


Combination of Immune-Related Network and Molecular Typing Analysis Defines a Three-Gene Signature for Predicting Prognosis of Triple-Negative Breast Cancer

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. TNBC Patients and Public Datasets
2.2. Differential Expression and Enrichment Analysis
2.3. Identification of High Immunogenicity Modules by WGCNA
2.4. Consensus Molecular Clustering of IRGs and Gene Set Variation Analysis (GSVA)
2.5. Generation of IRGs Gene Signature and Functional Enrichment Analysis
2.6. Construction and Validation of the Immune-Related Gene Panel (IRGP)
2.7. Comprehensive Analysis of Immune Characteristics and ICIs Therapy in Different IRGP Subgroups
2.8. Detection of the Expression of IRGP by qRT-PCR and Human Protein Atlas (HPA) Database
2.9. Statistical Analysis
3. Results
3.1. Identification of High Immunogenicity Modules in TNBC by WGCNA
3.2. Construction of Distinct IRGs Expression Patterns in TNBC
3.3. Generation of IRGs Gene Signatures and Construction of the Immune-Related Gene Panel (IRGP)
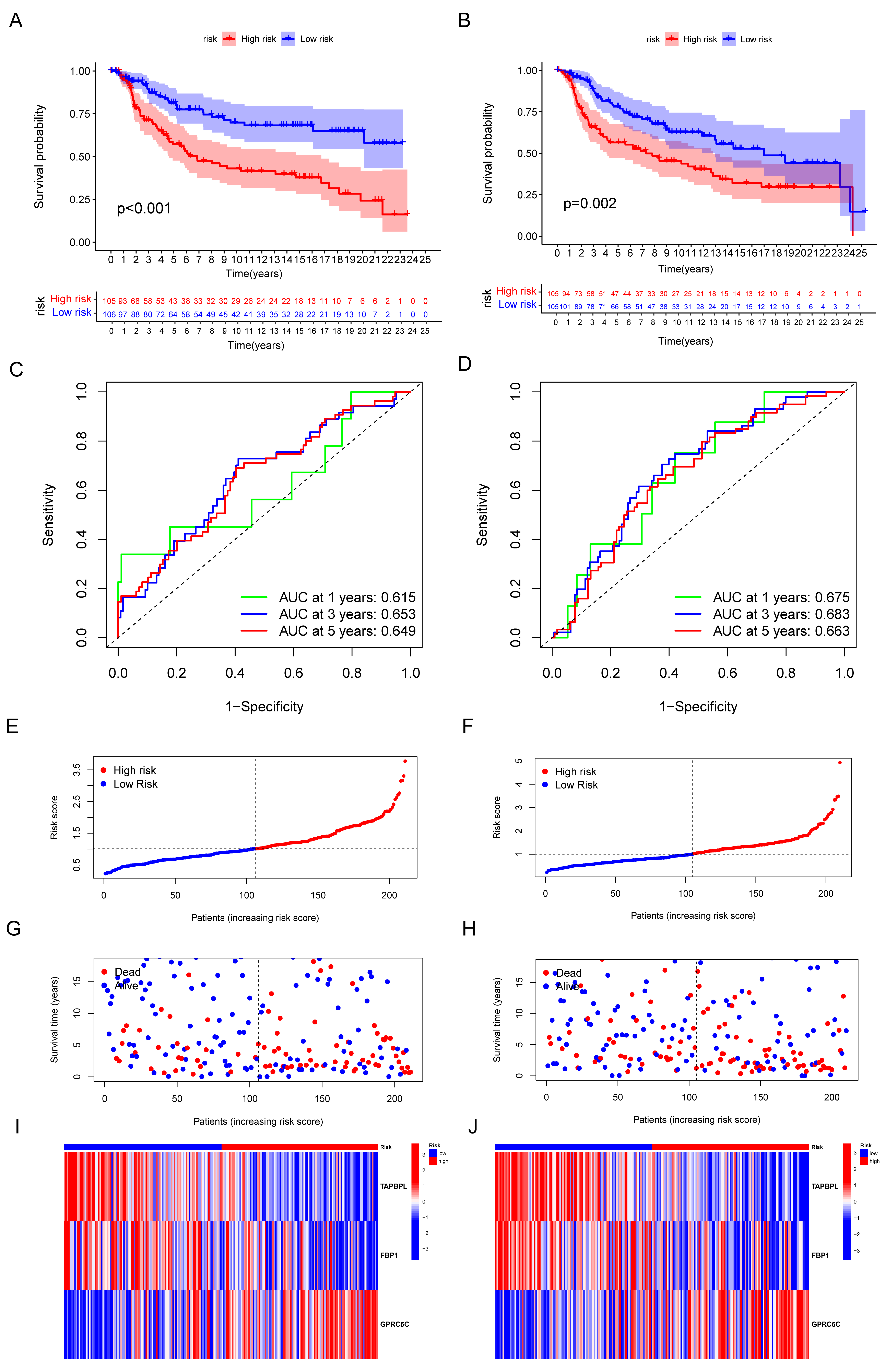
3.4. Validation of the Capacity of IRGP
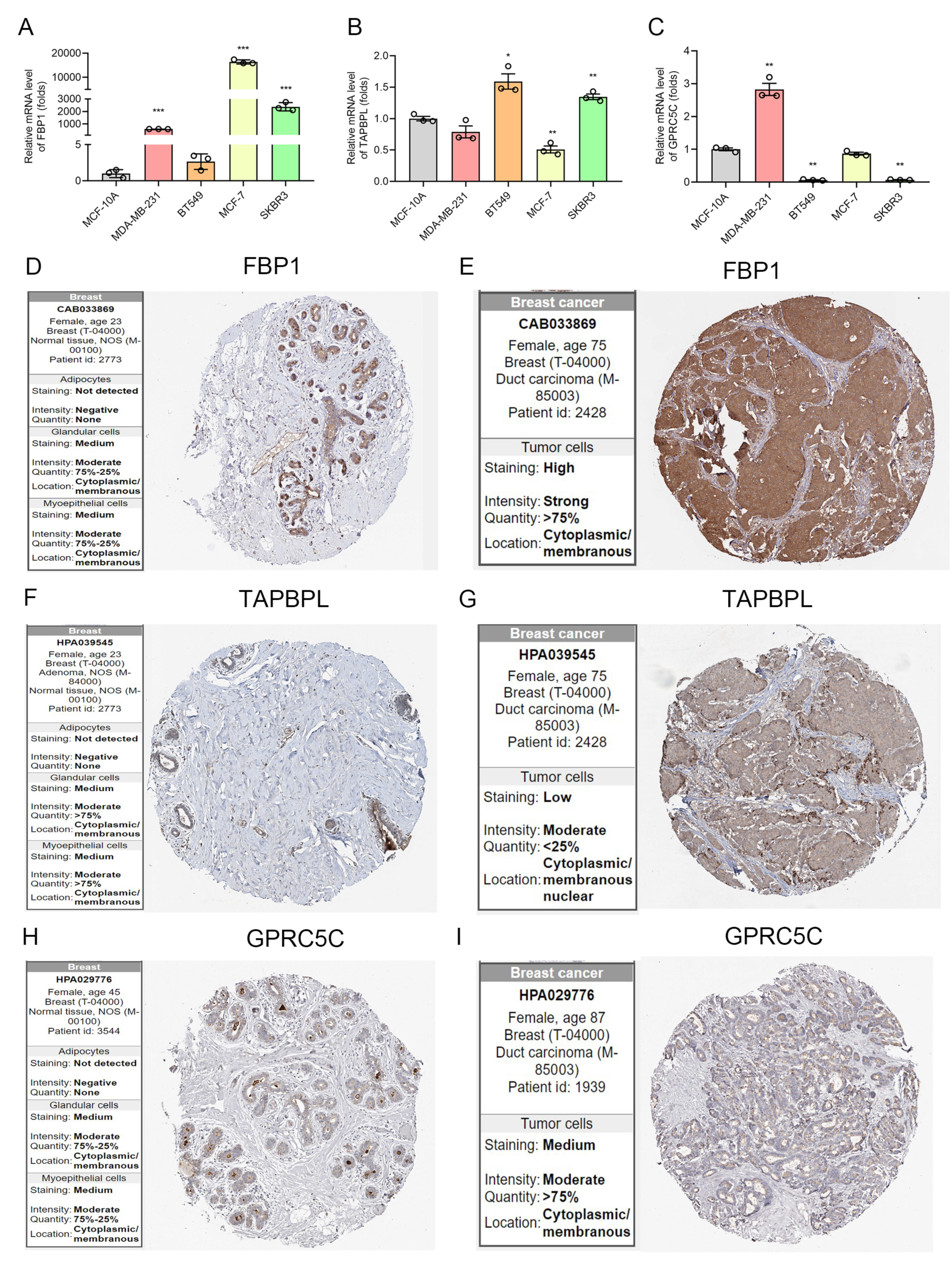
3.5. The mRNA and Protein Level of IRGP in BC
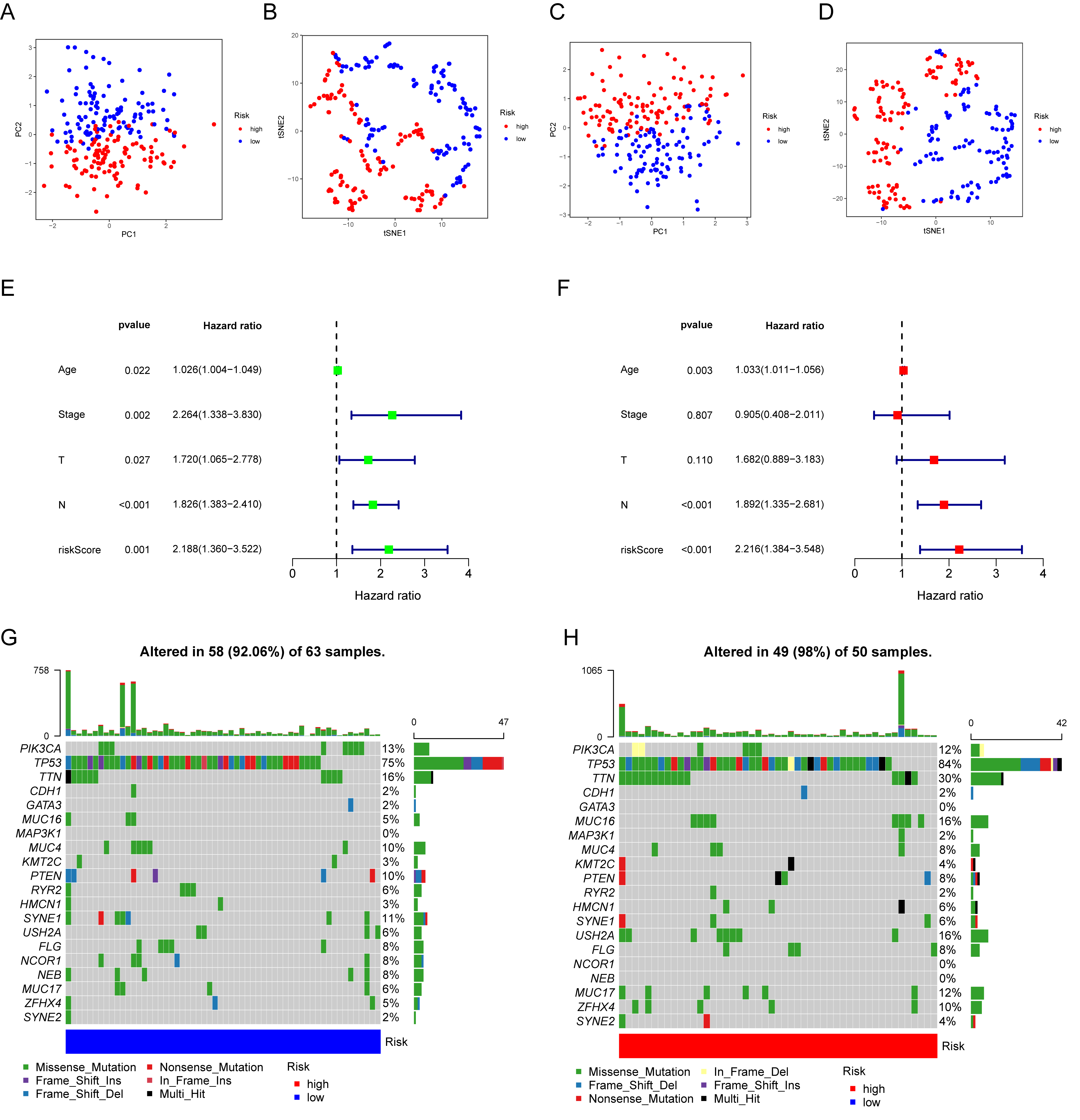
3.6. Prognostic Value and Genomic Features in Different IRGP Subgroups
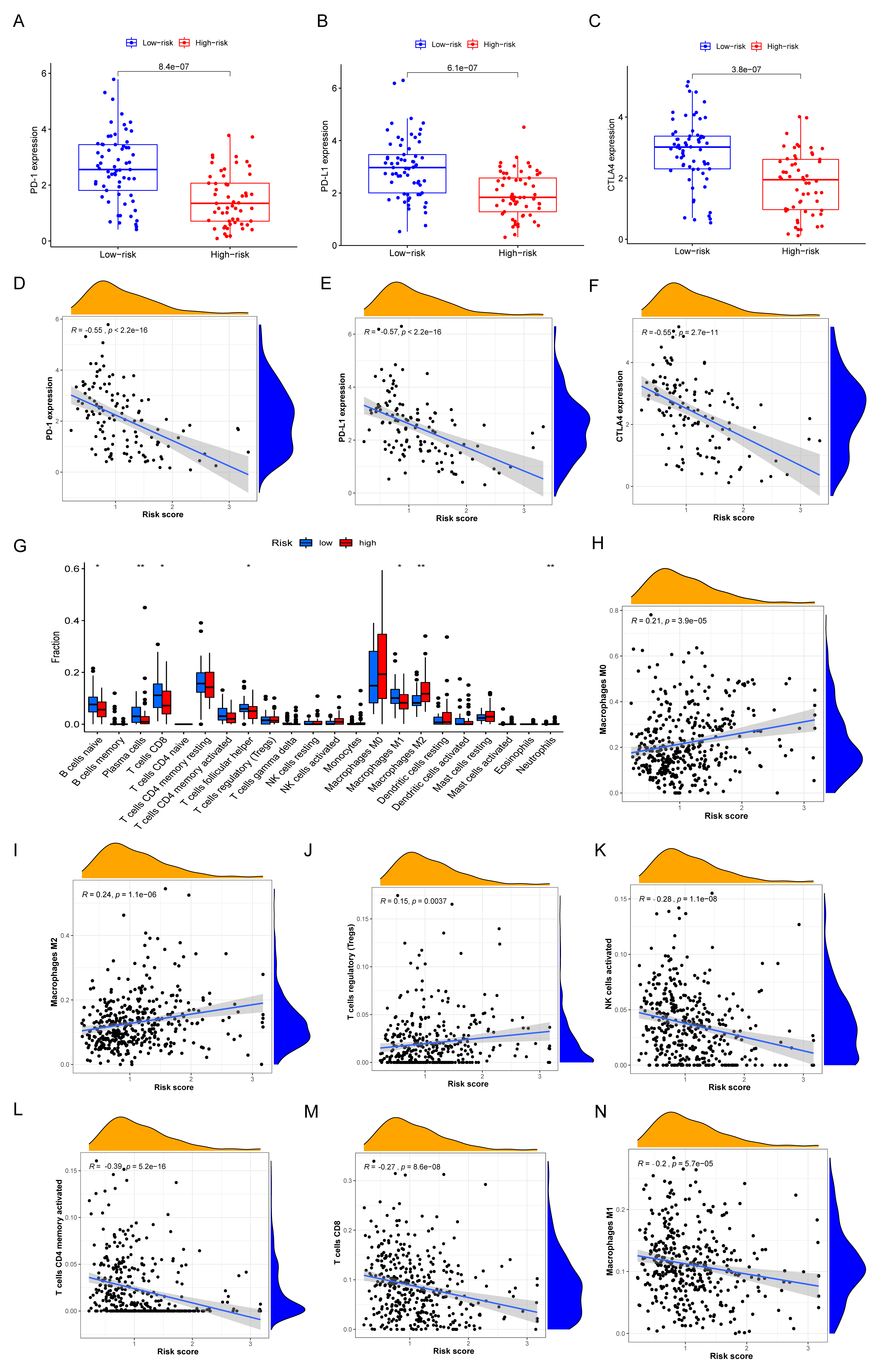
3.7. Immune Characteristics of Different IRGP Subgroups
3.8. Relationship of IRGP Subgroups with Immune Subtypes and IPS
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| APCs | Antigen presenting cells |
| BC | Breast cancer |
| BL1 | Basal-like 1 |
| BL2 | Basal-like 2 |
| CTLA-4 | Cytotoxic T-lymphocyte-associated antigen-4 |
| DEGs | Differentially expressed genes |
| FBP1 | Fructose-1,6-bisphosphatase |
| FDA | Food and Drug Administration |
| FDR | False discovery rate |
| GDPS | Genomics of Drug Sensitivity in Cancer |
| GO | Gene Ontology |
| GSVA | Gene Set Variation Analysis |
| GSEA | Gene set enrichment Analysis |
| ICIs | Immune checkpoint inhibitors |
| IM | Immunomodulatory |
| IPS | Immunophenoscore |
| IRGs | Immune-related genes |
| IRGP | Immune-related gene panel |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LAR | Luminal androgen receptor |
| LASSO | Least absolute shrinkage and selection operator |
| M | Mesenchymal |
| METABRIC | Molecular Taxonomy of Breast Cancer International Consortium |
| MHC | Major histocompatibility complex |
| MSL | Mesenchymal stem-like |
| OS | Overall survival |
| PARP | Poly (ADP-ribose) polymerase |
| PCA | Principal component analysis |
| PD-1 | Programmed cell death receptor 1 |
| PD-L1 | Programmed cell death 1 ligand 1 |
| ROC | Receiver operating characteristic |
| ssGSEA | Single-sample gene-set enrichment analysis |
| STAT3 | Signal transducer and activator of transcription 3 |
| TAPBPL | TAP binding protein like |
| TCGA | The Cancer Genome Atlas |
| TILs | Tumor infiltrating lymphocytes |
| TNBC | Triple-negative breast cancer |
| TOM | Topological overlap matrix |
| t-SNE | t-distributed stochastic neighbor embedding |
| WGCNA | Weighted gene co-expression network analysis |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.K. DNMT1: A key drug target in triple-negative breast cancer. Semin. Cancer Biol. 2021, 72, 198–213. [Google Scholar] [CrossRef]
- Yin, L.; Duan, J.J.; Bian, X.W.; Yu, S.C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. BCR 2020, 22, 61. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhu, X.; Tang, C.; Guan, X.; Zhang, W. Progress and challenges of immunotherapy in triple-negative breast cancer. Biochim. Biophys. Acta. Rev. Cancer 2021, 1876, 188593. [Google Scholar] [CrossRef] [PubMed]
- Jovanović, B.; Mayer, I.A.; Mayer, E.L.; Abramson, V.G.; Bardia, A.; Sanders, M.E.; Kuba, M.G.; Estrada, M.V.; Beeler, J.S.; Shaver, T.M.; et al. A Randomized Phase II Neoadjuvant Study of Cisplatin, Paclitaxel With or Without Everolimus in Patients with Stage II/III Triple-Negative Breast Cancer (TNBC): Responses and Long-term Outcome Correlated with Increased Frequency of DNA Damage Response Gene Mutations, TNBC Subtype, AR Status, and Ki67. Clin. Cancer Res. 2017, 23, 4035–4045. [Google Scholar] [PubMed]
- Emens, L.A. Breast Cancer Immunotherapy: Facts and Hopes. Clin. Cancer Res. 2018, 24, 511–520. [Google Scholar] [CrossRef]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef]
- Loi, S.; Michiels, S.; Adams, S.; Loibl, S.; Budczies, J.; Denkert, C.; Salgado, R. The journey of tumor-infiltrating lymphocytes as a biomarker in breast cancer: Clinical utility in an era of checkpoint inhibition. Ann. Oncol. 2021, 32, 1236–1244. [Google Scholar] [CrossRef]
- Emens, L.A.; Molinero, L.; Loi, S.; Rugo, H.S.; Schneeweiss, A.; Diéras, V.; Iwata, H.; Barrios, C.H.; Nechaeva, M.; Nguyen-Duc, A.; et al. Atezolizumab and nab-Paclitaxel in Advanced Triple-Negative Breast Cancer: Biomarker Evaluation of the IMpassion130 Study. J. Natl. Cancer Inst. 2021, 113, 1005–1016. [Google Scholar] [CrossRef]
- Cetin, B.; Gumusay, O. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, e108. [Google Scholar]
- Tarantino, P.; Antonarelli, G.; Ascione, L.; Curigliano, G. Investigational immunomodulatory drugs for enhancement of triple negative breast cancer (TNBC) immunotherapy: Early phase development. Expert Opin. Investig. Drugs 2022, 31, 499–513. [Google Scholar] [CrossRef]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Dunn, P.; Thomas, C.G.; Smith, B.; Schaefer, H.; Chen, J.; Hu, Z.; Zalocusky, K.A.; Shankar, R.D.; Shen-Orr, S.S.; et al. ImmPort, toward repurposing of open access immunological assay data for translational and clinical research. Sci. Data 2018, 5, 180015. [Google Scholar] [CrossRef]
- Breuer, K.; Foroushani, A.K.; Laird, M.R.; Chen, C.; Sribnaia, A.; Lo, R.; Winsor, G.L.; Hancock, R.E.; Brinkman, F.S.; Lynn, D.J. InnateDB: Systems biology of innate immunity and beyond—Recent updates and continuing curation. Nucleic Acids Res. 2013, 41, D1228–D1233. [Google Scholar] [CrossRef]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 1–25. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Zhang, J.; Han, X.; Lin, L.; Chen, J.; Wang, F.; Ding, Q.; Hao, L.; Wang, L.; Wei, J.; Wang, Y.; et al. Unraveling the Expression Patterns of Immune Checkpoints Identifies New Subtypes and Emerging Therapeutic Indicators in Lung Adenocarcinoma. Oxidative Med. Cell. Longev. 2022, 2022, 3583985. [Google Scholar] [CrossRef] [PubMed]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shan, B.; Lin, L.; Dong, J.; Sun, Q.; Zhou, Q.; Chen, J.; Han, X. Dissecting the Role of N6-Methylandenosine-Related Long Non-coding RNAs Signature in Prognosis and Immune Microenvironment of Breast Cancer. Front. Cell Dev. Biol. 2021, 9, 711859. [Google Scholar] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef]
- Geeleher, P.; Cox, N.; Huang, R.S. pRRophetic: An R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS ONE 2014, 9, e107468. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013, 41, D955–D961. [Google Scholar] [CrossRef]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e814. [Google Scholar] [CrossRef]
- Derakhshan, F.; Reis-Filho, J.S. Pathogenesis of Triple-Negative Breast Cancer. Annu. Rev. Pathol. 2022, 17, 181–204. [Google Scholar] [CrossRef]
- Gong, Y.; Ji, P.; Yang, Y.S.; Xie, S.; Yu, T.J.; Xiao, Y.; Jin, M.L.; Ma, D.; Guo, L.W.; Pei, Y.C.; et al. Metabolic-Pathway-Based Subtyping of Triple-Negative Breast Cancer Reveals Potential Therapeutic Targets. Cell Metab. 2021, 33, 51–64.e59. [Google Scholar] [CrossRef]
- Jiang, Y.Z.; Ma, D.; Suo, C.; Shi, J.; Xue, M.; Hu, X.; Xiao, Y.; Yu, K.D.; Liu, Y.R.; Yu, Y.; et al. Genomic and Transcriptomic Landscape of Triple-Negative Breast Cancers: Subtypes and Treatment Strategies. Cancer Cell 2019, 35, 428–440.e425. [Google Scholar] [CrossRef]
- Bianchini, G.; De Angelis, C.; Licata, L.; Gianni, L. Treatment landscape of triple-negative breast cancer—Expanded options, evolving needs. Nat. Rev. Clin. Oncol. 2022, 19, 91–113. [Google Scholar] [CrossRef]
- Zhang, J.; Pan, S.; Jian, C.; Hao, L.; Dong, J.; Sun, Q.; Jin, H.; Han, X. Immunostimulatory Properties of Chemotherapy in Breast Cancer: From Immunogenic Modulation Mechanisms to Clinical Practice. Front. Immunol. 2021, 12, 819405. [Google Scholar] [CrossRef]
- Gaikwad, S.; Agrawal, M.Y.; Kaushik, I.; Ramachandran, S.; Srivastava, S.K. Immune checkpoint proteins: Signaling mechanisms and molecular interactions in cancer immunotherapy. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2022. [Google Scholar]
- Yan, C.; Liu, Q.; Jia, R. Construction and Validation of a Prognostic Risk Model for Triple-Negative Breast Cancer Based on Autophagy-Related Genes. Front. Oncol. 2022, 12, 829045. [Google Scholar] [CrossRef]
- Yang, A.; Wu, M.; Ni, M.; Zhang, L.; Li, M.; Wei, P.; Yang, Y.; Xiao, W.; An, X. A risk scoring system based on tumor microenvironment cells to predict prognosis and immune activity in triple-negative breast cancer. Breast Cancer 2022, 29, 468–477. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, L.; Xu, X.; Li, X.; Guan, W.; Meng, T.; Xu, G. Transcriptome-Based Network Analysis Unveils Eight Immune-Related Genes as Molecular Signatures in the Immunomodulatory Subtype of Triple-Negative Breast Cancer. Front. Oncol. 2020, 10, 1787. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Pietenpol, J.A. Clinical implications of molecular heterogeneity in triple negative breast cancer. Breast 2015, 24 (Suppl. S2), S36–S40. [Google Scholar] [CrossRef]
- Jiang, Y.Z.; Liu, Y.; Xiao, Y.; Hu, X.; Jiang, L.; Zuo, W.J.; Ma, D.; Ding, J.; Zhu, X.; Zou, J.; et al. Molecular subtyping and genomic profiling expand precision medicine in refractory metastatic triple-negative breast cancer: The FUTURE trial. Cell Res. 2021, 31, 178–186. [Google Scholar] [CrossRef]
- Cao, K.; Liu, M.; Ma, K.; Jiang, X.; Ma, J.; Zhu, J. Prediction of prognosis and immunotherapy response with a robust immune-related lncRNA pair signature in lung adenocarcinoma. Cancer Immunol. Immunother. CII 2021, 71, 1295–1311. [Google Scholar] [CrossRef]
- Chen, Y.; Li, Z.Y.; Zhou, G.Q.; Sun, Y. An Immune-Related Gene Prognostic Index for Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2021, 27, 330–341. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Zhang, F.; Li, D.; Shi, X.; Xiong, Q.; Wei, Q.; Yang, L. Developing an immune-related gene prognostic index associated with progression and providing new insights into the tumor immune microenvironment of prostate cancer. Immunology 2022, 166, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Neerincx, A.; Boyle, L.H. Properties of the tapasin homologue TAPBPR. Curr. Opin. Immunol. 2017, 46, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Cui, C.; Su, M.; Silbart, L.K.; Liu, H.; Zhao, J.; He, L.; Huang, Y.; Xu, D.; Wei, X.; et al. Identification of TAPBPL as a novel negative regulator of T-cell function. EMBO Mol. Med. 2021, 13, e13404. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.Y.; Schwarz, H. CD137/CD137 ligand signalling regulates the immune balance: A potential target for novel immunotherapy of autoimmune diseases. J. Autoimmun. 2020, 112, 102499. [Google Scholar] [CrossRef]
- Yang, X.; Zhao, L.; Pei, J.; Wang, Z.; Zhang, J.; Wang, B. CELF6 modulates triple-negative breast cancer progression by regulating the stability of FBP1 mRNA. Breast Cancer Res. Treat. 2020, 183, 71–82. [Google Scholar] [CrossRef]
- Li, H.; Qi, Z.; Niu, Y.; Yang, Y.; Li, M.; Pang, Y.; Liu, M.; Cheng, X.; Xu, M.; Wang, Z. FBP1 regulates proliferation, metastasis, and chemoresistance by participating in C-MYC/STAT3 signaling axis in ovarian cancer. Oncogene 2021, 40, 5938–5949. [Google Scholar] [CrossRef]
- Zhao, W.; Yang, S.; Chen, J.; Zhao, J.; Dong, J. Forced overexpression of FBP1 inhibits proliferation and metastasis in cholangiocarcinoma cells via Wnt/β-catenin pathway. Life Sci. 2018, 210, 224–234. [Google Scholar] [CrossRef]
- Cong, J.; Wang, X.; Zheng, X.; Wang, D.; Fu, B.; Sun, R.; Tian, Z.; Wei, H. Dysfunction of Natural Killer Cells by FBP1-Induced Inhibition of Glycolysis during Lung Cancer Progression. Cell Metab. 2018, 28, 243–255.e245. [Google Scholar] [CrossRef]
- Liu, W.; Xiong, X.; Chen, W.; Li, X.; Hua, X.; Liu, Z.; Zhang, Z. High expression of FUSE binding protein 1 in breast cancer stimulates cell proliferation and diminishes drug sensitivity. Int. J. Oncol. 2020, 57, 488–499. [Google Scholar] [CrossRef]
- Robbins, M.J.; Michalovich, D.; Hill, J.; Calver, A.R.; Medhurst, A.D.; Gloger, I.; Sims, M.; Middlemiss, D.N.; Pangalos, M.N. Molecular cloning and characterization of two novel retinoic acid-inducible orphan G-protein-coupled receptors (GPRC5B and GPRC5C). Genomics 2000, 67, 8–18. [Google Scholar] [CrossRef]
- Yamaga, R.; Ikeda, K.; Boele, J.; Horie-Inoue, K.; Takayama, K.; Urano, T.; Kaida, K.; Carninci, P.; Kawai, J.; Hayashizaki, Y.; et al. Systemic identification of estrogen-regulated genes in breast cancer cells through cap analysis of gene expression mapping. Biochem. Biophys. Res. Commun. 2014, 447, 531–536. [Google Scholar] [CrossRef]
- Rajkumar, P.; Cha, B.; Yin, J.; Arend, L.J.; Păunescu, T.G.; Hirabayashi, Y.; Donowitz, M.; Pluznick, J.L. Identifying the localization and exploring a functional role for Gprc5c in the kidney. FASEB J. 2018, 32, 2046–2059. [Google Scholar] [CrossRef]
- Amisten, S.; Mohammad Al-Amily, I.; Soni, A.; Hawkes, R.; Atanes, P.; Persaud, S.J.; Rorsman, P.; Salehi, A. Anti-diabetic action of all-trans retinoic acid and the orphan G protein coupled receptor GPRC5C in pancreatic β-cells. Endocr. J. 2017, 64, 325–338. [Google Scholar] [CrossRef]
- Kumar, A.; Swain, C.A.; Shevde, L.A. Informing the new developments and future of cancer immunotherapy: Future of cancer immunotherapy. Cancer Metastasis Rev. 2021, 40, 549–562. [Google Scholar] [CrossRef]
- Kirtane, K.; Elmariah, H.; Chung, C.H.; Abate-Daga, D. Adoptive cellular therapy in solid tumor malignancies: Review of the literature and challenges ahead. J. Immunother. Cancer 2021, 9, e002723. [Google Scholar] [CrossRef]
- Park, J.; Kwon, M.; Kim, K.H.; Kim, T.S.; Hong, S.H.; Kim, C.G.; Kang, S.G.; Moon, J.H.; Kim, E.H.; Park, S.H.; et al. Immune Checkpoint Inhibitor-induced Reinvigoration of Tumor-infiltrating CD8+ T Cells is Determined by Their Differentiation Status in Glioblastoma. Clin. Cancer Res. 2019, 25, 2549–2559. [Google Scholar] [CrossRef]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): A randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020, 396, 1817–1828. [Google Scholar] [CrossRef]
- Emens, L.A.; Adams, S.; Barrios, C.H.; Diéras, V.; Iwata, H.; Loi, S.; Rugo, H.S.; Schneeweiss, A.; Winer, E.P.; Patel, S.; et al. First-line atezolizumab plus nab-paclitaxel for unresectable, locally advanced, or metastatic triple-negative breast cancer: IMpassion130 final overall survival analysis. Ann. Oncol. 2021, 32, 983–993. [Google Scholar] [CrossRef]
- Pan, Y.H.; Zhang, J.X.; Chen, X.; Liu, F.; Cao, J.Z.; Chen, Y.; Chen, W.; Luo, J.H. Predictive Value of the TP53/PIK3CA/ATM Mutation Classifier for Patients with Bladder Cancer Responding to Immune Checkpoint Inhibitor Therapy. Front. Immunol. 2021, 12, 643282. [Google Scholar] [CrossRef]
- Zhang, Y.; Lin, A.; Li, Y.; Ding, W.; Meng, H.; Luo, P.; Zhang, J. Age and Mutations as Predictors of the Response to Immunotherapy in Head and Neck Squamous Cell Cancer. Front. Cell Dev. Biol. 2020, 8, 608969. [Google Scholar] [CrossRef] [PubMed]
- Lyu, H.; Li, M.; Jiang, Z.; Liu, Z.; Wang, X. Correlate the TP53 Mutation and the HRAS Mutation with Immune Signatures in Head and Neck Squamous Cell Cancer. Comput. Struct. Biotechnol. J. 2019, 17, 1020–1030. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Pan, S.; Han, C.; Jin, H.; Sun, Q.; Du, J.; Han, X. Combination of Immune-Related Network and Molecular Typing Analysis Defines a Three-Gene Signature for Predicting Prognosis of Triple-Negative Breast Cancer. Biomolecules 2022, 12, 1556. https://doi.org/10.3390/biom12111556
Zhang J, Pan S, Han C, Jin H, Sun Q, Du J, Han X. Combination of Immune-Related Network and Molecular Typing Analysis Defines a Three-Gene Signature for Predicting Prognosis of Triple-Negative Breast Cancer. Biomolecules. 2022; 12(11):1556. https://doi.org/10.3390/biom12111556
Chicago/Turabian StyleZhang, Jinguo, Shuaikang Pan, Chaoqiang Han, Hongwei Jin, Qingqing Sun, Jun Du, and Xinghua Han. 2022. "Combination of Immune-Related Network and Molecular Typing Analysis Defines a Three-Gene Signature for Predicting Prognosis of Triple-Negative Breast Cancer" Biomolecules 12, no. 11: 1556. https://doi.org/10.3390/biom12111556
APA StyleZhang, J., Pan, S., Han, C., Jin, H., Sun, Q., Du, J., & Han, X. (2022). Combination of Immune-Related Network and Molecular Typing Analysis Defines a Three-Gene Signature for Predicting Prognosis of Triple-Negative Breast Cancer. Biomolecules, 12(11), 1556. https://doi.org/10.3390/biom12111556