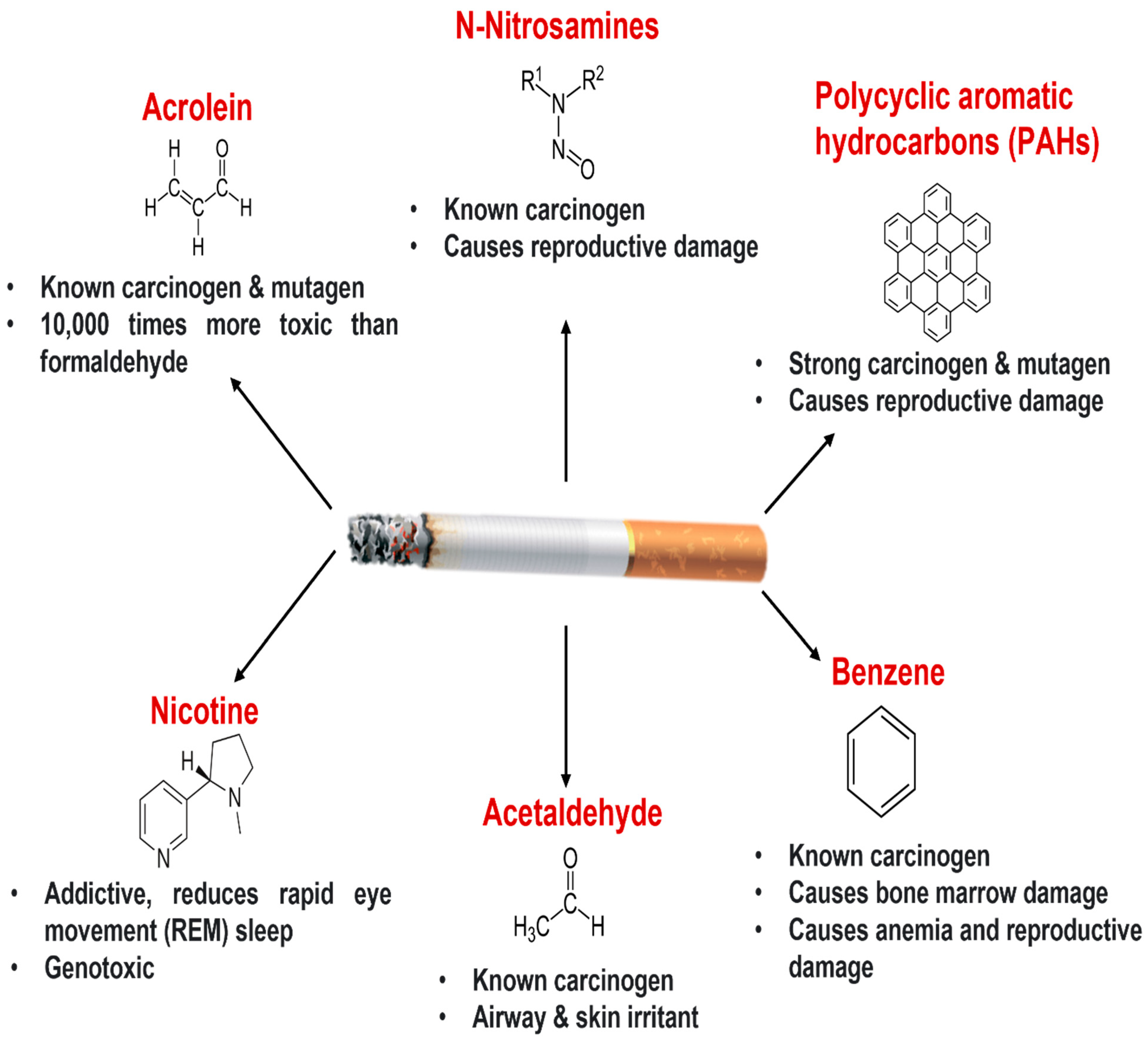
2. Cigarette Smoking and Lung Health

3. Lung Organoids: Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type Used | Media Composition | Matrigel Concentration | Duration of Culture | Reference |
|---|---|---|---|---|
| Basal cells from human trachea | Differentiation medium (Lonza) with insulin, bovine pituitary extract, hydrocortisone, GA1000, epinephrine, retinoic acid, transferrin, and triiodothyronine | 25% (growth-factor-reduced) on the bottom, 5% mixed with cells | 14 days | [63] |
| Human trachea and large airways | 50:50% DMEM: BEBM (Lonza) with BEGM supplements (minus gentamycin, amphotericin, triiodothyronine, retinoic acid) | 25% on the bottom, 5% mixed with cells | 14 days | [64] |
| Alveolar epithelial cells 2 (AEC2) Supported by PDGFRα+ lung lipofibroblasts | MTEC+ media: DMEM/F12 with bovine pituitary extract, insulin, epidermal growth factor, cholera toxin, 5% fetal bovine serum, antibiotics, Y-27632 (ROCK inhibitor) for the first 2 days, and retinoic acid | 1:1 ratio of Matrigel: MTEC+Plus media | 16–17 days | [65] |
| Alveolar epithelial cells 2 (AEC2) supported by lung mesenchymal cells (EpCAM− Sca1+) | DMEM/F12 supplemented with glutamine, ITS, sodium bicarbonate, 10% newborn calf serum, antibiotics, insulin, transferrin, and selenium. FGF7, FGF10, HGF, BMP-4, TGF-β1, and PDGF-AA were added according to requirement | 1:1 ratio of Matrigel: media | 14–16 days | [66] |
| Human pluripotent stem cells | Foregut media with noggin, fibroblast growth factor 4, SB431542, CHIR99021, and other growth factors | - | 15 days | [54] |
| Human pluripotent stem cells | Maintenance medium: DMEM/F12 (1:1) supplemented with—Primocin, FGF-2, β-mercaptoethanol, 20% knockout serum replacement Differentiation medium: IMDM/Ham’s F12 (3:1) supplemented with B27, N2, penicillin-streptomycin, bovine serum albumin, ascorbic acid, monothioglycerol, glutamax, and growth factor cocktails | 100% (Branching morphogenesis) | 20–25 days | [67] |
| Human-induced pluripotent stem cells | Directed differentiation medium: RPMI1640 media with HEPES, B27 supplement, glutamax, penicillin/streptomycin, human activin A, CHIR99021, and Y-27632 | - | 25 days | [68] |
| Human embryonic stem cells | First 3 days: RPMI1640 media with CHIR-99021 and activinA, Next 4 days: Advanced DMEM/F12 media with noggin, FGF4, SB431542, CHIR99021 After 7 days: Advanced DMEM/F12 media containing 1% fetal bovine serum | - | Up to 70 days | [69] |
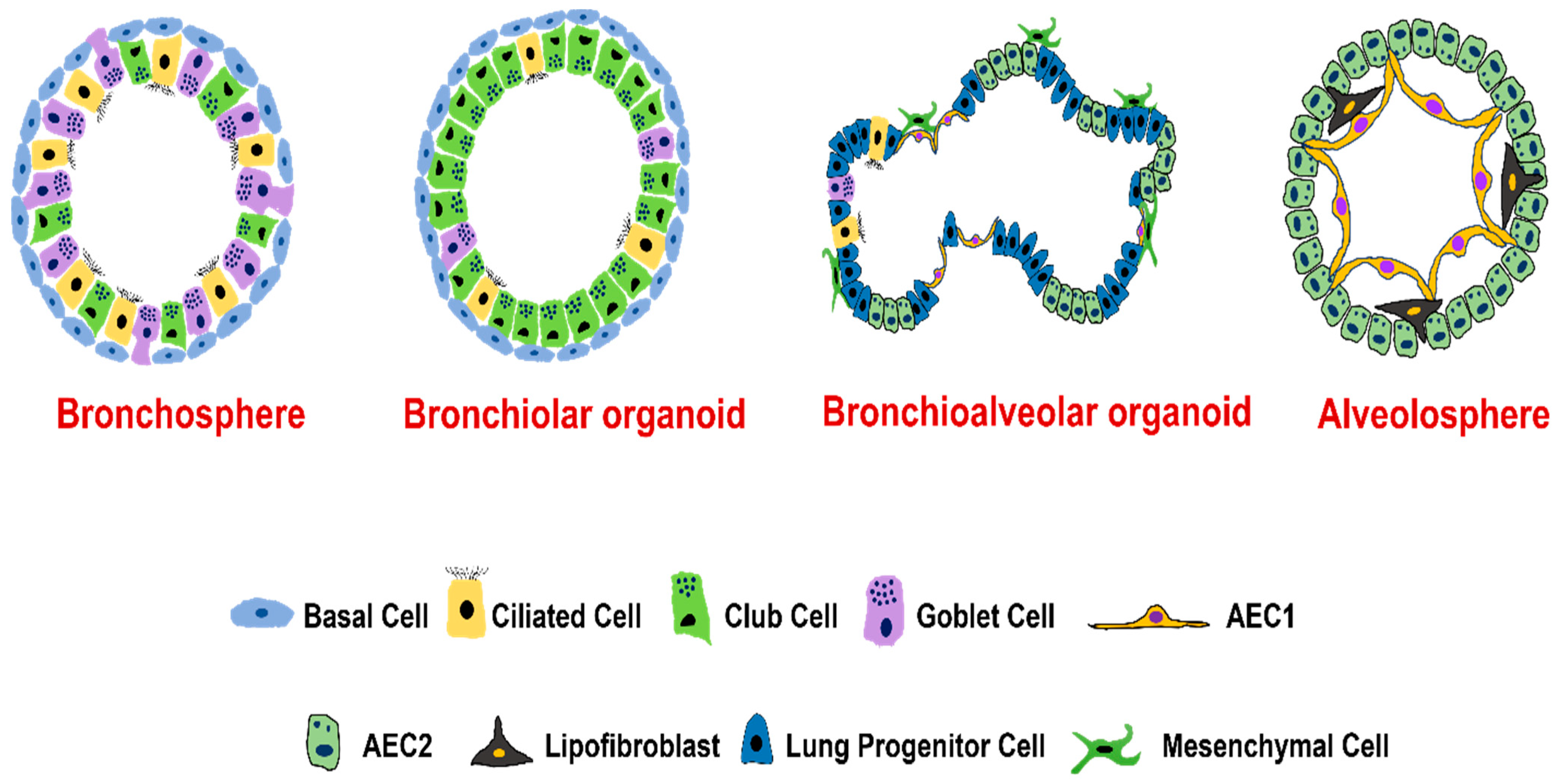
4. Types of Lung Organoids
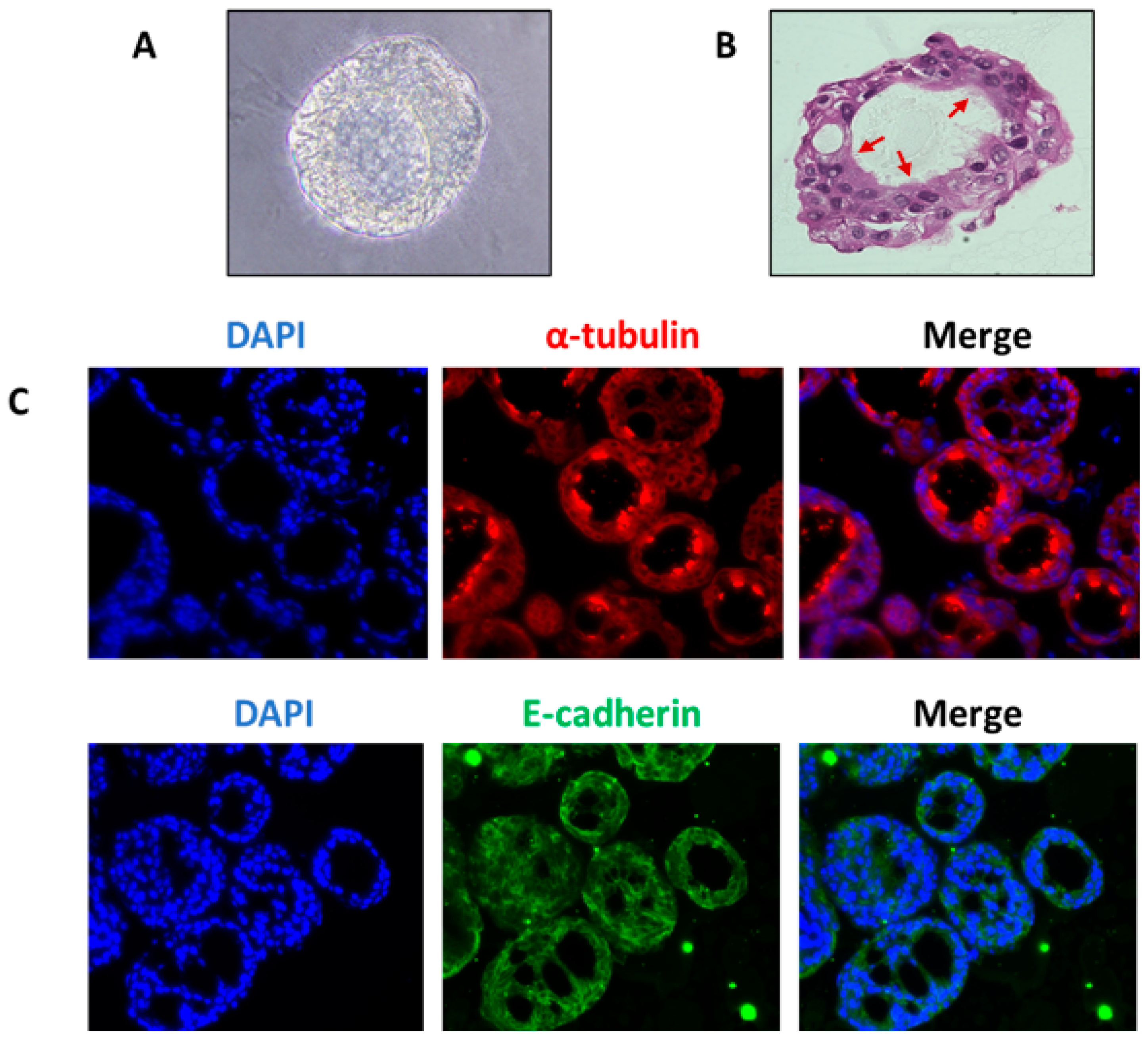
4.1. Organoids from Human Airway Basal Cells
4.2. Organoids from Human Airway Secretory Cells
4.3. Organoids from Human Alveolar Type II Epithelial Cells
4.4. Organoids from Distal Airway Multipotent Progenitor Cells
4.5. Organoids from Human Pluripotent Stem Cells (hPSCs)
4.6. Co-Culture Models of the Lung Organoids with Other Cell Types
5. Molecular Landscape of Epithelial Repair in Lung Organoid Models
5.1. Airway Epithelial Repair
5.2. Alveolar Epithelial Repair
6. Application of Organoids in Cigarette-Smoking-Associated Lung Diseases
6.1. COPD
6.2. Lung Cancer
6.3. Respiratory Viral Infections
6.4. Idiopathic Pulmonary Fibrosis (IPF)
7. Limitations of the Organoid Model
- Organoids are mostly derived from iPSCs. The cells from iPSCs are immature, imitate the second trimester of fetal development, and have similar gene expression profiles to the embryo, thus making it difficult to mimic adult diseases [138].
- Organoids are heterogeneous and have several variabilities that appear at many stages such as (1) between genotypes and different starting cell lines, (2) between batches of the organoids from the same starting material, (3) between different organoids in the same culture, and (4) between areas of a single organoid.
- The organoid model may be devoid of some of the cell types. During organogenesis, cells from different organs/origins (such as bone marrow and neural crest cells) are also present that aid in organ development which is not present in the organoid model. [139].
- Due to the lack of vascularization, the in vitro growth potential of organoids is restricted. As undefined mouse-derived ECM is required for organoid culture, batch-to-batch variation is a major concern. Also, solidified gel-ECM affects the penetration and availability of nutrients, drugs, and stimuli under investigation, causing variations in the results. Despite being heterogeneous in comparison to 2D cultures, the variable size, cell ratios, and morphology of organoids make them difficult for phenotypic screening. This limitation also makes it difficult to incorporate microsensors for critical control and functional parameters such as fluid pressure, oxygen, glucose, flow, cell migration, and barrier integrity. To overcome this limitation, lung-on-a-chip devices are now being developed where cells are cultured in a uniform manner and have a defined orientation [140].
- In addition to vasculature, the other important factor that affects organoid development and differentiation is the culture media. To date, none of the media used in organoid culture are chemically defined. The organoid media also contain some complex components such as bovine pituitary extract (BPE) or fetal bovine serum (FBS). Oxygen tension and glucose levels are also not tested in this system. Thus, we still do not have a defined list of growth factors, metabolites, or small molecules required for the renewal and differentiation of organoids [2]. Some recent studies reported chemically defined media with growth factors for the establishment of the organoid culture. Although, the ECM components and the mechanical forces need further assessment in detail [2,55,80,141].
- The organoid model is a single-organ model and cannot completely mimic the physiological niche provided by neural, immune, and stromal cells. Thus, how long these organoids can survive is a major limitation that makes them less complicated as compared to in vivo models [142]. To overcome these limitations, the lung organoid model is co-cultured with endothelial cells and immune components to make organoids better mimic the in vivo human lung physiology. Another approach being used is to combine organoids with an organ-on-a-chip which can prove to be an ideal research tool for high-throughput screening of drugs, regenerative biology, and understanding smoking-related disease pathophysiology [143,144]. Furthermore, using stem cell-derived organoids with an organ-on-a-chip approach can help develop patient-specific disease models that can open new avenues in personalized therapy.
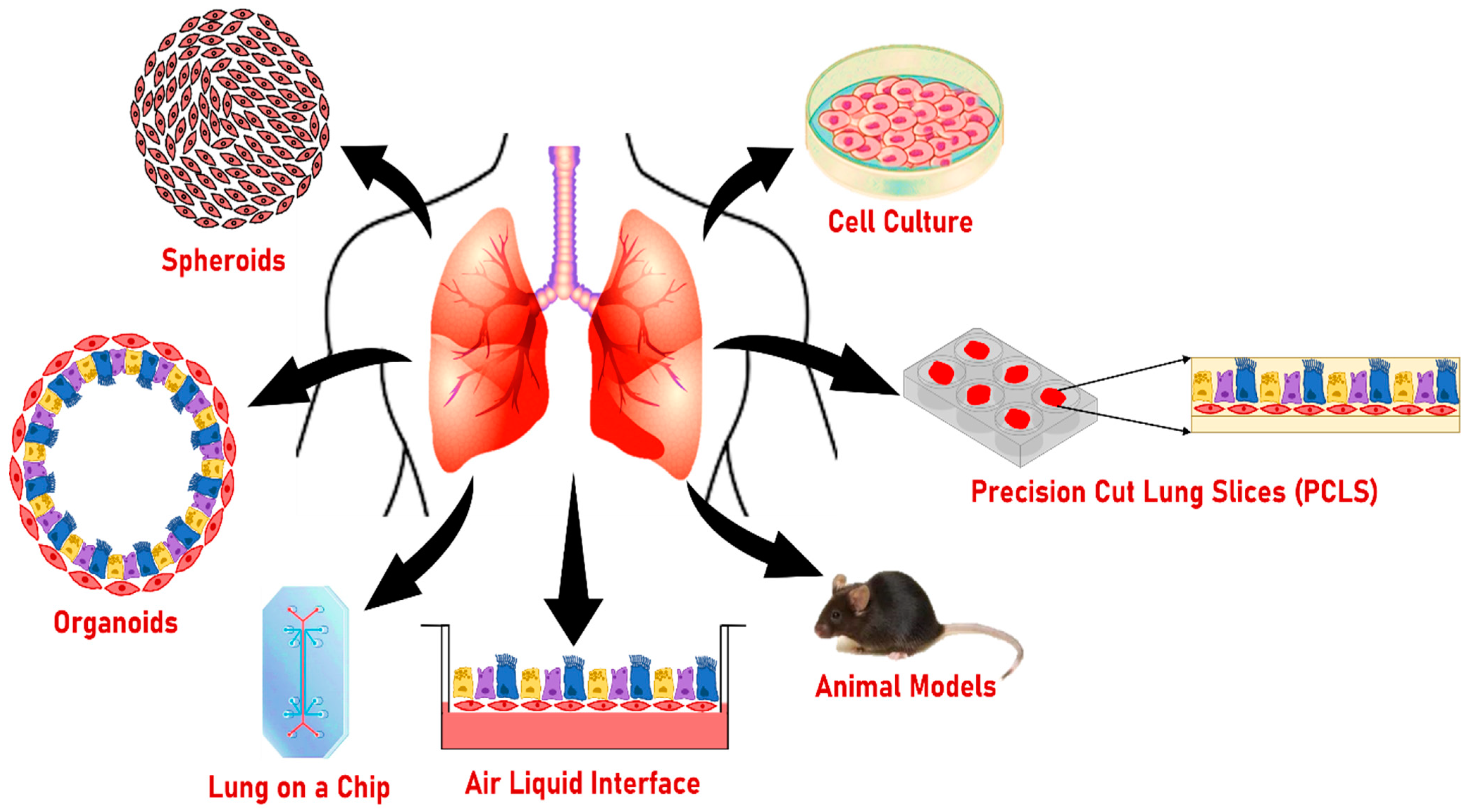
8. Other Models to Study the Effect of Smoking on Lung Health
8.1. Two-Dimensional Cultures
8.2. Air–Liquid Interphase Culture (ALI)
8.3. Lung on a Chip
8.4. Precision-Cut Lung Slices (PCLS)
8.5. Spheroids
8.6. Animal Models
9. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| COPD | Chronic obstructive pulmonary disease |
| ALI | Air liquid interface |
| PCLS | Precision cut lung slices |
| hPSCs | Human pluripotent stem cells |
| ASCs | Adult stem cells |
| iPSCs | Induced pluripotent stem cells |
| EMT | Epithelial–mesenchymal transition |
| CSE | Cigarette smoke extract |
| NNN | N-nitrosonornicotine |
| NNK | Nicotine-derived nitrosamine ketone |
| ECM | Extracellular matrix |
| PAHs | Polyaromatic hydrocarbons |
| AEC2 | Alveolar epithelial type II |
| AEC1 | Alveolar epithelial type I |
| SFTPC | Surfactant protein C |
| CF | Cystic fibrosis |
| HTBEC | Human tracheobronchial epithelial cells |
| FACS | Fluorescence-activated cell sorting |
| MACS | Magnetic-activated cell sorting |
| IPF | Idiopathic pulmonary fibrosis |
| BASCs | Bronchioalveolar stem cells |
| BMP4 | Bone morphogenetic protein 4 |
| NFATc1 | Nuclear factor of activated T cell c1 |
| TSP1 | Thrombospondin-1 |
| LBO | Lung bud organoid |
| Grhl2 | Grainyhead-like transcription factor 2 |
| STAT3 | Signal transducer and activator of the transcription 3 |
| DATPs | Damage-associated transient progenitors |
| TAZ | Transcriptional co-activator with PDZ-binding motif |
| YAP | Yes-associated protein |
| FGF | Fibroblast growth factor |
| BDNF | Brain-derived neurotrophic factor |
| TrkB | Receptor tropomyosin receptor kinase B |
| Th2 | T helper 2 |
| TGF-β | Transforming growth factor–β |
| FAM13A | Family with sequence similarity member 13A |
| AREG | Amphiregulin |
| EGFR | Epidermal growth factor receptor |
| FOXJ1 | Forkhead box protein J1 |
| FOXM1 | Forkhead box protein M1 |
| SPDEF | SAM pointed domain containing ETS transcription factor |
| PGE2 | Prostaglandin E2 |
| FAO | Fatty acid oxidation |
| RSV | Respiratory syncytial virus |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| HPSIP | Hermansky–Pudlak syndrome (HPS)-associated interstitial pneumonia |
| BPE | Bovine pituitary extract |
| HBEC | Human bronchial epithelial cells |
| HMOX-1 | Heme oxygenase-1 |
| MTS | Mainstream tobacco smoke |
| RA | Retinoic acid |
| FC | Fibrosis cocktail |
| DTA | Diphtheria toxin A-chain |
| EGF | Epidermal growth factor |
| CPT1A | Carnitine palmitoyltransferase 1A |
| FBS | Fetal bovine serum |
References
- Cunniff, B.; Druso, J.E.; van der Velden, J.L. Lung organoids: Advances in generation and 3D-visualization. Histochem. Cell Biol. 2021, 155, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Barkauskas, C.E.; Chung, M.I.; Fioret, B.; Gao, X.; Katsura, H.; Hogan, B.L. Lung organoids: Current uses and future promise. Development 2017, 144, 986–997. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Cao, Y.; Zhao, P.; Shen, S.; Xi, Y. Organoid: A powerful tool to study lung regeneration and disease. Cell Regen. 2021, 10, 21. [Google Scholar] [CrossRef] [PubMed]
- Nadkarni, R.R.; Abed, S.; Draper, J.S. Organoids as a model system for studying human lung development and disease. Biochem. Biophys. Res. Commun. 2016, 473, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Elicker, B.M.; Kallianos, K.G.; Jones, K.D.; Henry, T.S. Smoking-Related Lung Disease. Semin. Ultrasound CT MR 2019, 40, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Bracken, M.B. Why animal studies are often poor predictors of human reactions to exposure. J. R. Soc. Med. 2009, 102, 120–122. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Koo, B.-K.; Knoblich, J.A. Human organoids: Model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef]
- Choi, K.-Y.G.; Wu, B.C.; Lee, A.H.-Y.; Baquir, B.; Hancock, R.E.W. Utilizing Organoid and Air-Liquid Interface Models as a Screening Method in the Development of New Host Defense Peptides. Front. Cell. Infect. Microbiol. 2020, 10, 228. [Google Scholar] [CrossRef]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef]
- Jordan, R.E.; Cheng, K.K.; Miller, M.R.; Adab, P. Passive smoking and chronic obstructive pulmonary disease: Cross-sectional analysis of data from the Health Survey for England. BMJ Open 2011, 1, e000153. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Taneja, V.; Vassallo, R. Cigarette smoking and inflammation: Cellular and molecular mechanisms. J. Dent. Res. 2012, 91, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I.; van Schadewijk, A.A.; Crowther, A.J.; Hiemstra, P.S.; Stolk, J.; MacNee, W.; De Boer, W.I. 4-Hydroxy-2-nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2002, 166, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Liu, X.; Kobayashi, T.; Conner, H.; Kohyama, T.; Wen, F.Q.; Fang, Q.; Abe, S.; Bitterman, P.; Rennard, S.I. Reversible cigarette smoke extract-induced DNA damage in human lung fibroblasts. Am. J. Respir. Cell Mol. Biol. 2004, 31, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Kode, A.; Yang, S.R.; Rahman, I. Differential effects of cigarette smoke on oxidative stress and proinflammatory cytokine release in primary human airway epithelial cells and in a variety of transformed alveolar epithelial cells. Respir. Res. 2006, 7, 132. [Google Scholar] [CrossRef]
- Donohue, J.F. Ageing, smoking and oxidative stress. Thorax 2006, 61, 461–462. [Google Scholar] [CrossRef]
- Stampfli, M.R.; Anderson, G.P. How cigarette smoke skews immune responses to promote infection, lung disease and cancer. Nat. Rev. Immunol. 2009, 9, 377–384. [Google Scholar] [CrossRef]
- Tatsuta, M.; Kan, O.K.; Ishii, Y.; Yamamoto, N.; Ogawa, T.; Fukuyama, S.; Ogawa, A.; Fujita, A.; Nakanishi, Y.; Matsumoto, K. Effects of cigarette smoke on barrier function and tight junction proteins in the bronchial epithelium: Protective role of cathelicidin LL-37. Respir. Res. 2019, 20, 251. [Google Scholar] [CrossRef]
- Xavier, R.F.; Ramos, D.; Ito, J.T.; Rodrigues, F.M.; Bertolini, G.N.; Macchione, M.; de Toledo, A.C.; Ramos, E.M. Effects of cigarette smoking intensity on the mucociliary clearance of active smokers. Respiration 2013, 86, 479–485. [Google Scholar] [CrossRef]
- Broekema, M.; ten Hacken, N.H.; Volbeda, F.; Lodewijk, M.E.; Hylkema, M.N.; Postma, D.S.; Timens, W. Airway epithelial changes in smokers but not in ex-smokers with asthma. Am. J. Respir. Crit. Care Med. 2009, 180, 1170–1178. [Google Scholar] [CrossRef]
- Kato, Y.; Hirano, T.; Yoshida, K.; Yashima, K.; Akimoto, S.; Tsuji, K.; Ohira, T.; Tsuboi, M.; Ikeda, N.; Ebihara, Y.; et al. Frequent loss of E-cadherin and/or catenins in intrabronchial lesions during carcinogenesis of the bronchial epithelium. Lung Cancer 2005, 48, 323–330. [Google Scholar] [CrossRef]
- Takeyama, K.; Jung, B.; Shim, J.J.; Burgel, P.R.; Dao-Pick, T.; Ueki, I.F.; Protin, U.; Kroschel, P.; Nadel, J.A. Activation of epidermal growth factor receptors is responsible for mucin synthesis induced by cigarette smoke. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 280, L165–L172. [Google Scholar] [CrossRef] [PubMed]
- Lan, M.Y.; Ho, C.Y.; Lee, T.C.; Yang, A.H. Cigarette smoke extract induces cytotoxicity on human nasal epithelial cells. Am. J. Rhinol. 2007, 21, 218–223. [Google Scholar] [CrossRef]
- Luppi, F.; Aarbiou, J.; van Wetering, S.; Rahman, I.; de Boer, W.I.; Rabe, K.F.; Hiemstra, P.S. Effects of cigarette smoke condensate on proliferation and wound closure of bronchial epithelial cells in vitro: Role of glutathione. Respir. Res. 2005, 6, 140. [Google Scholar] [CrossRef] [PubMed]
- Sethi, S.; Murphy, T.F. Infection in the pathogenesis and course of chronic obstructive pulmonary disease. N. Engl. J. Med. 2008, 359, 2355–2365. [Google Scholar] [CrossRef] [PubMed]
- Cohen, N.A.; Zhang, S.; Sharp, D.B.; Tamashiro, E.; Chen, B.; Sorscher, E.J.; Woodworth, B.A. Cigarette smoke condensate inhibits transepithelial chloride transport and ciliary beat frequency. Laryngoscope 2009, 119, 2269–2274. [Google Scholar] [CrossRef] [PubMed]
- Charles, L.G.M.M.D. Cigarette Smoke Vapor-Phase Effects in the Rat Upper Respiratory Tract. Inhal. Toxicol. 2008, 10, 857–873. [Google Scholar] [CrossRef]
- Amatngalim, G.D.; Schrumpf, J.A.; Dishchekenian, F.; Mertens, T.C.J.; Ninaber, D.K.; van der Linden, A.C.; Pilette, C.; Taube, C.; Hiemstra, P.S.; van der Does, A.M. Aberrant epithelial differentiation by cigarette smoke dysregulates respiratory host defence. Eur. Respir. J. 2018, 51, 1701009. [Google Scholar] [CrossRef]
- Tamashiro, E.; Cohen, N.A.; Palmer, J.N.; Lima, W.T. Effects of cigarette smoking on the respiratory epithelium and its role in the pathogenesis of chronic rhinosinusitis. Braz. J. Otorhinolaryngol. 2009, 75, 903–907. [Google Scholar] [CrossRef]
- Verra, F.; Escudier, E.; Lebargy, F.; Bernaudin, J.F.; De Cremoux, H.; Bignon, J. Ciliary abnormalities in bronchial epithelium of smokers, ex-smokers, and nonsmokers. Am. J. Respir. Crit. Care Med. 1995, 151, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Churg, A.; Zay, K.; Shay, S.; Xie, C.; Shapiro, S.D.; Hendricks, R.; Wright, J.L. Acute cigarette smoke-induced connective tissue breakdown requires both neutrophils and macrophage metalloelastase in mice. Am. J. Respir. Cell Mol. Biol. 2002, 27, 368–374. [Google Scholar] [CrossRef]
- Dhami, R.; Gilks, B.; Xie, C.; Zay, K.; Wright, J.L.; Churg, A. Acute cigarette smoke-induced connective tissue breakdown is mediated by neutrophils and prevented by alpha1-antitrypsin. Am. J. Respir. Cell Mol. Biol. 2000, 22, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Milara, J.; Peiro, T.; Serrano, A.; Cortijo, J. Epithelial to mesenchymal transition is increased in patients with COPD and induced by cigarette smoke. Thorax 2013, 68, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Krimmer, D.I.; Burgess, J.K.; Wooi, T.K.; Black, J.L.; Oliver, B.G. Matrix proteins from smoke-exposed fibroblasts are pro-proliferative. Am. J. Respir. Cell Mol. Biol. 2012, 46, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Yamano, S.; Senoh, H.; Umeda, Y.; Hirai, S.; Saito, A.; Kasai, T.; Aiso, S. Carcinogenicity and chronic toxicity of acrolein in rats and mice by two-year inhalation study. Regul. Toxicol. Pharmacol. 2021, 121, 104863. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hu, W.; Hu, Y.; Tang, M.S. Acrolein is a major cigarette-related lung cancer agent: Preferential binding at p53 mutational hotspots and inhibition of DNA repair. Proc. Natl. Acad. Sci. USA 2006, 103, 15404–15409. [Google Scholar] [CrossRef]
- Balbo, S.; James-Yi, S.; O’Sullivan, M.G.; Stepanov, I.; Wang, M.; Zhang, S.; Kassie, F.; Carmella, S.; Wettlaufer, C.; Hohol, K.; et al. Abstract LB-63: (S)-N′-nitrosonornicotine, a constituent of smokeless tobacco, is a potent oral tumorigen in rats. Cancer Res. 2012, 72, LB-63. [Google Scholar] [CrossRef]
- Stepanov, I.; Sebero, E.; Wang, R.; Gao, Y.T.; Hecht, S.S.; Yuan, J.M. Tobacco-specific N-nitrosamine exposures and cancer risk in the Shanghai Cohort Study: Remarkable coherence with rat tumor sites. Int. J. Cancer 2014, 134, 2278–2283. [Google Scholar] [CrossRef] [PubMed]
- Goldman, R.; Enewold, L.; Pellizzari, E.; Beach, J.B.; Bowman, E.D.; Krishnan, S.S.; Shields, P.G. Smoking Increases Carcinogenic Polycyclic Aromatic Hydrocarbons in Human Lung Tissue1. Cancer Res. 2001, 61, 6367–6371. [Google Scholar] [PubMed]
- Meloche, J.; Renard, S.; Provencher, S.; Bonnet, S. Anti-inflammatory and immunosuppressive agents in PAH. Handb. Exp. Pharmacol. 2013, 218, 437–476. [Google Scholar] [CrossRef] [PubMed]
- Saad, A.A.; Hussein, T.; El-Sikaily, A.; Abdel-Mohsen, M.A.; Mokhamer, E.H.; Youssef, A.I.; Mohammed, J. Effect of Polycyclic Aromatic Hydrocarbons Exposure on Sperm DNA in Idiopathic Male Infertility. J. Health Pollut. 2019, 9, 190309. [Google Scholar] [CrossRef]
- Zhou, G.; Jiang, W.; Xia, G.; Wang, L.; Richardson, M.; Chu, C.; Moorthy, B. Attenuation of Polycyclic Aromatic Hydrocarbon (PAH)-Mediated Pulmonary DNA Adducts and Cytochrome P450 (CYP)1B1 by Dietary Antioxidants, Omega-3 Fatty Acids, in Mice. Antioxidants 2022, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.N.; Li, G.L.; Tain, F.D.; Fu, Z.I.; Jin, C.; Chen, Y.J.; Luo, S.J.; Ye, P.Z.; Zhang, J.Z.; Wang, G.C.; et al. A retrospective cohort study of leukemia and other cancers in benzene workers. Environ. Health Perspect. 1989, 82, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Farris, G.M.; Everitt, J.I.; Irons, R.D.; Popp, J.A. Carcinogenicity of Inhaled Benzene in CBA Mice. Toxicol. Sci. 1993, 20, 503–507. [Google Scholar] [CrossRef]
- Smith, M.T. Overview of benzene-induced aplastic anaemia. Eur. J. Haematol. Suppl. 1996, 60, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Katukam, V.; Kulakarni, M.; Syed, R.; Alharbi, K.; Naik, J. Effect of benzene exposure on fertility of male workers employed in bulk drug industries. Genet. Test. Mol. Biomark. 2012, 16, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Xu, K.; Ji, S.; Pu, Y.; Yu, L.; Yin, L.; Zhang, J.; Pu, Y. Toxicity in hematopoietic stem cells from bone marrow and peripheral blood in mice after benzene exposure: Single-cell transcriptome sequencing analysis. Ecotoxicol. Environ. Saf. 2021, 207, 111490. [Google Scholar] [CrossRef] [PubMed]
- Dellarco, V.L. A mutagenicity assessment of acetaldehyde. Mutat. Res. 1988, 195, 1–20. [Google Scholar] [CrossRef]
- Peterson, L.A.; Oram, M.K.; Flavin, M.; Seabloom, D.; Smith, W.E.; O’Sullivan, M.G.; Vevang, K.R.; Upadhyaya, P.; Stornetta, A.; Floeder, A.C.; et al. Coexposure to Inhaled Aldehydes or Carbon Dioxide Enhances the Carcinogenic Properties of the Tobacco-Specific Nitrosamine 4-Methylnitrosamino-1-(3-pyridyl)-1-butanone in the A/J Mouse Lung. Chem. Res. Toxicol. 2021, 34, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Pryor, W.A.; Stone, K. Oxidants in cigarette smoke. Radicals, hydrogen peroxide, peroxynitrate, and peroxynitrite. Ann. N. Y. Acad. Sci. 1993, 686, 12–27; discussion 27–28. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Wen, S.; Cao, W.; Yue, P.; Xu, X.; Zhang, Y.; Luo, L.; Chen, T.; Li, L.; Wang, F.; et al. Lung organoids, useful tools for investigating epithelial repair after lung injury. Stem. Cell Res. Ther. 2021, 12, 95. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Mun, H.; Sung, C.O.; Cho, E.J.; Jeon, H.J.; Chun, S.M.; Jung, D.J.; Shin, T.H.; Jeong, G.S.; Kim, D.K.; et al. Patient-derived lung cancer organoids as in vitro cancer models for therapeutic screening. Nat. Commun. 2019, 10, 3991. [Google Scholar] [CrossRef] [PubMed]
- Kopf-Maier, P.; Zimmermann, B. Organoid reorganization of human tumors under in vitro conditions. Cell Tissue Res. 1991, 264, 563–576. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, S.; Ito, I.; Nagasaki, T.; Yamamoto, Y.; Konishi, S.; Korogi, Y.; Matsumoto, H.; Muro, S.; Hirai, T.; Funato, M.; et al. Generation of alveolar epithelial spheroids via isolated progenitor cells from human pluripotent stem cells. Stem Cell Rep. 2014, 3, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Dye, B.R.; Hill, D.R.; Ferguson, M.A.; Tsai, Y.H.; Nagy, M.S.; Dyal, R.; Wells, J.M.; Mayhew, C.N.; Nattiv, R.; Klein, O.D.; et al. In vitro generation of human pluripotent stem cell derived lung organoids. Elife 2015, 4, e05098. [Google Scholar] [CrossRef]
- Sachs, N.; Papaspyropoulos, A.; Zomer-van Ommen, D.D.; Heo, I.; Bottinger, L.; Klay, D.; Weeber, F.; Huelsz-Prince, G.; Iakobachvili, N.; Amatngalim, G.D.; et al. Long-term expanding human airway organoids for disease modeling. EMBO J. 2019, 38, e100300. [Google Scholar] [CrossRef]
- Chen, Y.W.; Huang, S.X.; de Carvalho, A.; Ho, S.H.; Islam, M.N.; Volpi, S.; Notarangelo, L.D.; Ciancanelli, M.; Casanova, J.L.; Bhattacharya, J.; et al. A three-dimensional model of human lung development and disease from pluripotent stem cells. Nat. Cell Biol. 2017, 19, 542–549. [Google Scholar] [CrossRef]
- Jacob, A.; Morley, M.; Hawkins, F.; McCauley, K.B.; Jean, J.C.; Heins, H.; Na, C.L.; Weaver, T.E.; Vedaie, M.; Hurley, K.; et al. Differentiation of Human Pluripotent Stem Cells into Functional Lung Alveolar Epithelial Cells. Cell Stem. Cell 2017, 21, 472–488.e410. [Google Scholar] [CrossRef]
- McCauley, K.B.; Hawkins, F.; Serra, M.; Thomas, D.C.; Jacob, A.; Kotton, D.N. Efficient Derivation of Functional Human Airway Epithelium from Pluripotent Stem Cells via Temporal Regulation of Wnt Signaling. Cell Stem. Cell 2017, 20, 844–857.e846. [Google Scholar] [CrossRef]
- A549 Lung Cancer Cells. Available online: https://www.atcc.org/products/ccl-185 (accessed on 21 September 2022).
- Corro, C.; Novellasdemunt, L.; Li, V.S.W. A brief history of organoids. Am. J. Physiol. Cell Physiol. 2020, 319, C151–C165. [Google Scholar] [CrossRef]
- Fatehullah, A.; Tan, S.H.; Barker, N. Organoids as an in vitro model of human development and disease. Nat. Cell Biol. 2016, 18, 246–254. [Google Scholar] [CrossRef]
- Hsia, G.S.P.; Esposito, J.; da Rocha, L.A.; Ramos, S.L.G.; Okamoto, O.K. Clinical Application of Human Induced Pluripotent Stem Cell-Derived Organoids as an Alternative to Organ Transplantation. Stem Cells Int. 2021, 2021, 6632160. [Google Scholar] [CrossRef] [PubMed]
- Danahay, H.; Pessotti, A.D.; Coote, J.; Montgomery, B.E.; Xia, D.; Wilson, A.; Yang, H.; Wang, Z.; Bevan, L.; Thomas, C.; et al. Notch2 is required for inflammatory cytokine-driven goblet cell metaplasia in the lung. Cell Rep. 2015, 10, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Matsumoto, K.; Brockway, B.L.; Rackley, C.R.; Liang, J.; Lee, J.H.; Jiang, D.; Noble, P.W.; Randell, S.H.; Kim, C.F.; et al. Airway epithelial progenitors are region specific and show differential responses to bleomycin-induced lung injury. Stem Cells 2012, 30, 1948–1960. [Google Scholar] [CrossRef]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef]
- McQualter, J.L.; Yuen, K.; Williams, B.; Bertoncello, I. Evidence of an epithelial stem/progenitor cell hierarchy in the adult mouse lung. Proc. Natl. Acad. Sci. USA 2010, 107, 1414–1419. [Google Scholar] [CrossRef]
- Porotto, M.; Ferren, M.; Chen, Y.W.; Siu, Y.; Makhsous, N.; Rima, B.; Briese, T.; Greninger, A.L.; Snoeck, H.W.; Moscona, A. Authentic Modeling of Human Respiratory Virus Infection in Human Pluripotent Stem Cell-Derived Lung Organoids. mBio 2019, 10, e00723-19. [Google Scholar] [CrossRef] [PubMed]
- Leibel, S.L.; Winquist, A.; Tseu, I.; Wang, J.; Luo, D.; Shojaie, S.; Nathan, N.; Snyder, E.; Post, M. Reversal of Surfactant Protein B Deficiency in Patient Specific Human Induced Pluripotent Stem Cell Derived Lung Organoids by Gene Therapy. Sci. Rep. 2019, 9, 13450. [Google Scholar] [CrossRef]
- Chen, Y.; Feng, J.; Zhao, S.; Han, L.; Yang, H.; Lin, Y.; Rong, Z. Long-Term Engraftment Promotes Differentiation of Alveolar Epithelial Cells from Human Embryonic Stem Cell Derived Lung Organoids. Stem Cells Dev. 2018, 27, 1339–1349. [Google Scholar] [CrossRef]
- Crystal, R.G. Airway basal cells. The “smoking gun” of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2014, 190, 1355–1362. [Google Scholar] [CrossRef]
- Pardo-Saganta, A.; Law, B.M.; Tata, P.R.; Villoria, J.; Saez, B.; Mou, H.; Zhao, R.; Rajagopal, J. Injury induces direct lineage segregation of functionally distinct airway basal stem/progenitor cell subpopulations. Cell Stem. Cell 2015, 16, 184–197. [Google Scholar] [CrossRef]
- Rawlins, E.L.; Okubo, T.; Xue, Y.; Brass, D.M.; Auten, R.L.; Hasegawa, H.; Wang, F.; Hogan, B.L. The role of Scgb1a1+ Clara cells in the long-term maintenance and repair of lung airway, but not alveolar, epithelium. Cell Stem. Cell 2009, 4, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Basil, M.C.; Cardenas-Diaz, F.L.; Kathiriya, J.J.; Morley, M.P.; Carl, J.; Brumwell, A.N.; Katzen, J.; Slovik, K.J.; Babu, A.; Zhou, S.; et al. Human distal airways contain a multipotent secretory cell that can regenerate alveoli. Nature 2022, 604, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Tammela, T.; Hofree, M.; Choi, J.; Marjanovic, N.D.; Han, S.; Canner, D.; Wu, K.; Paschini, M.; Bhang, D.H.; et al. Anatomically and Functionally Distinct Lung Mesenchymal Populations Marked by Lgr5 and Lgr6. Cell 2017, 170, 1149–1163.e1112. [Google Scholar] [CrossRef] [PubMed]
- Brody, J.S.; Williams, M.C. Pulmonary alveolar epithelial cell differentiation. Annu. Rev. Physiol. 1992, 54, 351–371. [Google Scholar] [CrossRef]
- Fehrenbach, H. Alveolar epithelial type II cell: Defender of the alveolus revisited. Respir. Res. 2001, 2, 33–46. [Google Scholar] [CrossRef]
- Mason, R.J. Biology of alveolar type II cells. Respirology 2006, 11 (Suppl. 1), S12–S15. [Google Scholar] [CrossRef]
- Liao, D.; Li, H. Dissecting the Niche for Alveolar Type II Cells With Alveolar Organoids. Front. Cell Dev. Biol. 2020, 8, 419. [Google Scholar] [CrossRef]
- Sucre, J.M.S.; Jetter, C.S.; Loomans, H.; Williams, J.; Plosa, E.J.; Benjamin, J.T.; Young, L.R.; Kropski, J.A.; Calvi, C.L.; Kook, S.; et al. Successful Establishment of Primary Type II Alveolar Epithelium with 3D Organotypic Coculture. Am. J. Respir. Cell Mol. Biol. 2018, 59, 158–166. [Google Scholar] [CrossRef]
- Youk, J.; Kim, T.; Evans, K.V.; Jeong, Y.I.; Hur, Y.; Hong, S.P.; Kim, J.H.; Yi, K.; Kim, S.Y.; Na, K.J.; et al. Three-Dimensional Human Alveolar Stem Cell Culture Models Reveal Infection Response to SARS-CoV-2. Cell Stem Cell 2020, 27, 905–919.e910. [Google Scholar] [CrossRef]
- Kim, C.F.; Jackson, E.L.; Woolfenden, A.E.; Lawrence, S.; Babar, I.; Vogel, S.; Crowley, D.; Bronson, R.T.; Jacks, T. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 2005, 121, 823–835. [Google Scholar] [CrossRef]
- Hannan, N.R.; Sampaziotis, F.; Segeritz, C.P.; Hanley, N.A.; Vallier, L. Generation of Distal Airway Epithelium from Multipotent Human Foregut Stem Cells. Stem Cells Dev. 2015, 24, 1680–1690. [Google Scholar] [CrossRef] [PubMed]
- Salwig, I.; Spitznagel, B.; Vazquez-Armendariz, A.I.; Khalooghi, K.; Guenther, S.; Herold, S.; Szibor, M.; Braun, T. Bronchioalveolar stem cells are a main source for regeneration of distal lung epithelia in vivo. EMBO J. 2019, 38, e102099. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Liu, K.; Cui, G.; Huang, X.; Yao, S.; Guo, W.; Qin, Z.; Li, Y.; Yang, R.; Pu, W.; et al. Lung regeneration by multipotent stem cells residing at the bronchioalveolar-duct junction. Nat. Genet. 2019, 51, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Bhang, D.H.; Beede, A.; Huang, T.L.; Stripp, B.R.; Bloch, K.D.; Wagers, A.J.; Tseng, Y.H.; Ryeom, S.; Kim, C.F. Lung stem cell differentiation in mice directed by endothelial cells via a BMP4-NFATc1-thrombospondin-1 axis. Cell 2014, 156, 440–455. [Google Scholar] [CrossRef]
- Green, M.D.; Chen, A.; Nostro, M.C.; d’Souza, S.L.; Schaniel, C.; Lemischka, I.R.; Gouon-Evans, V.; Keller, G.; Snoeck, H.W. Generation of anterior foregut endoderm from human embryonic and induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 267–272. [Google Scholar] [CrossRef]
- Huang, S.X.; Green, M.D.; de Carvalho, A.T.; Mumau, M.; Chen, Y.W.; D’Souza, S.L.; Snoeck, H.W. The in vitro generation of lung and airway progenitor cells from human pluripotent stem cells. Nat. Protoc. 2015, 10, 413–425. [Google Scholar] [CrossRef]
- Tian, L.; Gao, J.; Garcia, I.M.; Chen, H.J.; Castaldi, A.; Chen, Y.W. Human pluripotent stem cell-derived lung organoids: Potential applications in development and disease modeling. Wiley Interdiscip Rev. Dev. Biol. 2021, 10, e399. [Google Scholar] [CrossRef]
- Lloyd, C.M.; Marsland, B.J. Lung Homeostasis: Influence of Age, Microbes, and the Immune System. Immunity 2017, 46, 549–561. [Google Scholar] [CrossRef]
- Zhao, X.; Xu, Z.; Xiao, L.; Shi, T.; Xiao, H.; Wang, Y.; Li, Y.; Xue, F.; Zeng, W. Review on the Vascularization of Organoids and Organoids-on-a-Chip. Front. Bioeng. Biotechnol. 2021, 9, 637048. [Google Scholar] [CrossRef]
- Leeman, K.T.; Pessina, P.; Lee, J.H.; Kim, C.F. Mesenchymal Stem Cells Increase Alveolar Differentiation in Lung Progenitor Organoid Cultures. Sci. Rep. 2019, 9, 6479. [Google Scholar] [CrossRef]
- Alvarez, D.F.; Huang, L.; King, J.A.; ElZarrad, M.K.; Yoder, M.C.; Stevens, T. Lung microvascular endothelium is enriched with progenitor cells that exhibit vasculogenic capacity. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L419–L430. [Google Scholar] [CrossRef] [PubMed]
- Lechner, A.J.; Driver, I.H.; Lee, J.; Conroy, C.M.; Nagle, A.; Locksley, R.M.; Rock, J.R. Recruited Monocytes and Type 2 Immunity Promote Lung Regeneration following Pneumonectomy. Cell Stem Cell 2017, 21, 120–134.e127. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Jang, Y.J.; Dabrowska, C.; Iich, E.; Evans, K.V.; Hall, H.; Janes, S.M.; Simons, B.D.; Koo, B.K.; Kim, J.; et al. Release of Notch activity coordinated by IL-1beta signalling confers differentiation plasticity of airway progenitors via Fosl2 during alveolar regeneration. Nat. Cell Biol. 2021, 23, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Ng-Blichfeldt, J.P.; de Jong, T.; Kortekaas, R.K.; Wu, X.; Lindner, M.; Guryev, V.; Hiemstra, P.S.; Stolk, J.; Konigshoff, M.; Gosens, R. TGF-beta activation impairs fibroblast ability to support adult lung epithelial progenitor cell organoid formation. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 317, L14–L28. [Google Scholar] [CrossRef] [PubMed]
- Tadokoro, T.; Wang, Y.; Barak, L.S.; Bai, Y.; Randell, S.H.; Hogan, B.L. IL-6/STAT3 promotes regeneration of airway ciliated cells from basal stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E3641–E3649. [Google Scholar] [CrossRef]
- Li, K.; Li, M.; Li, W.; Yu, H.; Sun, X.; Zhang, Q.; Li, Y.; Li, X.; Li, Y.; Abel, E.D.; et al. Airway epithelial regeneration requires autophagy and glucose metabolism. Cell Death Dis. 2019, 10, 875. [Google Scholar] [CrossRef]
- Sun, T.; Huang, Z.; Zhang, H.; Posner, C.; Jia, G.; Ramalingam, T.; Xu, M.; Brightbill, H.; Egen, J.; Dey, A. TAZ is required for lung alveolar epithelial cell differentiation after injury. JCI Insight 2019, 4, e128674. [Google Scholar] [CrossRef]
- Ng-Blichfeldt, J.P.; Schrik, A.; Kortekaas, R.K.; Noordhoek, J.A.; Heijink, I.H.; Hiemstra, P.S.; Stolk, J.; Konigshoff, M.; Gosens, R. Retinoic acid signaling balances adult distal lung epithelial progenitor cell growth and differentiation. EBioMedicine 2018, 36, 461–474. [Google Scholar] [CrossRef]
- Paris, A.J.; Hayer, K.E.; Oved, J.H.; Avgousti, D.C.; Toulmin, S.A.; Zepp, J.A.; Zacharias, W.J.; Katzen, J.B.; Basil, M.C.; Kremp, M.M.; et al. STAT3-BDNF-TrkB signalling promotes alveolar epithelial regeneration after lung injury. Nat. Cell Biol. 2020, 22, 1197–1210. [Google Scholar] [CrossRef]
- Glisinski, K.M.; Schlobohm, A.J.; Paramore, S.V.; Birukova, A.; Moseley, M.A.; Foster, M.W.; Barkauskas, C.E. Interleukin-13 disrupts type 2 pneumocyte stem cell activity. JCI Insight 2020, 5, e131232. [Google Scholar] [CrossRef]
- Hussain, M.; Xu, C.; Ahmad, M.; Yang, Y.; Lu, M.; Wu, X.; Tang, L.; Wu, X. Notch Signaling: Linking Embryonic Lung Development and Asthmatic Airway Remodeling. Mol. Pharmacol. 2017, 92, 676–693. [Google Scholar] [CrossRef]
- Hu, Y.; Ng-Blichfeldt, J.P.; Ota, C.; Ciminieri, C.; Ren, W.; Hiemstra, P.S.; Stolk, J.; Gosens, R.; Konigshoff, M. Wnt/beta-catenin signaling is critical for regenerative potential of distal lung epithelial progenitor cells in homeostasis and emphysema. Stem Cells 2020, 38, 1467–1478. [Google Scholar] [CrossRef]
- Lin, Y.; He, Z.; Ye, J.; Liu, Z.; She, X.; Gao, X.; Liang, R. Progress in Understanding the IL-6/STAT3 Pathway in Colorectal Cancer. OncoTargets Ther. 2020, 13, 13023–13032. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Park, J.E.; Tsagkogeorga, G.; Yanagita, M.; Koo, B.K.; Han, N.; Lee, J.H. Inflammatory Signals Induce AT2 Cell-Derived Damage-Associated Transient Progenitors that Mediate Alveolar Regeneration. Cell Stem Cell 2020, 27, 366–382.e367. [Google Scholar] [CrossRef]
- Zacharias, W.J.; Frank, D.B.; Zepp, J.A.; Morley, M.P.; Alkhaleel, F.A.; Kong, J.; Zhou, S.; Cantu, E.; Morrisey, E.E. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature 2018, 555, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Malpel, S.; Mendelsohn, C.; Cardoso, W.V. Regulation of retinoic acid signaling during lung morphogenesis. Development 2000, 127, 3057–3067. [Google Scholar] [CrossRef] [PubMed]
- Skronska-Wasek, W.; Mutze, K.; Baarsma, H.A.; Bracke, K.R.; Alsafadi, H.N.; Lehmann, M.; Costa, R.; Stornaiuolo, M.; Novellino, E.; Brusselle, G.G.; et al. Reduced Frizzled Receptor 4 Expression Prevents WNT/beta-Catenin-driven Alveolar Lung Repair in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2017, 196, 172–185. [Google Scholar] [CrossRef]
- Katsura, H.; Kobayashi, Y.; Tata, P.R.; Hogan, B.L.M. IL-1 and TNFalpha Contribute to the Inflammatory Niche to Enhance Alveolar Regeneration. Stem Cell Rep. 2019, 12, 657–666. [Google Scholar] [CrossRef]
- Durham, A.L.; Adcock, I.M. The relationship between COPD and lung cancer. Lung Cancer 2015, 90, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Li, Y.; Gong, L.; Yun, J.H.; Xu, S.; Tesfaigzi, Y.; Qiao, D.; Zhou, X. Tempo-spatial regulation of the Wnt pathway by FAM13A modulates the stemness of alveolar epithelial progenitors. EBioMedicine 2021, 69, 103463. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Lao, T.; Qiu, W.; Polverino, F.; Gupta, K.; Guo, F.; Mancini, J.D.; Naing, Z.Z.; Cho, M.H.; Castaldi, P.J.; et al. A Chronic Obstructive Pulmonary Disease Susceptibility Gene, FAM13A, Regulates Protein Stability of beta-Catenin. Am. J. Respir. Crit. Care Med. 2016, 194, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Lkhagvadorj, K.; Zeng, Z.; Song, J.; Reinders-Luinge, M.; Kooistra, W.; Song, S.; Krauss-Etschmann, S.; Melgert, B.N.; Cao, J.; Hylkema, M.N. Prenatal smoke exposure dysregulates lung epithelial cell differentiation in mouse offspring: Role for AREG-induced EGFR signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 319, L742–L751. [Google Scholar] [CrossRef] [PubMed]
- Zuo, W.L.; Yang, J.; Gomi, K.; Chao, I.; Crystal, R.G.; Shaykhiev, R. EGF-Amphiregulin Interplay in Airway Stem/Progenitor Cells Links the Pathogenesis of Smoking-Induced Lesions in the Human Airway Epithelium. Stem Cells 2017, 35, 824–837. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Bos, I.S.T.; Conlon, T.M.; Ansari, M.; Verschut, V.; Verkleij, L.A.; D’Ambrosi, A.; Matveyenko, A.; Schiller, H.B.; Königshoff, M.; et al. A transcriptomics-guided drug target discovery strategy identifies novel receptor ligands for lung regeneration. bioRxiv 2021. [Google Scholar] [CrossRef]
- Youlden, D.R.; Cramb, S.M.; Baade, P.D. The International Epidemiology of Lung Cancer: Geographical distribution and secular trends. J. Thorac. Oncol. 2008, 3, 819–831. [Google Scholar] [CrossRef]
- Shields, P.G. Molecular epidemiology of smoking and lung cancer. Oncogene 2002, 21, 6870–6876. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Xin, W.; Yan, C.; Shi, Y.; Li, Y.; Hu, Y.; Ying, K. Organoids in lung cancer: A teenager with infinite growth potential. Lung Cancer 2022, 172, 100–107. [Google Scholar] [CrossRef]
- Chen, J.H.; Chu, X.P.; Zhang, J.T.; Nie, Q.; Tang, W.F.; Su, J.; Yan, H.H.; Zheng, H.P.; Chen, Z.X.; Chen, X.; et al. Genomic characteristics and drug screening among organoids derived from non-small cell lung cancer patients. Thorac. Cancer 2020, 11, 2279–2290. [Google Scholar] [CrossRef]
- Schaal, C.M.; Bora-Singhal, N.; Kumar, D.M.; Chellappan, S.P. Regulation of Sox2 and stemness by nicotine and electronic-cigarettes in non-small cell lung cancer. Mol. Cancer 2018, 17, 149. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zhang, J.; Zhang, J.; Zhang, N.; Zeng, Y.; Tang, S.; Tao, Z.; Qu, X.; Jia, J.; Zhu, W.; et al. Nicotine-enhanced stemness and epithelial-mesenchymal transition of human umbilical cord mesenchymal stem cells promote tumor formation and growth in nude mice. Oncotarget 2018, 9, 591–606. [Google Scholar] [CrossRef] [PubMed]
- Kameyama, N.; Chubachi, S.; Hegab, A.E.; Yasuda, H.; Kagawa, S.; Tsutsumi, A.; Fukunaga, K.; Shimoda, M.; Kanai, Y.; Soejima, K.; et al. Intermittent Exposure to Cigarette Smoke Increases Lung Tumors and the Severity of Emphysema More than Continuous Exposure. Am. J. Respir. Cell Mol. Biol. 2018, 59, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Arcavi, L.; Benowitz, N.L. Cigarette smoking and infection. Arch. Intern. Med. 2004, 164, 2206–2216. [Google Scholar] [CrossRef] [PubMed]
- Irie, H.; Ozaki, M.; Chubachi, S.; Hegab, A.E.; Tsutsumi, A.; Kameyama, N.; Sakurai, K.; Nakayama, S.; Kagawa, S.; Wada, S.; et al. Short-term intermittent cigarette smoke exposure enhances alveolar type 2 cell stemness via fatty acid oxidation. Respir. Res. 2022, 23, 41. [Google Scholar] [CrossRef]
- Hewitt, R.; Farne, H.; Ritchie, A.; Luke, E.; Johnston, S.L.; Mallia, P. The role of viral infections in exacerbations of chronic obstructive pulmonary disease and asthma. Ther. Adv. Respir. Dis. 2016, 10, 158–174. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.S.; Lim, R.L.; Liu, J.; Ong, H.H.; Tan, V.J.; Lim, H.F.; Chung, K.F.; Adcock, I.M.; Chow, V.T.; Wang, Y. Respiratory Viral Infections in Exacerbation of Chronic Airway Inflammatory Diseases: Novel Mechanisms and Insights from the Upper Airway Epithelium. Front. Cell Dev. Biol. 2020, 8, 99. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Patel, K.B.; Booth, J.L.; Zhang, W.; Metcalf, J.P. Cigarette smoke extract suppresses the RIG-I-initiated innate immune response to influenza virus in the human lung. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L821–L830. [Google Scholar] [CrossRef]
- Noah, T.L.; Zhou, H.; Monaco, J.; Horvath, K.; Herbst, M.; Jaspers, I. Tobacco smoke exposure and altered nasal responses to live attenuated influenza virus. Environ. Health Perspect. 2011, 119, 78–83. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Feng, Y.; Kong, Y.; Barnes, P.F.; Huang, F.F.; Klucar, P.; Wang, X.; Samten, B.; Sengupta, M.; Machona, B.; Donis, R.; et al. Exposure to cigarette smoke inhibits the pulmonary T-cell response to influenza virus and Mycobacterium tuberculosis. Infect. Immun. 2011, 79, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Modestou, M.A.; Manzel, L.J.; El-Mahdy, S.; Look, D.C. Inhibition of IFN-gamma-dependent antiviral airway epithelial defense by cigarette smoke. Respir. Res. 2010, 11, 64. [Google Scholar] [CrossRef]
- Zhou, J.; Li, C.; Sachs, N.; Chiu, M.C.; Wong, B.H.; Chu, H.; Poon, V.K.; Wang, D.; Zhao, X.; Wen, L.; et al. Differentiated human airway organoids to assess infectivity of emerging influenza virus. Proc. Natl. Acad. Sci. USA 2018, 115, 6822–6827. [Google Scholar] [CrossRef] [PubMed]
- Lamers, M.M.; van der Vaart, J.; Knoops, K.; Riesebosch, S.; Breugem, T.I.; Mykytyn, A.Z.; Beumer, J.; Schipper, D.; Bezstarosti, K.; Koopman, C.D.; et al. An organoid-derived bronchioalveolar model for SARS-CoV-2 infection of human alveolar type II-like cells. EMBO J. 2021, 40, e105912. [Google Scholar] [CrossRef] [PubMed]
- Oh, C.K.; Murray, L.A.; Molfino, N.A. Smoking and idiopathic pulmonary fibrosis. Pulm. Med. 2012, 2012, 808260. [Google Scholar] [CrossRef]
- Fernandez, I.E.; Eickelberg, O. The impact of TGF-beta on lung fibrosis: From targeting to biomarkers. Proc. Am. Thorac. Soc. 2012, 9, 111–116. [Google Scholar] [CrossRef]
- Wilkinson, D.C.; Alva-Ornelas, J.A.; Sucre, J.M.; Vijayaraj, P.; Durra, A.; Richardson, W.; Jonas, S.J.; Paul, M.K.; Karumbayaram, S.; Dunn, B.; et al. Development of a Three-Dimensional Bioengineering Technology to Generate Lung Tissue for Personalized Disease Modeling. Stem Cells Transl. Med. 2017, 6, 622–633. [Google Scholar] [CrossRef] [PubMed]
- Strikoudis, A.; Cieslak, A.; Loffredo, L.; Chen, Y.W.; Patel, N.; Saqi, A.; Lederer, D.J.; Snoeck, H.W. Modeling of Fibrotic Lung Disease Using 3D Organoids Derived from Human Pluripotent Stem Cells. Cell Rep. 2019, 27, 3709–3723.e3705. [Google Scholar] [CrossRef] [PubMed]
- Ptasinski, V.; Monkley, S.; Tammia, M.; Öst, K.; Overed-Sayer, C.; Hazon, P.; Wagner, D.E.; Murray, L.A. Modeling idiopathic pulmonary fibrosis using induced pluripotent stem cell-derived alveolar epithelial organoids. ERJ Open Res. 2021, 7, 62. [Google Scholar] [CrossRef]
- Shankaran, A.; Prasad, K.; Chaudhari, S.; Brand, A.; Satyamoorthy, K. Advances in development and application of human organoids. 3 Biotech 2021, 11, 257. [Google Scholar] [CrossRef] [PubMed]
- Xinaris, C. Organoids for replacement therapy: Expectations, limitations and reality. Curr. Opin. Organ. Transpl. 2019, 24, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Gkatzis, K.; Taghizadeh, S.; Huh, D.; Stainier, D.Y.R.; Bellusci, S. Use of three-dimensional organoids and lung-on-a-chip methods to study lung development, regeneration and disease. Eur. Respir. J. 2018, 52, 1800876. [Google Scholar] [CrossRef] [PubMed]
- Salahudeen, A.A.; Choi, S.S.; Rustagi, A.; Zhu, J.; van Unen, V.; de la, O.S.; Flynn, R.A.; Margalef-Catala, M.; Santos, A.J.M.; Ju, J.; et al. Progenitor identification and SARS-CoV-2 infection in human distal lung organoids. Nature 2020, 588, 670–675. [Google Scholar] [CrossRef]
- Nagle, P.W.; Coppes, R.P. Current and Future Perspectives of the Use of Organoids in Radiobiology. Cells 2020, 9, 2649. [Google Scholar] [CrossRef] [PubMed]
- van Riet, S.; van Schadewijk, A.; Khedoe, P.; Limpens, R.; Barcena, M.; Stolk, J.; Hiemstra, P.S.; van der Does, A.M. Organoid-based expansion of patient-derived primary alveolar type 2 cells for establishment of alveolus epithelial Lung-Chip cultures. Am. J. Physiol. Lung Cell Mol. Physiol. 2022, 322, L526–L538. [Google Scholar] [CrossRef] [PubMed]
- Sokolowska, P.; Zuchowska, A.; Brzozka, Z. Why Can Organoids Improve Current Organ-on-Chip Platforms? Organoids 2022, 1, 69–84. [Google Scholar] [CrossRef]
- Agraval, H.; Sharma, J.R.; Prakash, N.; Yadav, U.C.S. Fisetin suppresses cigarette smoke extract-induced epithelial to mesenchymal transition of airway epithelial cells through regulating COX-2/MMPs/beta-catenin pathway. Chem. Biol. Interact. 2022, 351, 109771. [Google Scholar] [CrossRef]
- Hiemstra, P.S.; Grootaers, G.; van der Does, A.M.; Krul, C.A.M.; Kooter, I.M. Human lung epithelial cell cultures for analysis of inhaled toxicants: Lessons learned and future directions. Toxicol. Vitr. 2018, 47, 137–146. [Google Scholar] [CrossRef]
- Chen, C.H.; Li, Y.R.; Lin, S.H.; Chang, H.H.; Chai, W.H.; Chan, P.C.; Lin, C.H. Tiotropium/Olodaterol treatment reduces cigarette smoke extract-induced cell death in BEAS-2B bronchial epithelial cells. BMC Pharmacol. Toxicol. 2020, 21, 74. [Google Scholar] [CrossRef]
- Heijink, I.H.; Brandenburg, S.M.; Postma, D.S.; van Oosterhout, A.J. Cigarette smoke impairs airway epithelial barrier function and cell-cell contact recovery. Eur. Respir. J. 2012, 39, 419–428. [Google Scholar] [CrossRef]
- Upadhyay, S.; Palmberg, L. Air-Liquid Interface: Relevant In Vitro Models for Investigating Air Pollutant-Induced Pulmonary Toxicity. Toxicol. Sci. 2018, 164, 21–30. [Google Scholar] [CrossRef]
- Ahmad, S.; Ahmad, A.; Neeves, K.B.; Hendry-Hofer, T.; Loader, J.E.; White, C.W.; Veress, L. In vitro cell culture model for toxic inhaled chemical testing. J. Vis. Exp. 2014, 87, e51539. [Google Scholar] [CrossRef]
- Clippinger, A.J.; Ahluwalia, A.; Allen, D.; Bonner, J.C.; Casey, W.; Castranova, V.; David, R.M.; Halappanavar, S.; Hotchkiss, J.A.; Jarabek, A.M.; et al. Expert consensus on an in vitro approach to assess pulmonary fibrogenic potential of aerosolized nanomaterials. Arch. Toxicol. 2016, 90, 1769–1783. [Google Scholar] [CrossRef]
- Gminski, R.; Tang, T.; Mersch-Sundermann, V. Cytotoxicity and genotoxicity in human lung epithelial A549 cells caused by airborne volatile organic compounds emitted from pine wood and oriented strand boards. Toxicol. Lett. 2010, 196, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Lenz, A.G.; Karg, E.; Brendel, E.; Hinze-Heyn, H.; Maier, K.L.; Eickelberg, O.; Stoeger, T.; Schmid, O. Inflammatory and oxidative stress responses of an alveolar epithelial cell line to airborne zinc oxide nanoparticles at the air-liquid interface: A comparison with conventional, submerged cell-culture conditions. BioMed Res. Int. 2013, 2013, 652632. [Google Scholar] [CrossRef]
- Jiang, D.; Schaefer, N.; Chu, H.W. Air-Liquid Interface Culture of Human and Mouse Airway Epithelial Cells. Methods Mol. Biol. 2018, 1809, 91–109. [Google Scholar] [CrossRef]
- Schamberger, A.C.; Staab-Weijnitz, C.A.; Mise-Racek, N.; Eickelberg, O. Cigarette smoke alters primary human bronchial epithelial cell differentiation at the air-liquid interface. Sci. Rep. 2015, 5, 8163. [Google Scholar] [CrossRef] [PubMed]
- Eenjes, E.; van Riet, S.; Kroon, A.A.; Slats, A.M.; Khedoe, P.; Boerema-de Munck, A.; Buscop-van Kempen, M.; Ninaber, D.K.; Reiss, I.K.M.; Clevers, H.; et al. Disease modeling following organoid-based expansion of airway epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2021, 321, L775–L786. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef] [PubMed]
- Benam, K.H.; Dauth, S.; Hassell, B.; Herland, A.; Jain, A.; Jang, K.J.; Karalis, K.; Kim, H.J.; MacQueen, L.; Mahmoodian, R.; et al. Engineered in vitro disease models. Annu. Rev. Pathol. 2015, 10, 195–262. [Google Scholar] [CrossRef]
- Shrestha, J.; Razavi Bazaz, S.; Aboulkheyr Es, H.; Yaghobian Azari, D.; Thierry, B.; Ebrahimi Warkiani, M.; Ghadiri, M. Lung-on-a-chip: The future of respiratory disease models and pharmacological studies. Crit. Rev. Biotechnol. 2020, 40, 213–230. [Google Scholar] [CrossRef]
- Benam, K.H.; Novak, R.; Nawroth, J.; Hirano-Kobayashi, M.; Ferrante, T.C.; Choe, Y.; Prantil-Baun, R.; Weaver, J.C.; Bahinski, A.; Parker, K.K.; et al. Matched-Comparative Modeling of Normal and Diseased Human Airway Responses Using a Microengineered Breathing Lung Chip. Cell Syst. 2016, 3, 456–466.e454. [Google Scholar] [CrossRef]
- Hou, W.; Hu, S.; Yong, K.-t.; Zhang, J.; Ma, H. Cigarette smoke-induced malignant transformation via STAT3 signalling in pulmonary epithelial cells in a lung-on-a-chip model. Bio-Des. Manuf. 2020, 3, 383–395. [Google Scholar] [CrossRef]
- Halappanavar, S.; Russell, M.; Stampfli, M.R.; Williams, A.; Yauk, C.L. Induction of the interleukin 6/ signal transducer and activator of transcription pathway in the lungs of mice sub-chronically exposed to mainstream tobacco smoke. BMC Med. Genom. 2009, 2, 56. [Google Scholar] [CrossRef] [PubMed]
- Majorova, D.; Atkins, E.; Martineau, H.; Vokral, I.; Oosterhuis, D.; Olinga, P.; Wren, B.; Cuccui, J.; Werling, D. Use of Precision-Cut Tissue Slices as a Translational Model to Study Host-Pathogen Interaction. Front. Vet. Sci. 2021, 8, 686088. [Google Scholar] [CrossRef] [PubMed]
- Bailey, K.E.; Pino, C.; Lennon, M.L.; Lyons, A.; Jacot, J.G.; Lammers, S.R.; Konigshoff, M.; Magin, C.M. Embedding of Precision-Cut Lung Slices in Engineered Hydrogel Biomaterials Supports Extended Ex Vivo Culture. Am. J. Respir. Cell Mol. Biol. 2020, 62, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Betts, C.; Cunoosamy, D.M.; Aberg, P.M.; Hornberg, J.J.; Sivars, K.B.; Cohen, T.S. Use of precision cut lung slices as a translational model for the study of lung biology. Respir. Res. 2019, 20, 162. [Google Scholar] [CrossRef]
- Preuss, E.B.; Schubert, S.; Werlein, C.; Stark, H.; Braubach, P.; Hofer, A.; Plucinski, E.K.J.; Shah, H.R.; Geffers, R.; Sewald, K.; et al. The Challenge of Long-Term Cultivation of Human Precision-Cut Lung Slices. Am. J. Pathol. 2022, 192, 239–253. [Google Scholar] [CrossRef]
- Obernolte, H.; Ritter, D.; Knebel, J.; Braubach, P.; Jonigk, D.; Warnecke, G.; Krüger, M.; Fieguth, H.; Pfennig, O.; Braun, A. Cigarette smoke and cigarette smoke condensate induce inflammation and cytotoxicity in precision-cut lung slices (PCLS). Pneumologie 2016, 70, P42. [Google Scholar] [CrossRef]
- Bialkowska, K.; Komorowski, P.; Bryszewska, M.; Milowska, K. Spheroids as a Type of Three-Dimensional Cell Cultures-Examples of Methods of Preparation and the Most Important Application. Int. J. Mol. Sci. 2020, 21, 6225. [Google Scholar] [CrossRef]
- Pampaloni, F.; Stelzer, E. Three-dimensional cell cultures in toxicology. Biotechnol. Genet. Eng. Rev. 2010, 26, 117–138. [Google Scholar] [CrossRef]
- Jimenez-Valdes, R.J.; Can, U.I.; Niemeyer, B.F.; Benam, K.H. Where We Stand: Lung Organotypic Living Systems That Emulate Human-Relevant Host-Environment/Pathogen Interactions. Front. Bioeng. Biotechnol. 2020, 8, 989. [Google Scholar] [CrossRef]
- Barre-Sinoussi, F.; Montagutelli, X. Animal models are essential to biological research: Issues and perspectives. Future Sci. OA 2015, 1, FSO63. [Google Scholar] [CrossRef]
- Wang, G.; Mohammadtursun, N.; Sun, J.; Lv, Y.; Jin, H.; Lin, J.; Kong, L.; Zhao, Z.; Zhang, H.; Dong, J. Establishment and Evaluation of a Rat Model of Sidestream Cigarette Smoke-Induced Chronic Obstructive Pulmonary Disease. Front. Physiol. 2018, 9, 58. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Arcos, I.; Geraghty, P.; Baumlin, N.; Campos, M.; Dabo, A.J.; Jundi, B.; Cummins, N.; Eden, E.; Grosche, A.; Salathe, M.; et al. Chronic electronic cigarette exposure in mice induces features of COPD in a nicotine-dependent manner. Thorax 2016, 71, 1119–1129. [Google Scholar] [CrossRef] [PubMed]
- Ergorul, C.; Levin, L.A. Solving the lost in translation problem: Improving the effectiveness of translational research. Curr. Opin. Pharmacol. 2013, 13, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Van Norman, G.A. Limitations of Animal Studies for Predicting Toxicity in Clinical Trials: Is it Time to Rethink Our Current Approach? JACC Basic Transl. Sci. 2019, 4, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Aghapour, M.; Raee, P.; Moghaddam, S.J.; Hiemstra, P.S.; Heijink, I.H. Airway Epithelial Barrier Dysfunction in Chronic Obstructive Pulmonary Disease: Role of Cigarette Smoke Exposure. Am. J. Respir. Cell Mol. Biol. 2018, 58, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, S.; Sajjan, U.S. Repair and Remodeling of airway epithelium after injury in Chronic Obstructive Pulmonary Disease. Curr. Respir. Care Rep. 2013, 2, 145–154. [Google Scholar] [CrossRef]
- Gohy, S.T.; Hupin, C.; Fregimilicka, C.; Detry, B.R.; Bouzin, C.; Gaide Chevronay, H.; Lecocq, M.; Weynand, B.; Ladjemi, M.Z.; Pierreux, C.E.; et al. Imprinting of the COPD airway epithelium for dedifferentiation and mesenchymal transition. Eur. Respir. J. 2015, 45, 1258–1272. [Google Scholar] [CrossRef]
- Li, Y.; Wu, Q.; Sun, X.; Shen, J.; Chen, H. Organoids as a Powerful Model for Respiratory Diseases. Stem Cells Int. 2020, 2020, 5847876. [Google Scholar] [CrossRef]
- Kathiriya, J.J.; Wang, C.; Zhou, M.; Brumwell, A.; Cassandras, M.; Le Saux, C.J.; Cohen, M.; Alysandratos, K.D.; Wang, B.; Wolters, P.; et al. Human alveolar type 2 epithelium transdifferentiates into metaplastic KRT5(+) basal cells. Nat. Cell Biol. 2022, 24, 10–23. [Google Scholar] [CrossRef]
- Cassandras, M.; Wang, C.; Kathiriya, J.; Tsukui, T.; Matatia, P.; Matthay, M.; Wolters, P.; Molofsky, A.; Sheppard, D.; Chapman, H.; et al. Gli1(+) mesenchymal stromal cells form a pathological niche to promote airway progenitor metaplasia in the fibrotic lung. Nat. Cell Biol. 2020, 22, 1295–1306. [Google Scholar] [CrossRef]
- Weiner, A.I.; Jackson, S.R.; Zhao, G.; Quansah, K.K.; Farshchian, J.N.; Neupauer, K.M.; Littauer, E.Q.; Paris, A.J.; Liberti, D.C.; Scott Worthen, G.; et al. Mesenchyme-free expansion and transplantation of adult alveolar progenitor cells: Steps toward cell-based regenerative therapies. NPJ Regen. Med. 2019, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Dye, B.R.; Dedhia, P.H.; Miller, A.J.; Nagy, M.S.; White, E.S.; Shea, L.D.; Spence, J.R. A bioengineered niche promotes in vivo engraftment and maturation of pluripotent stem cell derived human lung organoids. Elife 2016, 5, e19732. [Google Scholar] [CrossRef] [PubMed]



| Model | Species | Cells Used to form Organoids | Signaling Pathway | Biological Function | Link between Biological Function and Disease | References |
|---|---|---|---|---|---|---|
| Lung organoid model to study airway epithelial repair | ||||||
| Airway organoids | Human | Secretory cells and AEC2 cells | IL-1β-NOTCH-FOSL2 axis | Acquisition of differentiation plasticity; differentiation of the secretory cells to AEC2 cells | To understand the role of NOTCH and IL-1β in repair and regeneration signaling pathways in response to lung injury | [94] |
| Airway organoids | Human | iPSCs | WNT signaling | Repair and regeneration of airway epithelium | To understand the role of WNT signaling in human airway patterning which can aid in lung disease modeling | [58] |
| Adult lung epithelial progenitor cell organoid | Human and mouse | Human lung fibroblasts with adult mouse lung epithelial cell adhesion molecule-positive cells (EpCAM+) | TGF-β- WNT/β-catenin signaling | TGF-β activation impairs fibroblast ability to support adult lung epithelial progenitor cell organoid formation | To understand the aberrant mesenchymal–epithelial signaling during COPD and IPF pathophysiology | [95] |
| Airway organoids | Mouse | Basal cells | IL-6-STAT3 pathway | Differentiation of basal cells to ciliated cells and secretory cells | To evaluate the IL-6/STAT3 signaling in multi-ciliogenesis and airway repair process in response to lung injury | [96] |
| Airway organoids | Mouse | Airway stem-such as vClub cells | Glucose uptake through endocytosis/recycling of GLUT1 | Autophagy-regulated lung epithelial proliferation and regeneration | To understand the role of autophagy in repair of injured epithelium in response to allergens or other types of lung injury | [97] |
| Lung organoid model to study alveolar epithelial repair | ||||||
| Alveolar organoids | Mouse | AEC2 cells | PDZ-binding motif (TAZ) | Lung alveolar epithelial cell differentiation | To understand the role of lung alveolar epithelial cell differentiation in response to lung injury during the pathogenesis of IPF | [98] |
| Adult lung organoids | Human and mouse | Primary mouse and human lung epithelial cells (airway and alveolar epithelial cells) | Retinoic acid (RA)–yes-associated protein (YAP) pathway | Balances adult distal lung epithelial progenitor cell growth and differentiation | To understand the role of the retinoic acid–yes-associated protein pathway in endogenous lung regeneration during COPD pathogenesis | [99] |
| Alveolar organoids | Human | AEC2 cells | STAT3–BDNF–TrkB signaling pathway | Alveolar–epithelial regeneration and repair | To evaluate the role of the STAT3–BDNF–TrkB signaling pathway in alveolar–epithelial regeneration during viral infections and acute respiratory distress syndrome | [100] |
| Alveolar organoids | Human | AEC2 cells | WNT/β-catenin signaling | Proliferation and transdifferentiation of AEC2 to AEC1 cells and maintenance of stemness | To understand the regulation of WNT signaling in the alveolar epithelial progenitor cells of COPD patients and discover new treatment strategies | [50] |
| Alveolar organoids | Mouse | AEC2 cells | IL-1 -TNFα—NF-κB signaling pathway | Increased alveolar proliferation, differentiation, regeneration, and repair | To understand the role of IL-1/TNFα-NF-κB signaling axis in tissue recovery following injury (e.g., influenza-induced injury) | [50] |
| Alveolosphere organoids | Human and mouse | AEC2 cells | IL-13/STAT6 pathway | Decrease expression of the alveolar epithelial cell markers; sustain the inflammatory response | To understand the IL-13-mediated chemokine and inflammation-driven responses in COPD and pulmonary fibrosis pathogenesis | [101] |
| Model | Advantages | Disadvantages |
|---|---|---|
| Cell lines |
|
|
| ALI |
|
|
| Lung on a chip |
|
|
| PCLS |
|
|
| Spheroids |
|
|
| Animal models |
|
|
| Organoids |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agraval, H.; Chu, H.W. Lung Organoids in Smoking Research: Current Advances and Future Promises. Biomolecules 2022, 12, 1463. https://doi.org/10.3390/biom12101463
Agraval H, Chu HW. Lung Organoids in Smoking Research: Current Advances and Future Promises. Biomolecules. 2022; 12(10):1463. https://doi.org/10.3390/biom12101463
Chicago/Turabian StyleAgraval, Hina, and Hong Wei Chu. 2022. "Lung Organoids in Smoking Research: Current Advances and Future Promises" Biomolecules 12, no. 10: 1463. https://doi.org/10.3390/biom12101463
APA StyleAgraval, H., & Chu, H. W. (2022). Lung Organoids in Smoking Research: Current Advances and Future Promises. Biomolecules, 12(10), 1463. https://doi.org/10.3390/biom12101463