
Differential Gene Expression and DNA Methylation in the Risk of Depression in LOAD Patients



Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Cohort
2.2. Differential Expression
2.3. Differential DNA-Methylation
3. Results
3.1. Differential Expressed Genes Are Associated with Depression Symptoms in LOAD Patients in a Sex-Specific Manner
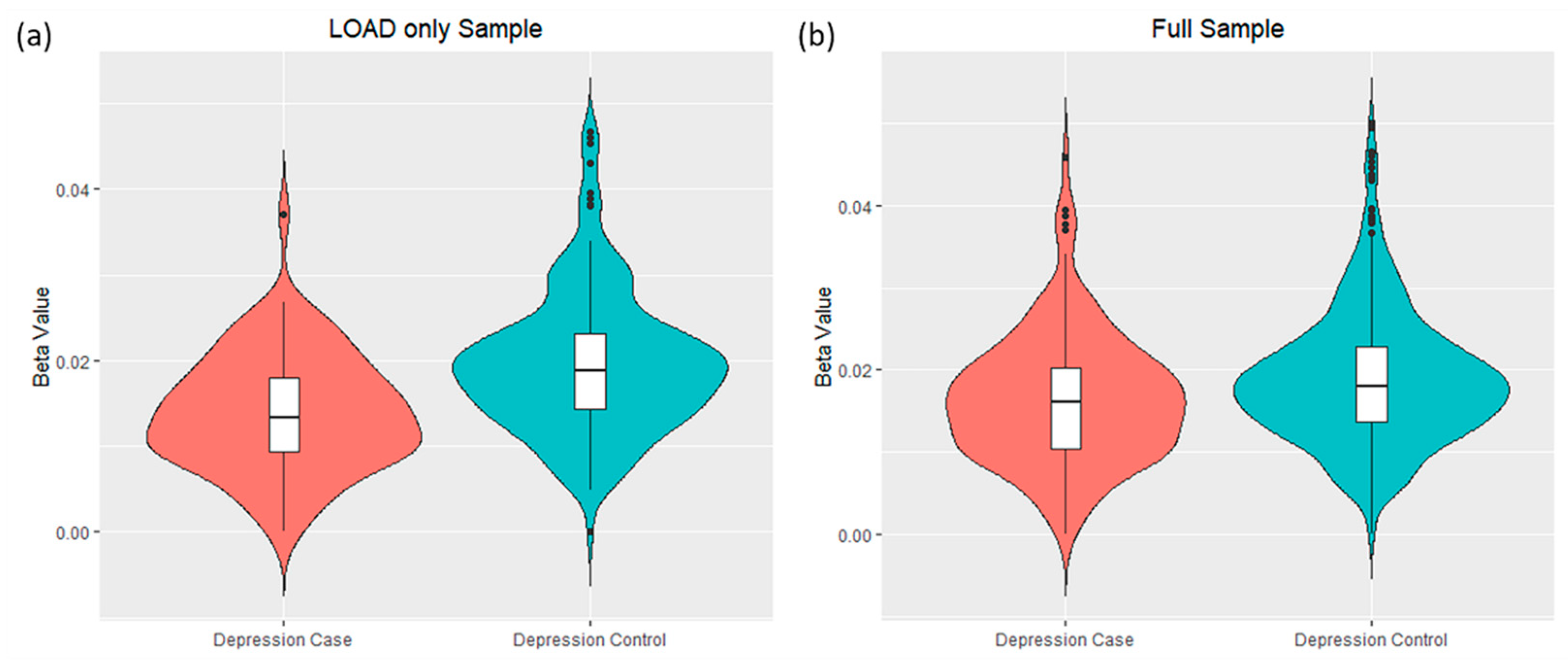
3.2. Differential DNA-Methylation Sites Are Associated with Depression Symptoms in LOAD Patients
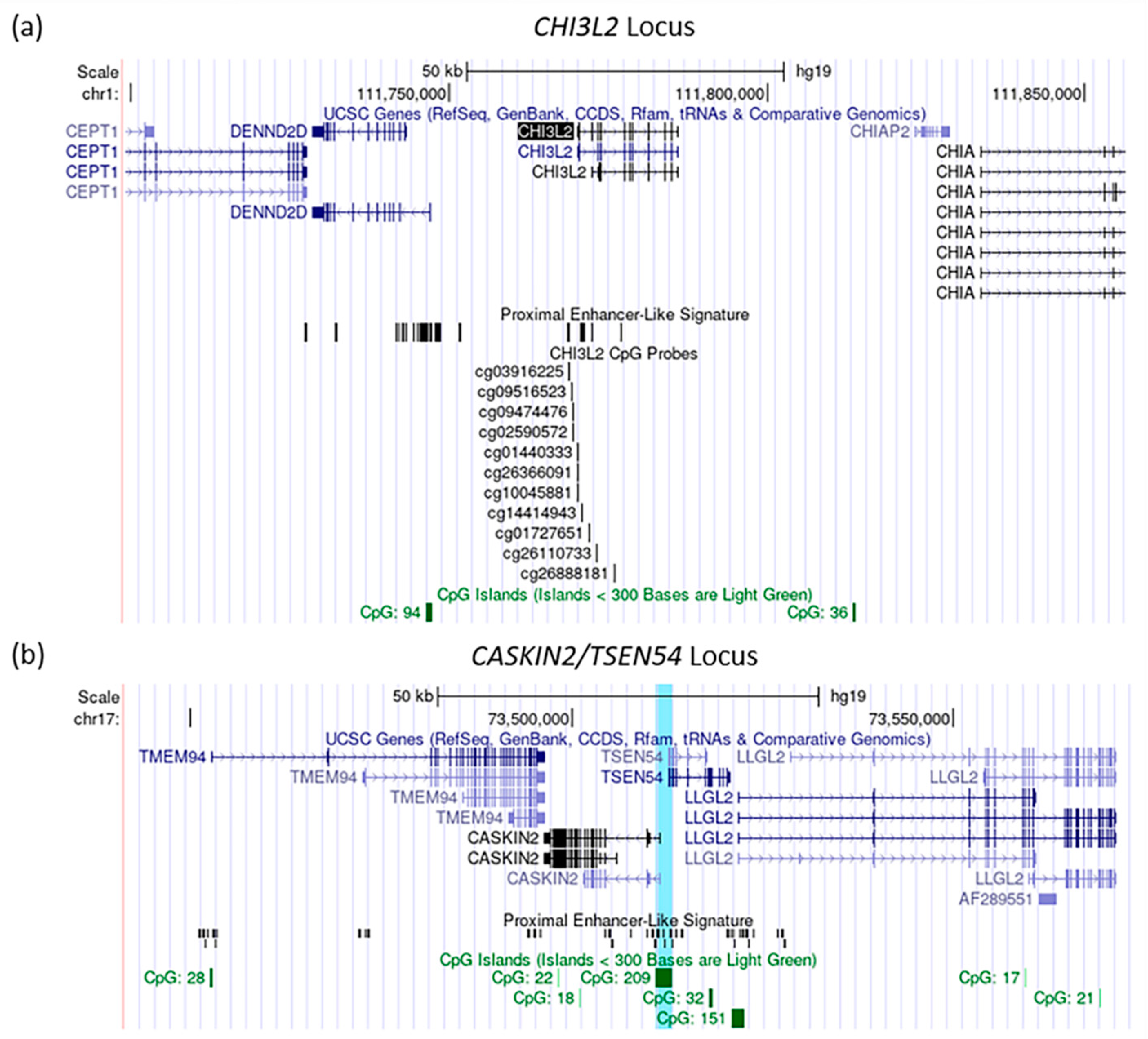
3.3. Integration Analysis of the Gene Expression and the DNA-Methylation Associations with Depression Symptoms in LOAD
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Altomari, N.; Bruno, F.; Laganà, V.; Smirne, N.; Colao, R.; Curcio, S.; Di Lorenzo, R.; Frangipane, F.; Maletta, R.; Puccio, G.; et al. A Comparison of Behavioral and Psychological Symptoms of Dementia (BPSD) and BPSD Sub-Syndromes in Early-Onset and Late-Onset Alzheimer’s Disease. J. Alzheimer’s Dis. 2022, 85, 691–699. [Google Scholar] [CrossRef]
- Hallikainen, I.; Hongisto, K.; Välimäki, T.; Hänninen, T.; Martikainen, J.; Koivisto, A.M. The Progression of Neuropsychiatric Symptoms in Alzheimer’s Disease during a Five-Year Follow-Up: Kuopio ALSOVA Study. J. Alzheimer’s Dis. 2018, 61, 1367–1376. [Google Scholar] [CrossRef] [PubMed]
- Laganà, V.; Bruno, F.; Altomari, N.; Bruni, G.; Smirne, N.; Curcio, S.; Mirabelli, M.; Colao, R.; Puccio, G.; Frangipane, F.; et al. Neuropsychiatric or Behavioral and Psychological Symptoms of Dementia (BPSD): Focus on Prevalence and Natural History in Alzheimer’s Disease and Frontotemporal Dementia. Front. Neurol. 2022, 13, 832199. [Google Scholar] [CrossRef] [PubMed]
- Lyketsos, C.G. Neuropsychiatric Symptoms in Dementia: Overview and Measurement Challenges. J. Prev. Alzheimer’s Dis. 2015, 2, 155–156. [Google Scholar] [CrossRef]
- Lyketsos, C.G.; Carrillo, M.C.; Ryan, J.M.; Khachaturian, A.S.; Trzepacz, P.; Amatniek, J.; Cedarbaum, J.; Brashear, R.; Miller, D.S. Neuropsychiatric symptoms in Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 532–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masters, M.C.; Morris, J.C.; Roe, C.M. “Noncognitive” symptoms of early Alzheimer disease: A longitudinal analysis. Neurology 2015, 84, 617–622. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.F.; Tan, L.; Wang, H.F.; Jiang, T.; Tan, M.S.; Tan, L.; Xu, W.; Li, J.Q.; Wang, J.; Lai, T.J.; et al. The prevalence of neuropsychiatric symptoms in Alzheimer’s disease: Systematic review and meta-analysis. J. Affect. Disord. 2016, 190, 264–271. [Google Scholar] [CrossRef]
- Banning, L.C.P.; Ramakers, I.; Rosenberg, P.B.; Lyketsos, C.G.; Leoutsakos, J.S. Alzheimer’s disease biomarkers as predictors of trajectories of depression and apathy in cognitively normal individuals, mild cognitive impairment, and Alzheimer’s disease dementia. Int. J. Geriatr. Psychiatry 2021, 36, 224–234. [Google Scholar] [CrossRef]
- Lutz, M.W.; Sprague, D.; Barrera, J.; Chiba-Falek, O. Shared genetic etiology underlying Alzheimer’s disease and major depressive disorder. Transl. Psychiatry 2020, 10, 88. [Google Scholar] [CrossRef] [Green Version]
- Upadhya, S.; Liu, H.; Luo, S.; Lutz, M.W.; Chiba-Falek, O. Polygenic Risk Score Effectively Predicts Depression Onset in Alzheimer’s Disease Based on Major Depressive Disorder Risk Variants. Front. Neurosci. 2022, 16, 827447. [Google Scholar] [CrossRef]
- Grubman, A.; Chew, G.; Ouyang, J.F.; Sun, G.; Choo, X.Y.; McLean, C.; Simmons, R.K.; Buckberry, S.; Vargas-Landin, D.B.; Poppe, D.; et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 2019, 22, 2087–2097. [Google Scholar] [CrossRef]
- Marques-Coelho, D.; Iohan, L.d.C.C.; Melo de Farias, A.R.; Flaig, A.; Letournel, F.; Martin-Négrier, M.-L.; Chapon, F.; Faisant, M.; Godfraind, C.; Maurage, C.-A.; et al. Differential transcript usage unravels gene expression alterations in Alzheimer’s disease human brains. npj Aging Mech. Dis. 2021, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Chen, S.; Zheng, C.; Wei, H.; Song, X. Meta-Analysis of Gene Expression and Identification of Biological Regulatory Mechanisms in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Jager, P.L.; Srivastava, G.; Lunnon, K.; Burgess, J.; Schalkwyk, L.C.; Yu, L.; Eaton, M.L.; Keenan, B.T.; Ernst, J.; McCabe, C.; et al. Alzheimer’s disease: Early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat. Neurosci. 2014, 17, 1156–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Silva, T.C.; Young, J.I.; Gomez, L.; Schmidt, M.A.; Hamilton-Nelson, K.L.; Kunkle, B.W.; Chen, X.; Martin, E.R.; Wang, L. Epigenome-wide meta-analysis of DNA methylation differences in prefrontal cortex implicates the immune processes in Alzheimer’s disease. Nat. Commun. 2020, 11, 6114. [Google Scholar] [CrossRef] [PubMed]
- Gomez Rueda, H.; Bustillo, J. Brain differential gene expression and blood cross-validation of a molecular signature of patients with major depressive disorder. Psychiatr. Genet. 2022, 32, 105–115. [Google Scholar] [CrossRef]
- Jansen, R.; Penninx, B.W.J.H.; Madar, V.; Xia, K.; Milaneschi, Y.; Hottenga, J.J.; Hammerschlag, A.R.; Beekman, A.; van der Wee, N.; Smit, J.H.; et al. Gene expression in major depressive disorder. Mol. Psychiatry 2016, 21, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Verma, P.; Shakya, M. Transcriptomics and sequencing analysis of gene expression profiling for major depressive disorder. Indian J. Psychiatry 2021, 63, 549–553. [Google Scholar] [CrossRef]
- Wittenberg, G.M.; Greene, J.; Vértes, P.E.; Drevets, W.C.; Bullmore, E.T. Major Depressive Disorder Is Associated With Differential Expression of Innate Immune and Neutrophil-Related Gene Networks in Peripheral Blood: A Quantitative Review of Whole-Genome Transcriptional Data From Case-Control Studies. Biol. Psychiatry 2020, 88, 625–637. [Google Scholar] [CrossRef]
- Cole, J.J.; McColl, A.; Shaw, R.; Lynall, M.-E.; Cowen, P.J.; de Boer, P.; Drevets, W.C.; Harrison, N.; Pariante, C.; Pointon, L.; et al. No evidence for differential gene expression in major depressive disorder PBMCs, but robust evidence of elevated biological ageing. Transl. Psychiatry 2021, 11, 404. [Google Scholar] [CrossRef]
- Story Jovanova, O.; Nedeljkovic, I.; Spieler, D.; Walker, R.M.; Liu, C.; Luciano, M.; Bressler, J.; Brody, J.; Drake, A.J.; Evans, K.L.; et al. DNA Methylation Signatures of Depressive Symptoms in Middle-aged and Elderly Persons: Meta-analysis of Multiethnic Epigenome-wide Studies. JAMA Psychiatry 2018, 75, 949–959. [Google Scholar] [CrossRef] [Green Version]
- Bennett, D.A.; Buchman, A.S.; Boyle, P.A.; Barnes, L.L.; Wilson, R.S.; Schneider, J.A. Religious Orders Study and Rush Memory and Aging Project. J. Alzheimer’s Dis. 2018, 64, S161–S189. [Google Scholar] [CrossRef]
- Bennett, D.A.; Schneider, J.A.; Arvanitakis, Z.; Wilson, R.S. Overview and findings from the religious orders study. Curr. Alzheimer Res. 2012, 9, 628–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, D.A.; Schneider, J.A.; Buchman, A.S.; Barnes, L.L.; Boyle, P.A.; Wilson, R.S. Overview and findings from the rush Memory and Aging Project. Curr. Alzheimer Res. 2012, 9, 646–663. [Google Scholar] [CrossRef] [PubMed]
- Bennett, D.A.; Wilson, R.S.; Schneider, J.A.; Bienias, J.L.; Arnold, S.E. Cerebral infarctions and the relationship of depression symptoms to level of cognitive functioning in older persons. Am. J. Geriatr. Psychiatry 2004, 12, 211–219. [Google Scholar] [CrossRef]
- Association, A.P. Diagnostic and Statistical Manual of Mental Disorders, 3rd ed.; American Psychiatric Publishing: Washington, DC, USA, 1987. [Google Scholar]
- Arevalo-Rodriguez, I.; Smailagic, N.; Roqué, I.F.M.; Ciapponi, A.; Sanchez-Perez, E.; Giannakou, A.; Pedraza, O.L.; Bonfill Cosp, X.; Cullum, S. Mini-Mental State Examination (MMSE) for the detection of Alzheimer’s disease and other dementias in people with mild cognitive impairment (MCI). Cochrane Database Syst. Rev. 2015, 2015, Cd010783. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Mostafavi, S.; Gaiteri, C.; Sullivan, S.E.; White, C.C.; Tasaki, S.; Xu, J.; Taga, M.; Klein, H.U.; Patrick, E.; Komashko, V.; et al. A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer’s disease. Nat. Neurosci. 2018, 21, 811–819. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Du, P.; Zhang, X.; Huang, C.-C.; Jafari, N.; Kibbe, W.A.; Hou, L.; Lin, S.M. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinform. 2010, 11, 587. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Kasher, P.R.; Namavar, Y.; van Tijn, P.; Fluiter, K.; Sizarov, A.; Kamermans, M.; Grierson, A.J.; Zivkovic, D.; Baas, F. Impairment of the tRNA-splicing endonuclease subunit 54 (tsen54) gene causes neurological abnormalities and larval death in zebrafish models of pontocerebellar hypoplasia. Hum. Mol. Genet. 2011, 20, 1574–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwan, J.J.; Donaldson, L.W. A lack of peptide binding and decreased thermostability suggests that the CASKIN2 scaffolding protein SH3 domain may be vestigial. BMC Struct. Biol. 2016, 16, 14. [Google Scholar] [CrossRef] [Green Version]
- Devi, G.; Scheltens, P. Heterogeneity of Alzheimer’s disease: Consequence for drug trials? Alzheimer’s Res. Ther. 2018, 10, 122. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, D.; Wahlund, L.O.; Westman, E. The heterogeneity within Alzheimer’s disease. Aging (Albany NY) 2018, 10, 3058–3060. [Google Scholar] [CrossRef]
- Komarova, N.L.; Thalhauser, C.J. High degree of heterogeneity in Alzheimer’s disease progression patterns. PLoS Comput. Biol. 2011, 7, e1002251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, B.; Masellis, M.; Freedman, M.; Stuss, D.T.; Black, S.E. Clinical, imaging, and pathological heterogeneity of the Alzheimer’s disease syndrome. Alzheimer’s Res. Ther. 2013, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallier, P.N.; Ferrara, M.; Romagnolo, F.; Ferretti, M.T.; Soreq, H.; Cerase, A. Chromosomal and environmental contributions to sex differences in the vulnerability to neurological and neuropsychiatric disorders: Implications for therapeutic interventions. Prog. Neurobiol. 2022, 219, 102353. [Google Scholar] [CrossRef]
- Albert, P.R. Why is depression more prevalent in women? J. Psychiatry Neurosci. 2015, 40, 219–221. [Google Scholar] [CrossRef]
- Shi, P.; Yang, A.; Zhao, Q.; Chen, Z.; Ren, X.; Dai, Q. A Hypothesis of Gender Differences in Self-Reporting Symptom of Depression: Implications to Solve Under-Diagnosis and Under-Treatment of Depression in Males. Front. Psychiatry 2021, 12, 589687. [Google Scholar] [CrossRef]
- Gamache, J.; Yun, Y.; Chiba-Falek, O. Sex-dependent effect of APOE on Alzheimer’s disease and other age-related neurodegenerative disorders. Dis. Model. Mech. 2020, 13, dmm045211. [Google Scholar] [CrossRef] [PubMed]
- Podcasy, J.L.; Epperson, C.N. Considering sex and gender in Alzheimer disease and other dementias. Dialogues Clin. Neurosci. 2016, 18, 437–446. [Google Scholar] [CrossRef]
- Eikelboom, W.S.; Pan, M.; Ossenkoppele, R.; Coesmans, M.; Gatchel, J.R.; Ismail, Z.; Lanctôt, K.L.; Fischer, C.E.; Mortby, M.E.; van den Berg, E.; et al. Sex differences in neuropsychiatric symptoms in Alzheimer’s disease dementia: A meta-analysis. Alzheimer’s Res. Ther. 2022, 14, 48. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yang, Y.; Duan, H.; He, J.; Sun, L.; Hu, W.; Zeng, J. CHI3L2 Is a Novel Prognostic Biomarker and Correlated With Immune Infiltrates in Gliomas. Front. Oncol. 2021, 11, 611038. [Google Scholar] [CrossRef]
- Moreno-Rodriguez, M.; Perez, S.E.; Nadeem, M.; Malek-Ahmadi, M.; Mufson, E.J. Frontal cortex chitinase and pentraxin neuroinflammatory alterations during the progression of Alzheimer’s disease. J. Neuroinflamm. 2020, 17, 58. [Google Scholar] [CrossRef] [Green Version]
- Ciccocioppo, F.; Bologna, G.; Ercolino, E.; Pierdomenico, L.; Simeone, P.; Lanuti, P.; Pieragostino, D.; Del Boccio, P.; Marchisio, M.; Miscia, S. Neurodegenerative diseases as proteinopathies-driven immune disorders. Neural Regen. Res. 2020, 15, 850–856. [Google Scholar] [CrossRef]
- Feng, T.; Tripathi, A.; Pillai, A. Inflammatory Pathways in Psychiatric Disorders: The case of Schizophrenia and Depression. Curr. Behav. Neurosci. Rep. 2020, 7, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Burgess, S.; Suckling, J.; Lalousis, P.A.; Batool, F.; Griffiths, S.L.; Palmer, E.; Karwath, A.; Barsky, A.; Gkoutos, G.V.; et al. Inflammation and Brain Structure in Schizophrenia and Other Neuropsychiatric Disorders: A Mendelian Randomization Study. JAMA Psychiatry 2022, 79, 498–507. [Google Scholar] [CrossRef]
- Sanfilippo, C.; Castrogiovanni, P.; Imbesi, R.; Di Rosa, M. CHI3L2 Expression Levels Are Correlated with AIF1, PECAM1, and CALB1 in the Brains of Alzheimer’s Disease Patients. J. Mol. Neurosci. 2020, 70, 1598–1610. [Google Scholar] [CrossRef] [PubMed]
- Hennings, J.M.; Uhr, M.; Klengel, T.; Weber, P.; Pütz, B.; Touma, C.; Czamara, D.; Ising, M.; Holsboer, F.; Lucae, S. RNA expression profiling in depressed patients suggests retinoid-related orphan receptor alpha as a biomarker for antidepressant response. Transl. Psychiatry 2015, 5, e538. [Google Scholar] [CrossRef]
- Sahakyan, A.; Yang, Y.; Plath, K. The Role of Xist in X-Chromosome Dosage Compensation. Trends Cell Biol. 2018, 28, 999–1013. [Google Scholar] [CrossRef] [PubMed]
- Ji, B.; Higa, K.K.; Kelsoe, J.R.; Zhou, X. Over-expression of XIST, the Master Gene for X Chromosome Inactivation, in Females With Major Affective Disorders. eBioMedicine 2015, 2, 909–918. [Google Scholar] [CrossRef] [Green Version]
- Chanda, K.; Mukhopadhyay, D. LncRNA Xist, X-chromosome Instability and Alzheimer’s Disease. Curr. Alzheimer Res. 2020, 17, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.-W.; Liu, H.-J.; Hong, Y.-X.; Meng, T.; Du, J.; Chang, C. lncRNA XIST induces Aβ accumulation and neuroinflammation by the epigenetic repression of NEP in Alzheimer’s disease. J. Neurogenet. 2022, 36, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Yue, D.; Guanqun, G.; Jingxin, L.; Sen, S.; Shuang, L.; Yan, S.; Minxue, Z.; Ping, Y.; Chong, L.; Zhuobo, Z.; et al. Silencing of long noncoding RNA XIST attenuated Alzheimer’s disease-related BACE1 alteration through miR-124. Cell Biol. Int. 2020, 44, 630–636. [Google Scholar] [CrossRef]
- Kuehner, J.N.; Bruggeman, E.C.; Wen, Z.; Yao, B. Epigenetic Regulations in Neuropsychiatric Disorders. Front. Genet. 2019, 10, 268. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Lee, K.; Kim, K.; Yi, S.-J. The role of histone modifications: From neurodevelopment to neurodiseases. Signal Transduct. Target. Ther. 2022, 7, 217. [Google Scholar] [CrossRef]
- Lutz, M.W.; Chiba-Falek, O. Bioinformatics pipeline to guide late-onset Alzheimer’s disease (LOAD) post-GWAS studies: Prioritizing transcription regulatory variants within LOAD-associated regions. Alzheimer’s Dement (N. Y.) 2022, 8, e12244. [Google Scholar] [CrossRef]
- Mulvey, B.; Dougherty, J.D. Transcriptional-regulatory convergence across functional MDD risk variants identified by massively parallel reporter assays. Transl. Psychiatry 2021, 11, 403. [Google Scholar] [CrossRef]
- Walgrave, H.; Zhou, L.; De Strooper, B.; Salta, E. The promise of microRNA-based therapies in Alzheimer’s disease: Challenges and perspectives. Mol. Neurodegener. 2021, 16, 76. [Google Scholar] [CrossRef]
- Le François, B.; Zhang, L.; Mahajan, G.J.; Stockmeier, C.A.; Friedman, E.; Albert, P.R. A Novel Alternative Splicing Mechanism That Enhances Human 5-HT1A Receptor RNA Stability Is Altered in Major Depression. J. Neurosci. 2018, 38, 8200–8210. [Google Scholar] [CrossRef] [Green Version]
- Love, J.E.; Hayden, E.J.; Rohn, T.T. Alternative Splicing in Alzheimer’s Disease. J. Parkinsons Dis. Alzheimers Dis. 2015, 2, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiba-Falek, O.; Barrera, J.; Song, L.; Safi, A.; Yu, Y.J.; Garrett, M.; Gamache, J.; Chipman, D.; Ashley-Koch, A.; Crawford, G. Cell-type-specific and sex-dependent changes in the chromatin accessibility landscape in late-onset Alzheimer’s disease brains. Alzheimer’s Dement. 2020, 16, e037712. [Google Scholar] [CrossRef]
- Hüls, A.; Robins, C.; Conneely, K.N.; De Jager, P.L.; Bennett, D.A.; Epstein, M.P.; Wingo, T.S.; Wingo, A.P. Association between DNA methylation levels in brain tissue and late-life depression in community-based participants. Transl. Psychiatry 2020, 10, 262. [Google Scholar] [CrossRef]
- Edgar, R.D.; Jones, M.J.; Robinson, W.P.; Kobor, M.S. An empirically driven data reduction method on the human 450K methylation array to remove tissue specific non-variable CpGs. Clin. Epigenetics 2017, 9, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauber, C.; Gerl, M.J.; Klose, C.; Ottosson, F.; Melander, O.; Simons, K. Lipidomic risk scores are independent of polygenic risk scores and can predict incidence of diabetes and cardiovascular disease in a large population cohort. PLoS Biol. 2022, 20, e3001561. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| RNA-Seq Sample LOAD Only | RNA-Seq Sample All | Methylation Sample LOAD Only | Methylation Sample All | |
|---|---|---|---|---|
| Subjects | 166 | 424 | 252 | 603 |
| Depression Cases/Controls | 50/116 | 99/325 | 79/173 | 145/458 |
| Mean age at death (SD) | 91.0 (6.1) | 88.5 (6.6) | 873.80 (3.66.5) | 86.583.0 (4.56.5) |
| Percent Female | 66% | 63% | 66% | 63% |
| Mean Years of Education (SD) | 16.6 (3.4) | 16.5 (3.5) | 16.3 (3.5) | 16.4 (3.5) |
| APOEe4 Count: | ||||
| 0 | 102 | 309 | 160 | 435 |
| 1 | 6 | 111 | 86 | 159 |
| 2 | 4 | 4 | 6 | 9 |
| Mean PMI 1 in hours (SD) | 6.8 (4.1) | 7.0 (4.9) | 6.8 (4.7) | 7.5 (6.0) |
| Percent Braak Stage ≥ 4 | 67% | 52% | 69% | 52% |
| Mean MMSE 2 at Last Visit (SD) | 13.6 (8.7) | 21.6 (8.9) | 12.8 (8.6) | 20.9 (9.3) |
| Male | Female | LOAD | ||||
|---|---|---|---|---|---|---|
| Gene | Log2FC | p | Log2FC | p | Log2FC | p |
| APLNR | 1.652 | 0.018 | −0.333 | 0.999 | 0.084 | 0.999 |
| BEST3 | 0.997 | 0.0003 | −0.093 | 0.999 | 0.213 | 0.999 |
| BIRC3 | 0.995 | 0.025 | −0.193 | 0.999 | 0.173 | 0.999 |
| CHI3L1 | 1.736 | 0.03 | −0.447 | 0.999 | 0.286 | 0.999 |
| CHI3L2 | 4.246 | 1.84 × 10−8 | −0.715 | 0.999 | 1.191 | 0.999 |
| ENSG00000232306.1 | −8.116 | 0.005 | NA | NA | −0.441 | 0.999 |
| ENSG00000273259.2 | 2.669 | 0.002 | −1.424 | 0.999 | −0.175 | 0.999 |
| FP236383.12 | −0.556 | 0.999 | 2.289 | 0.002 | 1.863 | 0.064 |
| GBP3 | 0.922 | 0.034 | −0.06 | 0.999 | 0.26 | 0.999 |
| GBPI | 1.343 | 0.0003 | −0.522 | 0.999 | 0.09 | 0.999 |
| GPLIR2 | 0.673 | 0.026 | −0.04 | 0.999 | 0.116 | 0.999 |
| HSPA6 | 0.868 | 0.999 | −1.79 | 0.049 | −0.617 | 0.999 |
| IL18BP | 0.727 | 0.002 | −0.188 | 0.999 | 0.072 | 0.999 |
| LRRC55 | 0.78 | 0.034 | −0.087 | 0.999 | 0.129 | 0.999 |
| MLST8 | −0.243 | 0.043 | 0.06 | 0.999 | −0.001 | 0.999 |
| PDPN | 0.829 | 0.005 | −0.225 | 0.999 | 0.045 | 0.999 |
| PLEKHA4 | 0.946 | 0.0007 | −0.241 | 0.999 | 0.082 | 0.999 |
| PLPP4 | 0.619 | 0.038 | −0.111 | 0.999 | 0.095 | 0.999 |
| RP11-364B14.3 | −0.745 | 2.67 × 10−5 | 0.096 | 0.999 | −0.084 | 0.999 |
| S100A3 | 2.496 | 2.67 × 10−5 | −0.306 | 0.999 | 0.678 | 0.999 |
| SELE | 1.935 | 0.688 | −1.988 | 0.049 | −0.794 | 0.999 |
| SFN | 2.049 | 0.041 | −0.508 | 0.999 | 0.39 | 0.999 |
| SLAMF8 | 1.155 | 0.017 | −0.826 | 0.999 | 0.117 | 0.999 |
| SPOCD1 | 1.368 | 0.002 | −0.3 | 0.999 | 0.095 | 0.999 |
| TIMP1 | 0.87 | 0.03 | −0.456 | 0.999 | −0.037 | 0.999 |
| TKTL1 | 2.234 | 0.0006 | −0.583 | 0.999 | 0.362 | 0.999 |
| TNFAIP2 | 1.098 | 0.014 | −0.185 | 0.999 | 0.235 | 0.999 |
| VAMP5 | 0.452 | 0.048 | −0.119 | 0.999 | 0.041 | 0.999 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Upadhya, S.; Gingerich, D.; Lutz, M.W.; Chiba-Falek, O. Differential Gene Expression and DNA Methylation in the Risk of Depression in LOAD Patients. Biomolecules 2022, 12, 1679. https://doi.org/10.3390/biom12111679
Upadhya S, Gingerich D, Lutz MW, Chiba-Falek O. Differential Gene Expression and DNA Methylation in the Risk of Depression in LOAD Patients. Biomolecules. 2022; 12(11):1679. https://doi.org/10.3390/biom12111679
Chicago/Turabian StyleUpadhya, Suraj, Daniel Gingerich, Michael William Lutz, and Ornit Chiba-Falek. 2022. "Differential Gene Expression and DNA Methylation in the Risk of Depression in LOAD Patients" Biomolecules 12, no. 11: 1679. https://doi.org/10.3390/biom12111679
APA StyleUpadhya, S., Gingerich, D., Lutz, M. W., & Chiba-Falek, O. (2022). Differential Gene Expression and DNA Methylation in the Risk of Depression in LOAD Patients. Biomolecules, 12(11), 1679. https://doi.org/10.3390/biom12111679