
Structure and Dynamics of Human Chemokine CCL16—Implications for Biological Activity
 , , , ,
, , , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. RNA Isolation and Real-Time PCR
2.4. Immunohistochemistry
2.5. Cytokine Analysis
2.6. Heparin Affinity Chromatography
2.7. Immunoblotting
2.8. Statistical Analysis
2.9. Protein Expression and Purification
2.10. Crystallisation and Diffraction Data Collection
2.11. Structure Determination
2.12. Mass Spectrometry (MS)
2.13. Molecular Dynamics (MD) Simulations
2.14. Figure Preparation
3. Results and Discussion
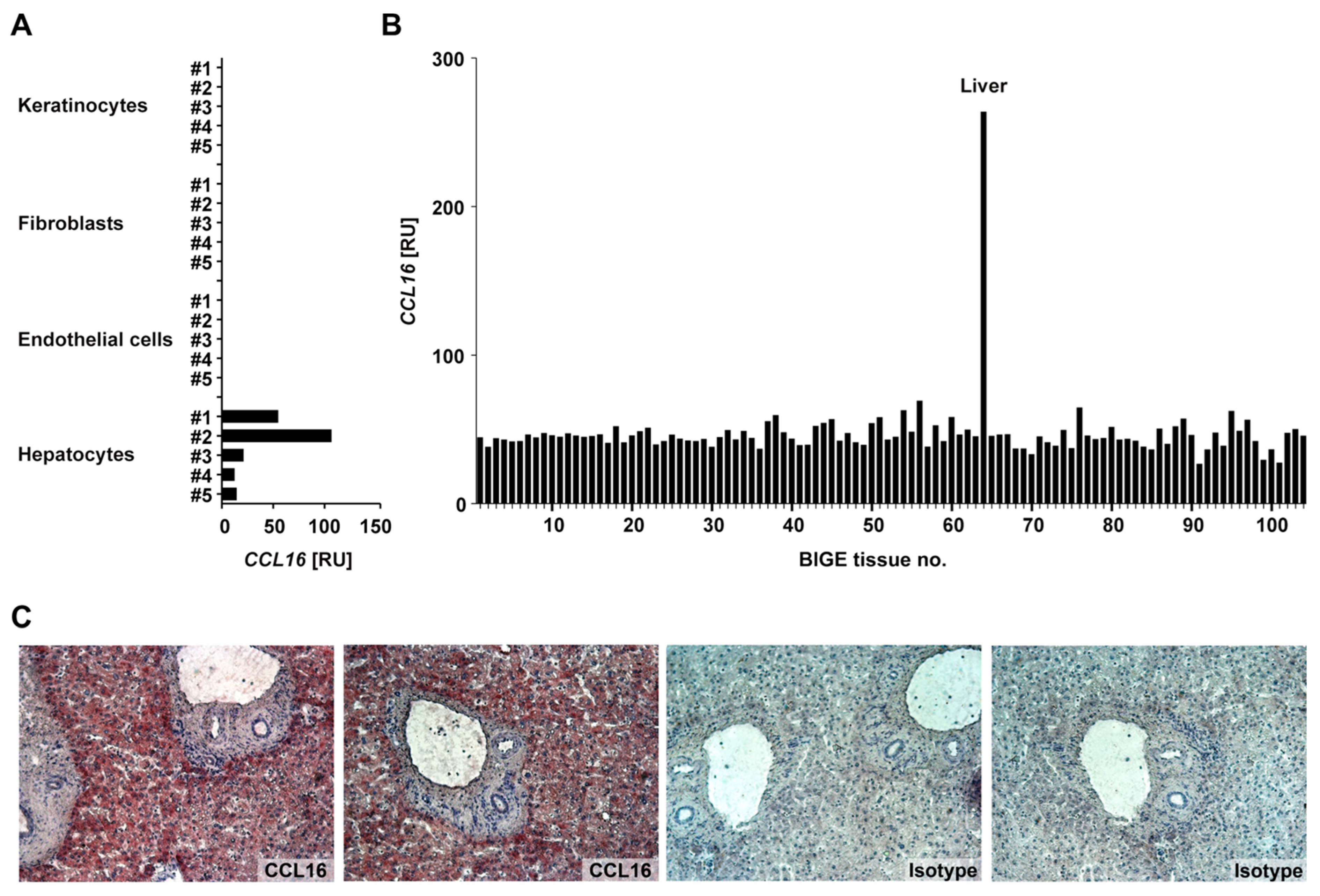
3.1. Characteristics of CCL16 Expression
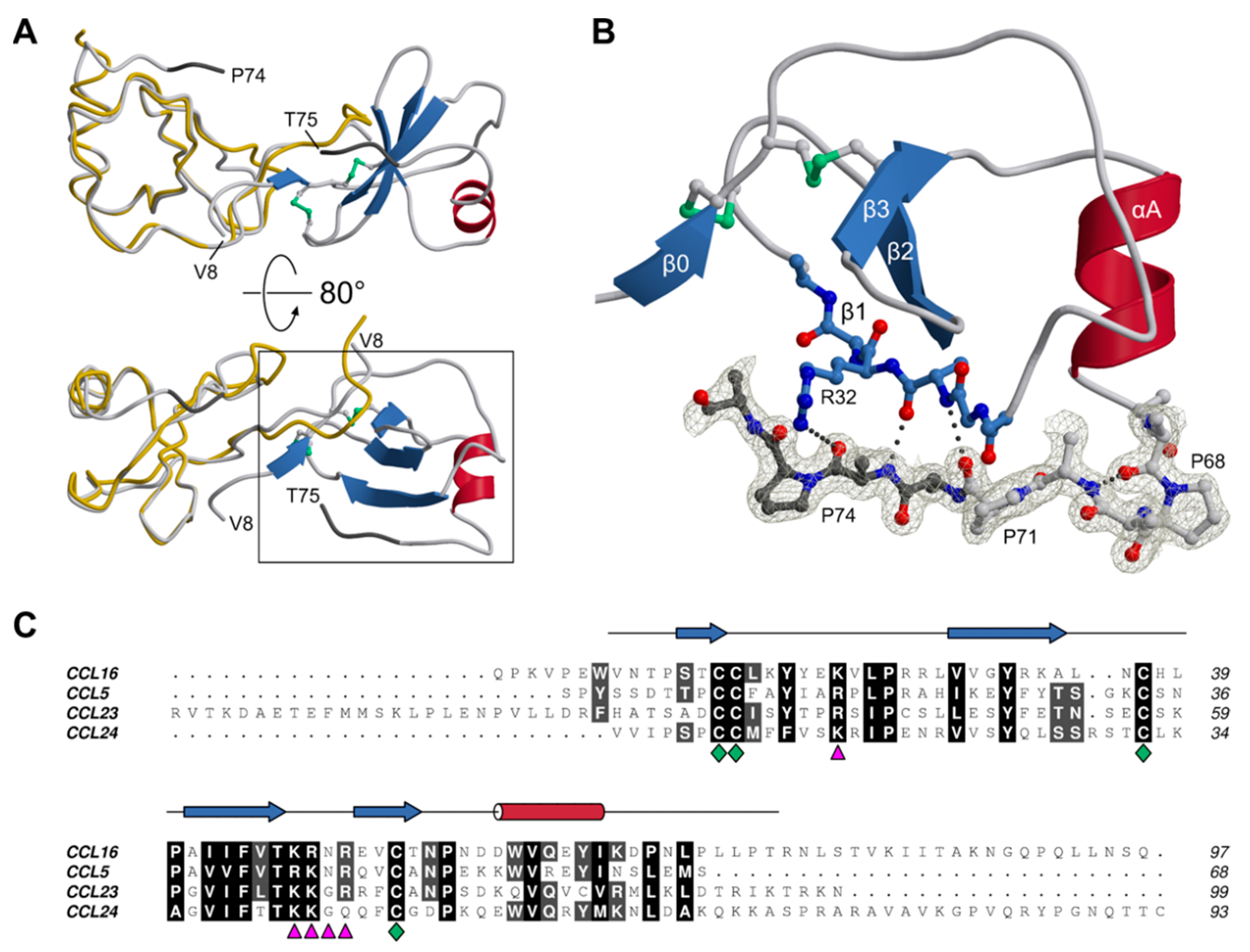
3.2. Structure of CCL16
3.3. C-Terminal Dynamics
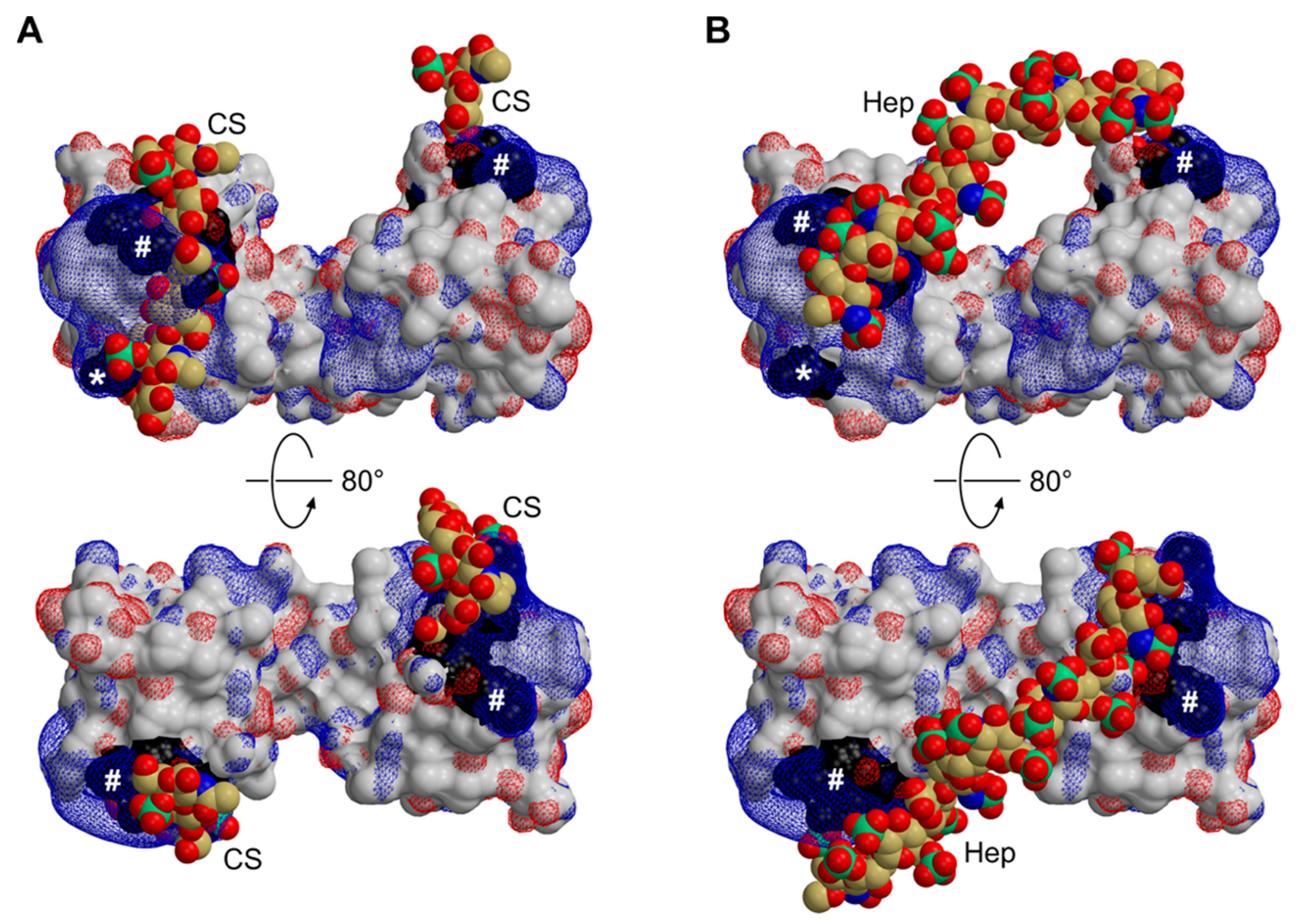
3.4. Functional Implications
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Allen, S.J.; Crown, S.E.; Handel, T.M. Chemokine: Receptor structure, interactions, and antagonism. Annu. Rev. Immunol. 2007, 25, 787–820. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Kufareva, I.; Holden, L.G.; Wang, C.; Zheng, Y.; Zhao, C.; Fenalti, G.; Wu, H.; Han, G.W.; Cherezov, V.; et al. Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science 2015, 347, 1117–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proudfoot, A.E.; Handel, T.M.; Johnson, Z.; Lau, E.K.; LiWang, P.; Clark-Lewis, I.; Borlat, F.; Wells, T.N.; Kosco-Vilbois, M.H. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc. Natl. Acad. Sci. USA 2003, 100, 1885–1890. [Google Scholar] [CrossRef] [Green Version]
- Handel, T.M.; Johnson, Z.; Crown, S.E.; Lau, E.K.; Proudfoot, A.E. Regulation of protein function by glycosaminoglycans-As exemplified by chemokines. Annu. Rev. Biochem. 2005, 74, 385–410. [Google Scholar] [CrossRef]
- Dyer, D.P.; Salanga, C.L.; Volkman, B.F.; Kawamura, T.; Handel, T.M. The dependence of chemokine-glycosaminoglycan interactions on chemokine oligomerization. Glycobiology 2016, 26, 312–326. [Google Scholar] [CrossRef] [Green Version]
- Mortier, A.; Gouwy, M.; Van Damme, J.; Proost, P. Effect of posttranslational processing on the in vitro and in vivo activity of chemokines. Exp. Cell Res. 2011, 317, 642–654. [Google Scholar] [CrossRef] [PubMed]
- Shoudai, K.; Hieshima, K.; Fukuda, S.; Iio, M.; Miura, R.; Imai, T.; Yoshie, O.; Nomiyama, H. Isolation of cDNA encoding a novel human CC chemokine NCC-4/LEC. Biochim. Biophys. Acta 1998, 1396, 273–277. [Google Scholar] [CrossRef]
- Hedrick, J.A.; Helms, A.; Vicari, A.; Zlotnik, A. Characterization of a novel CC chemokine, HCC-4, whose expression is increased by interleukin-10. Blood 1998, 91, 4242–4247. [Google Scholar] [CrossRef]
- Youn, B.S.; Zhang, S.; Broxmeyer, H.E.; Antol, K.; Fraser, M.J., Jr.; Hangoc, G.; Kwon, B.S. Isolation and characterization of LMC, a novel lymphocyte and monocyte chemoattractant human CC chemokine, with myelosuppressive activity. Biochem. Biophys. Res. Commun. 1998, 247, 217–222. [Google Scholar] [CrossRef]
- Starr, A.E.; Dufour, A.; Maier, J.; Overall, C.M. Biochemical analysis of matrix metalloproteinase activation of chemokines CCL15 and CCL23 and increased glycosaminoglycan binding of CCL16. J. Biol. Chem. 2012, 287, 5848–5860. [Google Scholar] [CrossRef]
- Pannellini, T.; Iezzi, M.; Di Carlo, E.; Eleuterio, E.; Coletti, A.; Modesti, A.; Rosini, S.; Neri, M.; Musiani, P. The expression of LEC/CCL16, a powerful inflammatory chemokine, is upregulated in ulcerative colitis. Int. J. Immunopathol. Pharmacol. 2004, 17, 171–180. [Google Scholar] [CrossRef]
- Del Valle-Pinero, A.Y.; Martino, A.C.; Taylor, T.J.; Majors, B.L.; Patel, N.S.; Heitkemper, M.M.; Henderson, W.A. Pro-inflammatory chemokine C-C motif ligand 16 (CCL-16) dysregulation in irritable bowel syndrome (IBS): A pilot study. Neurogastroenterol. Motil. 2011, 23, 1092–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nureki, S.; Miyazaki, E.; Usagawa, Y.; Ueno, T.; Ando, M.; Takenaka, R.; Ito, T.; Ishii, T.; Kumamoto, T. Elevated concentrations of liver-Expressed chemokine/CC chemokine ligand 16 in bronchoalveolar lavage fluid from patients with eosinophilic pneumonia. Int. Arch. Allergy Immunol. 2009, 150, 282–290. [Google Scholar] [CrossRef]
- Mäkikallio, K.; Kaukola, T.; Tuimala, J.; Kingsmore, S.F.; Hallman, M.; Ojaniemi, M. Umbilical artery chemokine CCL16 is associated with preterm preeclampsia and fetal growth restriction. Cytokine 2012, 60, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Wu, N.; Xiang, Y.; Wu, L.; Li, C.; Yuan, Z.; Jia, X.; Zhang, Z.; Zhong, L.; Li, Y. Prognostic value of chemokines in patients with newly diagnosed atrial fibrillation. Int. J. Cardiol. 2020, 320, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Tavener, S.K.; Jewell, D.E.; Panickar, K.S. The increase in circulating levels of pro-inflammatory chemokines, cytokines, and complement C5 in canines with impaired kidney function. Curr. Issues Mol. Biol. 2022, 44, 1664–1676. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Hever, A.; Willhite, D.; Zlotnik, A.; Hevezi, P. Effects of RNA degradation on gene expression analysis of human postmortem tissues. FASEB J. 2005, 19, 1356–1358. [Google Scholar] [CrossRef]
- Gerber, P.A.; Hevezi, P.; Buhren, B.A.; Martinez, C.; Schrumpf, H.; Gasis, M.; Grether-Beck, S.; Krutmann, J.; Homey, B.; Zlotnik, A. Systematic identification and characterization of novel human skin-Associated genes encoding membrane and secreted proteins. PLoS ONE 2013, 8, e63949. [Google Scholar] [CrossRef]
- Homey, B.; Dieu-Nosjean, M.C.; Wiesenborn, A.; Massacrier, C.; Pin, J.J.; Oldham, E.; Catron, D.; Buchanan, M.E.; Müller, A.; de Waal, M.R.; et al. Up-Regulation of macrophage inflammatory protein-3 alpha/CCL20 and CC chemokine receptor 6 in psoriasis. J. Immunol. 2000, 164, 6621–6632. [Google Scholar] [CrossRef] [Green Version]
- Pivarcsi, A.; Müller, A.; Hippe, A.; Rieker, J.; van Lierop, A.; Steinhoff, M.; Seeliger, S.; Kubitza, R.; Pippirs, U.; Meller, S.; et al. Tumor immune escape by the loss of homeostatic chemokine expression. Proc. Natl. Acad. Sci. USA 2007, 104, 19055–19060. [Google Scholar] [CrossRef]
- Albrecht, U.; Yang, X.; Asselta, R.; Keitel, V.; Tenchini, M.L.; Ludwig, S.; Heinrich, P.C.; Häussinger, D.; Schaper, F.; Bode, J.G. Activation of NF-Kappab by IL-1beta blocks IL-6-Induced sustained STAT3 activation and STAT3-Dependent gene expression of the human gamma-Fibrinogen gene. Cell Signal 2007, 19, 1866–1878. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Vagin, A.; Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Blain, K.Y.; Kwiatkowski, W.; Zhao, Q.; La Fleur, D.; Naik, C.; Chun, T.W.; Tsareva, T.; Kanakaraj, P.; Laird, M.W.; Shah, R.; et al. Structural and functional characterization of CC chemokine CCL14. Biochemistry 2007, 46, 10008–10015. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-Atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Meixner, M. Optimization of Protein Separation by Size Sieving Capillary Electrophoresis. Master’s Thesis, Eberhard Karls Universität Tübingen, Tübingen, Germany, 2015. [Google Scholar]
- Frohnhöfer, H.G.; Geiger-Rudolph, S.; Pattky, M.; Meixner, M.; Huhn, C.; Maischein, H.M.; Geisler, R.; Gehring, I.; Maderspacher, F.; Nüsslein-Volhard, C.; et al. Spermidine, but not spermine, is essential for pigment pattern formation in zebrafish. Biol. Open 2016, 5, 736–744. [Google Scholar] [CrossRef] [Green Version]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Hao, G.F.; Xu, W.F.; Yang, S.G.; Yang, G.F. Multiple simulated annealing-Molecular dynamics (MSA-MD) for conformational space search of peptide and miniprotein. Sci. Rep. 2015, 5, 15568. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar] [CrossRef]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [Green Version]
- Bonomi, M.; Branduardi, D.; Bussi, G.; Camilloni, C.; Provasi, D.; Raiteri, P.; Donadio, D.; Marinelli, F.; Pietrucci, F.; Broglia, R.A.; et al. PLUMED: A portable plugin for free-energy calculations with molecular dynamics. Comput. Phys. Commun. 2009, 180, 1961–1972. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G. Hamiltonian replica exchange in GROMACS: A flexible implementation. Mol. Phys. 2014, 112, 379–384. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182. [Google Scholar] [CrossRef]
- Feenstra, K.A.; Hess, B.; Berendsen, H.J.C. Improving efficiency of large time-scale molecular dynamics simulations of hydrogen-rich systems. J. Comput. Chem. 1999, 20, 786–798. [Google Scholar] [CrossRef]
- Hess, B. P-LINCS: A parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Fenn, T.D.; Ringe, D.; Petsko, G.A. POVScript+: A program for model and data visualization using persistence of vision ray-tracing. J. Appl. Cryst. 2003, 36, 944–947. [Google Scholar] [CrossRef] [Green Version]
- Merritt, E.A.; Bacon, D.J. Raster3D: Photorealistic molecular graphics. Methods Enzymol. 1997, 277, 505–524. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Sanner, M.F.; Olson, A.J.; Spehner, J.C. Reduced surface: An efficient way to compute molecular surfaces. Biopolymers 1996, 38, 305–320. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, J.; Grishin, N.V. PROMALS3D: Multiple protein sequence alignment enhanced with evolutionary and three-dimensional structural information. Methods Mol. Biol. 2014, 1079, 263–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theobald, D.L.; Steindel, P.A. Optimal simultaneous superpositioning of multiple structures with missing data. Bioinformatics 2012, 28, 1972–1979. [Google Scholar] [CrossRef] [Green Version]
- Kleywegt, G.J. Use of non-crystallographic symmetry in protein structure refinement. Acta Crystallogr. D Biol. Crystallogr. 1996, 52, 842–857. [Google Scholar] [CrossRef]
- Roth, R.B.; Hevezi, P.; Lee, J.; Willhite, D.; Lechner, S.M.; Foster, A.C.; Zlotnik, A. Gene expression analyses reveal molecular relationships among 20 regions of the human CNS. Neurogenetics 2006, 7, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Nomiyama, H.; Hieshima, K.; Nakayama, T.; Sakaguchi, T.; Fujisawa, R.; Tanase, S.; Nishiura, H.; Matsuno, K.; Takamori, H.; Tabira, Y.; et al. Human CC chemokine liver-expressed chemokine/CCL16 is a functional ligand for CCR1, CCR2 and CCR5, and constitutively expressed by hepatocytes. Int. Immunol. 2001, 13, 1021–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, W.G.; Triandafillou, C.G.; Huang, T.Y.; Zulueta, M.M.; Banerjee, S.; Dinner, A.R.; Hung, S.C.; Tang, W.J. Structural basis for oligomerization and glycosaminoglycan binding of CCL5 and CCL3. Proc. Natl. Acad. Sci. USA 2016, 113, 5000–5005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clore, G.M.; Gronenborn, A.M. Three-Dimensional structures of alpha and beta chemokines. FASEB J. 1995, 9, 57–62. [Google Scholar] [CrossRef]
- Shaw, J.P.; Johnson, Z.; Borlat, F.; Zwahlen, C.; Kungl, A.; Roulin, K.; Harrenga, A.; Wells, T.N.; Proudfoot, A.E. The X-ray structure of RANTES: Heparin-Derived disaccharides allows the rational design of chemokine inhibitors. Structure 2004, 12, 2081–2093. [Google Scholar] [CrossRef] [Green Version]
- Cardin, A.D.; Weintraub, H.J. Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis 1989, 9, 21–32. [Google Scholar] [CrossRef]
- Graham, G.J.; Wilkinson, P.C.; Nibbs, R.J.; Lowe, S.; Kolset, S.O.; Parker, A.; Freshney, M.G.; Tsang, M.L.; Pragnell, I.B. Uncoupling of stem cell inhibition from monocyte chemoattraction in MIP-1alpha by mutagenesis of the proteoglycan binding site. EMBO J. 1996, 15, 6506–6515. [Google Scholar] [CrossRef] [PubMed]
- Laurence, J.S.; Blanpain, C.; De Leener, A.; Parmentier, M.; LiWang, P.J. Importance of basic residues and quaternary structure in the function of MIP-1 beta: CCR5 binding and cell surface sugar interactions. Biochemistry 2001, 40, 4990–4999. [Google Scholar] [CrossRef]
- Proudfoot, A.E.; Fritchley, S.; Borlat, F.; Shaw, J.P.; Vilbois, F.; Zwahlen, C.; Trkola, A.; Marchant, D.; Clapham, P.R.; Wells, T.N. The BBXB motif of RANTES is the principal site for heparin binding and controls receptor selectivity. J. Biol. Chem. 2001, 276, 10620–10626. [Google Scholar] [CrossRef] [Green Version]
- Deshauer, C.; Morgan, A.M.; Ryan, E.O.; Handel, T.M.; Prestegard, J.H.; Wang, X. Interactions of the chemokine CCL5/RANTES with medium-Sized chondroitin sulfate ligands. Structure 2015, 23, 1066–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Kett, W.C.; Severin, I.C.; Agyekum, I.; Duan, J.; Amster, I.J.; Proudfoot, A.E.I.; Coombe, D.R.; Woods, R.J. The interaction of heparin tetrasaccharides with chemokine CCL5 is modulated by sulfation pattern and pH. J. Biol. Chem. 2015, 290, 15421–15436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Watson, C.; Sharp, J.S.; Handel, T.M.; Prestegard, J.H. Oligomeric structure of the chemokine CCL5/RANTES from NMR, MS, and SAXS data. Structure 2011, 19, 1138–1148. [Google Scholar] [CrossRef] [Green Version]
- Joseph, P.R.; Mosier, P.D.; Desai, U.R.; Rajarathnam, K. Solution NMR characterization of chemokine CXCL8/IL-8 monomer and dimer binding to glycosaminoglycans: Structural plasticity mediates differential binding interactions. Biochem. J. 2015, 472, 121–133. [Google Scholar] [CrossRef] [Green Version]
- Zhuo, J.Y.; Lu, D.; Lin, Z.Y.; Cen, B.N.; Wei, X.Y.; Xie, H.Y.; Zheng, S.S.; Xu, X. CC motif chemokine ligand 16 inhibits the progression of liver cirrhosis via inactivating hepatic stellate cells. Hepatobiliary Pancreat. Dis. Int. 2020, 19, 440–448. [Google Scholar] [CrossRef]
- She, S.; Ren, L.; Chen, P.; Wang, M.; Chen, D.; Wang, Y.; Chen, H. Functional roles of chemokine receptor CCR2 and its ligands in liver disease. Front. Immunol. 2022, 13, 812431. [Google Scholar] [CrossRef]
- Suhre, K.; Sarwath, H.; Engelke, R.; Sohail, M.U.; Cho, S.J.; Whalen, W.; Alvarez-Mulett, S.; Krumsiek, J.; Choi, A.M.K.; Schmidt, F. Identification of robust protein associations with COVID-19 disease based on five clinical studies. Front. Immunol. 2022, 12, 781100. [Google Scholar] [CrossRef]
- Kaltschmidt, B.; Fitzek, A.D.E.; Schaedler, J.; Förster, C.; Kaltschmidt, C.; Hansen, T.; Steinfurth, F.; Windmöller, B.A.; Pilger, C.; Kong, C.; et al. Hepatic vasculopathy and regenerative responses of the liver in fatal cases of COVID-19. Clin. Gastroenterol. Hepatol. 2021, 19, 1726–1729. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Human CCL16 (PDB-ID 5LTL) | |
| Data Collection | |
| Space group | C 1 2 1 |
| Cell dimensions | |
| a, b, c {Å} | 136.35, 24.59, 37.97 |
| α, β, γ {°} | 90.00, 93.12, 90.00 |
| Resolution range | 37.91–1.45 (1.49–1.45) |
| Wilson B factor {Å2} | 20.4 |
| CC1/2 | 99.8 (88.3) |
| Rmeas {%} | 6.3 (59.1) |
| I/σI | 12.6 (2.3) |
| Completeness {%} | 99.5 (99.6) |
| Multiplicity | 5.0 (5.1) |
| Refinement | |
| Resolution range | 37.91–1.45 |
| No. reflections | 22,637 |
| Rwork/Rfree | 0.171/0.198 |
| Mean B {Å2} (no. atoms) | |
| Protein | 29.3 (1140) |
| Ions/ligands | 55.1 (7) |
| Water | 39.1 (91) |
| R.m.s. deviation | |
| Bond lengths {Å} | 0.006 |
| Bond angles {°} | 1.049 |
| Ramachandran plot {% res.} | |
| Favoured | 97.7 |
| Allowed | 2.3 |
| Outliers | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weiergräber, O.H.; Petrović, D.; Kislat, A.; Pattky, M.; Fabig, J.; Batra-Safferling, R.; Schulte am Esch, J.; Hänel, K.; Huhn, C.; Strodel, B.; et al. Structure and Dynamics of Human Chemokine CCL16—Implications for Biological Activity. Biomolecules 2022, 12, 1588. https://doi.org/10.3390/biom12111588
Weiergräber OH, Petrović D, Kislat A, Pattky M, Fabig J, Batra-Safferling R, Schulte am Esch J, Hänel K, Huhn C, Strodel B, et al. Structure and Dynamics of Human Chemokine CCL16—Implications for Biological Activity. Biomolecules. 2022; 12(11):1588. https://doi.org/10.3390/biom12111588
Chicago/Turabian StyleWeiergräber, Oliver H., Dušan Petrović, Andreas Kislat, Martin Pattky, Judith Fabig, Renu Batra-Safferling, Jan Schulte am Esch, Karen Hänel, Carolin Huhn, Birgit Strodel, and et al. 2022. "Structure and Dynamics of Human Chemokine CCL16—Implications for Biological Activity" Biomolecules 12, no. 11: 1588. https://doi.org/10.3390/biom12111588