
Tenascin-C: A Key Regulator in Angiogenesis during Wound Healing

Abstract
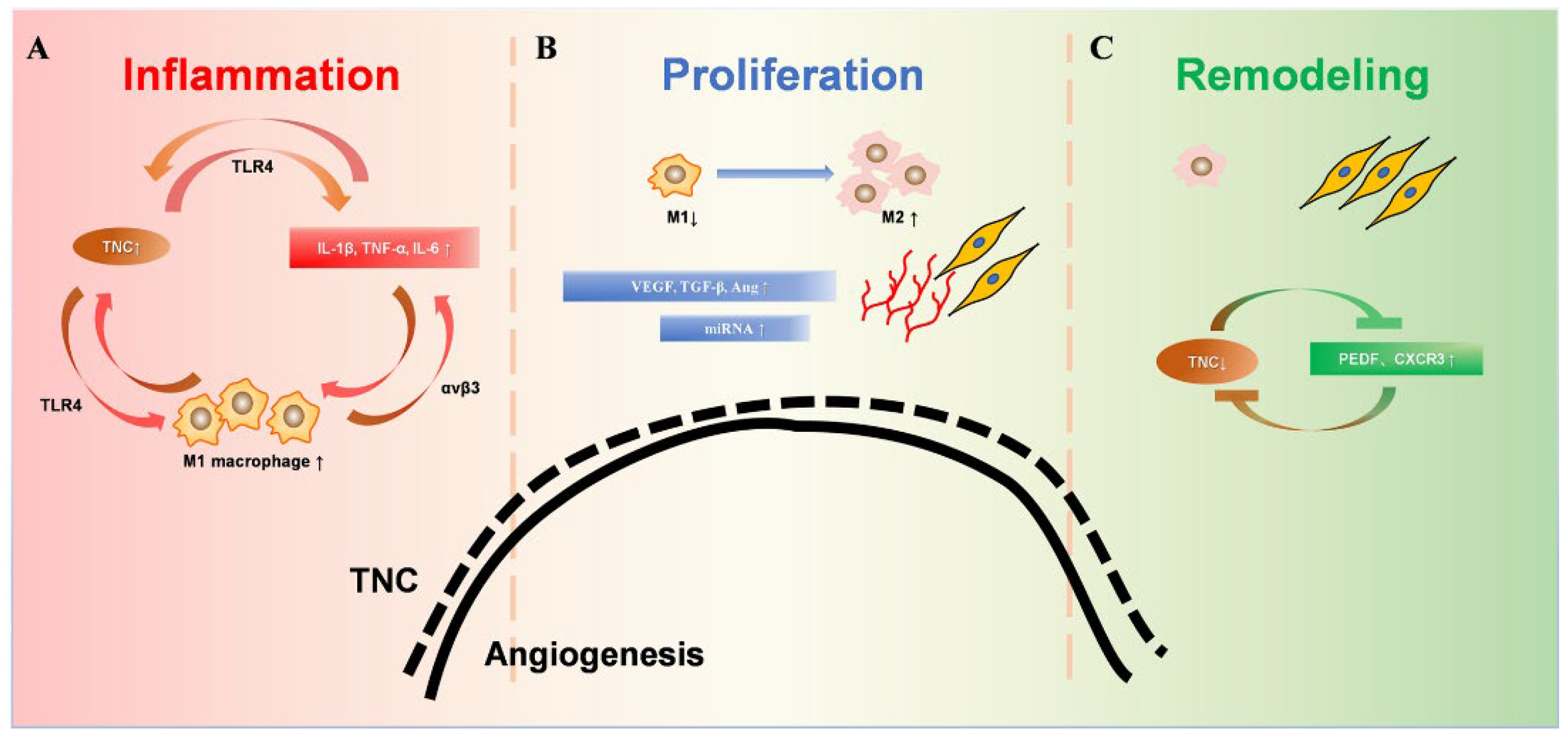
:1. Introduction
2. Results
2.1. The Construction and Expression Signature of TNC
2.2. TNC in Healing Wound
2.3. Initial Activation of TNC in the Inflammation Phase
2.4. Peak Expression of TNC in the Proliferation Phase
2.4.1. TNC Promotes the M2 Polarization of Macrophages
2.4.2. Associations between TNC and VEGF and TGF-β and Angiopoietin
2.4.3. TNC and miRNA Cluster
2.5. TNC in the Remodeling Phase
2.5.1. TNC and PEDF
2.5.2. TNC and CXCR3
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alghamdi, M.A.; Wallace, H.J.; Melton, P.E.; Moses, E.K.; Stevenson, A.; Al-Eitan, L.N.; Rea, S.; Duke, J.M.; Danielsen, P.L.; Prêle, C.M.; et al. Identification of Differentially Methylated CpG Sites in Fibroblasts from Keloid Scars. Biomedicines 2020, 8, 181. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K.; Gordillo, G.M.; Roy, S.; Kirsner, R.; Lambert, L.; Hunt, T.K.; Gottrup, F.; Gurtner, G.C.; Longaker, M.T. Human skin wounds: A major and snowballing threat to public health and the economy. Wound Repair Regen. Off. Publ. Wound Heal. Soc. Eur. Tissue Repair Soc. 2009, 17, 763–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, D.Y.; Lovicu, F.J. Myofibroblast transdifferentiation: The dark force in ocular wound healing and fibrosis. Prog. Retin. Eye Res. 2017, 60, 44–65. [Google Scholar] [CrossRef] [PubMed]
- Sylakowski, K.; Wells, A. ECM-regulation of autophagy: The yin and the yang of autophagy during wound healing. Matrix Biol. J. Int. Soc. Matrix Biol. 2021, 100–101, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Giblin, S.P.; Midwood, K.S. Tenascin-C: Form versus function. Cell Adhes. Migr. 2015, 9, 48–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imanaka-Yoshida, K. Tenascin-C in Heart Diseases-The Role of Inflammation. Int. J. Mol. Sci. 2021, 22, 5828. [Google Scholar] [CrossRef]
- Midwood, K.S.; Chiquet, M.; Tucker, R.P.; Orend, G. Tenascin-C at a glance. J. Cell Sci. 2016, 129, 4321–4327. [Google Scholar] [CrossRef] [Green Version]
- Tucker, R.P.; Chiquet-Ehrismann, R. Tenascin-C: Its functions as an integrin ligand. Int. J. Biochem. Cell Biol. 2015, 65, 165–168. [Google Scholar] [CrossRef]
- Sumioka, T.; Fujita, N.; Kitano, A.; Okada, Y.; Saika, S. Impaired angiogenic response in the cornea of mice lacking tenascin C. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2462–2467. [Google Scholar] [CrossRef] [Green Version]
- Kubo, Y.; Ishikawa, K.; Mori, K.; Kobayashi, Y.; Nakama, T.; Arima, M.; Nakao, S.; Hisatomi, T.; Haruta, M.; Sonoda, K.H.; et al. Periostin and tenascin-C interaction promotes angiogenesis in ischemic proliferative retinopathy. Sci. Rep. 2020, 10, 9299. [Google Scholar] [CrossRef]
- Kim, Y.H.; Jung, E.; Im, G.B.; Kim, Y.J.; Kim, S.W.; Jeong, G.J.; Jang, Y.C.; Park, K.M.; Kim, D.I.; Yu, T.; et al. Regulation of intracellular transition metal ion level with a pH-sensitive inorganic nanocluster to improve therapeutic angiogenesis by enriching conditioned medium retrieved from human adipose derived stem cells. Nano Converg. 2020, 7, 34. [Google Scholar] [CrossRef]
- Castellon, R.; Caballero, S.; Hamdi, H.K.; Atilano, S.R.; Aoki, A.M.; Tarnuzzer, R.W.; Kenney, M.C.; Grant, M.B.; Ljubimov, A.V. Effects of tenascin-C on normal and diabetic retinal endothelial cells in culture. Investig. Ophthalmol. Vis. Sci. 2002, 43, 2758–2766. [Google Scholar]
- Reinke, J.M.; Sorg, H. Wound repair and regeneration. Eur. Surg. Res. Eur. Chir. Forschung. Rech. Chir. Eur. 2012, 49, 35–43. [Google Scholar] [CrossRef]
- Wallace, H.A.; Basehore, B.M.; Zito, P.M. Wound Healing Phases. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2022. [Google Scholar]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv. Drug Deliv. Rev. 2016, 97, 4–27. [Google Scholar] [CrossRef]
- Hussey, G.S.; Keane, T.J.; Badylak, S.F. The extracellular matrix of the gastrointestinal tract: A regenerative medicine platform. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 540–552. [Google Scholar] [CrossRef]
- Mazini, L.; Rochette, L.; Admou, B.; Amal, S.; Malka, G. Hopes and Limits of Adipose-Derived Stem Cells (ADSCs) and Mesenchymal Stem Cells (MSCs) in Wound Healing. Int. J. Mol. Sci. 2020, 21, 1306. [Google Scholar] [CrossRef] [Green Version]
- Tucker, R.P.; Drabikowski, K.; Hess, J.F.; Ferralli, J.; Chiquet-Ehrismann, R.; Adams, J.C. Phylogenetic analysis of the tenascin gene family: Evidence of origin early in the chordate lineage. BMC Evol. Biol. 2006, 6, 60. [Google Scholar] [CrossRef] [Green Version]
- Chiovaro, F.; Chiquet-Ehrismann, R.; Chiquet, M. Transcriptional regulation of tenascin genes. Cell Adhes. Migr. 2015, 9, 34–47. [Google Scholar] [CrossRef] [Green Version]
- Rathjen, F.G.; Hodge, R. Early Days of Tenascin-R Research: Two Approaches Discovered and Shed Light on Tenascin-R. Front. Immunol. 2020, 11, 612482. [Google Scholar] [CrossRef]
- Miller, W.L. Tenascin-X-Discovery and Early Research. Front. Immunol. 2020, 11, 612497. [Google Scholar] [CrossRef]
- Degen, M.; Scherberich, A.; Tucker, R.P. Tenascin-W: Discovery, Evolution, and Future Prospects. Front. Immunol. 2020, 11, 623305. [Google Scholar] [CrossRef] [PubMed]
- Imanaka-Yoshida, K.; Yoshida, T.; Miyagawa-Tomita, S. Tenascin-C in development and disease of blood vessels. Anat. Rec. 2014, 297, 1747–1757. [Google Scholar] [CrossRef] [PubMed]
- Imanaka-Yoshida, K.; Tawara, I.; Yoshida, T. Tenascin-C in cardiac disease: A sophisticated controller of inflammation, repair, and fibrosis. Am. J. Physiol. Cell Physiol. 2020, 319, C781–C796. [Google Scholar] [CrossRef] [PubMed]
- Mighell, A.J.; Thompson, J.; Hume, W.J.; Markham, A.F.; Robinson, P.A. Human tenascin-C: Identification of a novel type III repeat in oral cancer and of novel splice variants in normal, malignant and reactive oral mucosae. Int. J. Cancer 1997, 72, 236–240. [Google Scholar] [CrossRef]
- Lingasamy, P.; Laarmann, A.H.; Teesalu, T. Tumor Penetrating Peptide-Functionalized Tenascin-C Antibody for Glioblastoma Targeting. Curr. Cancer Drug Targets 2021, 21, 70–79. [Google Scholar] [CrossRef]
- Fujita, M.; Sasada, M.; Iyoda, T.; Nagai, R.; Kudo, C.; Yamamoto, T.; Osada, S.; Kodama, H.; Fukai, F. Anoikis resistance conferred by tenascin-C-derived peptide TNIIIA2 and its disruption by integrin inactivation. Biochem. Biophys. Res. Commun. 2021, 536, 14–19. [Google Scholar] [CrossRef]
- Hasegawa, M.; Yoshida, T.; Sudo, A. Tenascin-C in Osteoarthritis and Rheumatoid Arthritis. Front. Immunol. 2020, 11, 577015. [Google Scholar] [CrossRef]
- Golledge, J.; Clancy, P.; Maguire, J.; Lincz, L.; Koblar, S. The role of tenascin C in cardiovascular disease. Cardiovasc. Res. 2011, 92, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Saika, S.; Sumioka, T.; Okada, Y.; Yamanaka, O.; Kitano, A.; Miyamoto, T.; Shirai, K.; Kokado, H. Wakayama symposium: Modulation of wound healing response in the corneal stroma by osteopontin and tenascin-C. Ocul. Surf. 2013, 11, 12–15. [Google Scholar] [CrossRef]
- Suzuki, H.; Fujimoto, M.; Kawakita, F.; Liu, L.; Nakatsuka, Y.; Nakano, F.; Nishikawa, H.; Okada, T.; Kanamaru, H.; Imanaka-Yoshida, K.; et al. Tenascin-C in brain injuries and edema after subarachnoid hemorrhage: Findings from basic and clinical studies. J. Neurosci. Res. 2020, 98, 42–56. [Google Scholar] [CrossRef] [Green Version]
- Dueck, M.; Riedl, S.; Hinz, U.; Tandara, A.; Möller, P.; Herfarth, C.; Faissner, A. Detection of tenascin-C isoforms in colorectal mucosa, ulcerative colitis, carcinomas and liver metastases. Int. J. Cancer 1999, 82, 477–483. [Google Scholar] [CrossRef]
- Latijnhouwers, M.A.; de Jongh, G.J.; Bergers, M.; de Rooij, M.J.; Schalkwijk, J. Expression of tenascin-C splice variants by human skin cells. Arch. Dermatol. Res. 2000, 292, 446–454. [Google Scholar] [CrossRef]
- Chiquet, M. Tenascin-C: From Discovery to Structure-Function Relationships. Front. Immunol. 2020, 11, 611789. [Google Scholar] [CrossRef]
- Irintchev, A.; Rollenhagen, A.; Troncoso, E.; Kiss, J.Z.; Schachner, M. Structural and functional aberrations in the cerebral cortex of tenascin-C deficient mice. Cereb. Cortex 2005, 15, 950–962. [Google Scholar] [CrossRef]
- Gurevicius, K.; Kuang, F.; Stoenica, L.; Irintchev, A.; Gureviciene, I.; Dityatev, A.; Schachner, M.; Tanila, H. Genetic ablation of tenascin-C expression leads to abnormal hippocampal CA1 structure and electrical activity in vivo. Hippocampus 2009, 19, 1232–1246. [Google Scholar] [CrossRef]
- Martin, P.; Nunan, R. Cellular and molecular mechanisms of repair in acute and chronic wound healing. Br. J. Dermatol. 2015, 173, 370–378. [Google Scholar] [CrossRef]
- Banda, M.J.; Knighton, D.R.; Hunt, T.K.; Werb, Z. Isolation of a nonmitogenic angiogenesis factor from wound fluid. Proc. Natl. Acad. Sci. USA 1982, 79, 7773–7777. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, T.X.; Leethanakul, C.; Patel, V.; Mitola, D.; Lund, L.R.; Danø, K.; Johnsen, M.; Gutkind, J.S.; Bugge, T.H. Laser capture microdissection-based in vivo genomic profiling of wound keratinocytes identifies similarities and differences to squamous cell carcinoma. Oncogene 2003, 22, 3964–3976. [Google Scholar] [CrossRef] [Green Version]
- DiPietro, L.A. Angiogenesis and wound repair: When enough is enough. J. Leukoc. Biol. 2016, 100, 979–984. [Google Scholar] [CrossRef] [Green Version]
- Nagy, J.A.; Benjamin, L.; Zeng, H.; Dvorak, A.M.; Dvorak, H.F. Vascular permeability, vascular hyperpermeability and angiogenesis. Angiogenesis 2008, 11, 109–119. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.; Rafii, S.; Wu, M.H.; Wijelath, E.S.; Yu, C.; Ishida, A.; Fujita, Y.; Kothari, S.; Mohle, R.; Sauvage, L.R.; et al. Evidence for circulating bone marrow-derived endothelial cells. Blood 1998, 92, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Risau, W.; Flamme, I. Vasculogenesis. Annu. Rev. Cell Dev. Biol. 1995, 11, 73–91. [Google Scholar] [CrossRef] [PubMed]
- Marchand, M.; Monnot, C.; Muller, L.; Germain, S. Extracellular matrix scaffolding in angiogenesis and capillary homeostasis. Semin. Cell Dev. Biol. 2019, 89, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Vlodavsky, I.; Korner, G.; Ishai-Michaeli, R.; Bashkin, P.; Bar-Shavit, R.; Fuks, Z. Extracellular matrix-resident growth factors and enzymes: Possible involvement in tumor metastasis and angiogenesis. Cancer Metastasis Rev. 1990, 9, 203–226. [Google Scholar] [CrossRef] [PubMed]
- Branski, L.K.; Gauglitz, G.G.; Herndon, D.N.; Jeschke, M.G. A review of gene and stem cell therapy in cutaneous wound healing. Burns 2009, 35, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Shibuya, M. Vascular endothelial growth factor and its receptor system: Physiological functions in angiogenesis and pathological roles in various diseases. J. Biochem. 2013, 153, 13–19. [Google Scholar] [CrossRef] [Green Version]
- Smigiel, K.S.; Parks, W.C. Macrophages, Wound Healing, and Fibrosis: Recent Insights. Curr. Rheumatol. Rep. 2018, 20, 17. [Google Scholar] [CrossRef]
- Li, Z.; Chen, S.; Cui, H.; Li, X.; Chen, D.; Hao, W.; Wang, J.; Li, Z.; Zheng, Z.; Zhang, Z.; et al. Tenascin-C-mediated suppression of extracellular matrix adhesion force promotes entheseal new bone formation through activation of Hippo signalling in ankylosing spondylitis. Ann. Rheum. Dis. 2021, 80, 891–902. [Google Scholar] [CrossRef]
- Martin, J.F.; Bradley, A.; Olson, E.N. The paired-like homeo box gene MHox is required for early events of skeletogenesis in multiple lineages. Genes Dev. 1995, 9, 1237–1249. [Google Scholar] [CrossRef] [Green Version]
- Ihida-Stansbury, K.; McKean, D.M.; Gebb, S.A.; Martin, J.F.; Stevens, T.; Nemenoff, R.; Akeson, A.; Vaughn, J.; Jones, P.L. Paired-related homeobox gene Prx1 is required for pulmonary vascular development. Circ. Res. 2004, 94, 1507–1514. [Google Scholar] [CrossRef] [Green Version]
- Jones, F.S.; Chalepakis, G.; Gruss, P.; Edelman, G.M. Activation of the cytotactin promoter by the homeobox-containing gene Evx-1. Proc. Natl. Acad. Sci. USA 1992, 89, 2091–2095. [Google Scholar] [CrossRef]
- Copertino, D.W.; Edelman, G.M.; Jones, F.S. Multiple promoter elements differentially regulate the expression of the mouse tenascin gene. Proc. Natl. Acad. Sci. USA 1997, 94, 1846–1851. [Google Scholar] [CrossRef] [Green Version]
- Gherzi, R.; Briata, P.; Boncinelli, E.; Ponassi, M.; Querzè, G.; Viti, F.; Corte, G.; Zardi, L. The human homeodomain protein OTX2 binds to the human tenascin-C promoter and trans-represses its activity in transfected cells. DNA Cell Biol. 1997, 16, 559–567. [Google Scholar] [CrossRef]
- Yeo, S.Y.; Lee, K.W.; Shin, D.; An, S.; Cho, K.H.; Kim, S.H. A positive feedback loop bi-stably activates fibroblasts. Nat. Commun. 2018, 9, 3016. [Google Scholar] [CrossRef] [Green Version]
- Haan, J.; Smeets, M.B.; Pasterkamp, G.; Arslan, F.J.M.I. Danger Signals in the Initiation of the Inflammatory Response after Myocardial Infarction. Mediat. Inflamm. 2013, 2013, 206039. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.A.J.J.o.M.; Cardiology, C. Inflammatory and fibrotic responses of cardiac fibroblasts to myocardial damage associated molecular patterns (DAMPs). J. Mol. Cell. Cardiol. 2016, 94, 189–200. [Google Scholar] [CrossRef]
- Marzeda, A.M.; Midwood, K.S. Internal Affairs: Tenascin-C as a Clinically Relevant, Endogenous Driver of Innate Immunity. J. Histochem. Cytochem. 2018, 66, 289–304. [Google Scholar] [CrossRef] [Green Version]
- Frangogiannis, N.G.J.P.R. The immune system and cardiac repair. Pharmacol. Res. 2008, 58, 88–111. [Google Scholar] [CrossRef] [Green Version]
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair After Myocardial Infarction From Inflammation to Fibrosis. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef]
- Midwood, K.; Sacre, S.; Piccinini, A.M.; Inglis, J.; Trebaul, A.; Chan, E.; Drexler, S.; Sofat, N.; Kashiwagi, M.; Orend, G.J.N.M. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat. Med. 2009, 15, 774–780. [Google Scholar] [CrossRef]
- Zuliani-Alvarez, L.; Marzeda, A.M.; Deligne, C.; Schwenzer, A.; McCann, F.E.; Marsden, B.D.; Piccinini, A.M.; Midwood, K.S. Mapping tenascin-C interaction with toll-like receptor 4 reveals a new subset of endogenous inflammatory triggers. Nat. Commun. 2017, 8, 1595. [Google Scholar] [CrossRef] [PubMed]
- Machino-Ohtsuka, T.; Tajiri, K.; Kimura, T.; Sakai, S.; Sato, A.; Yoshida, T.; Hiroe, M.; Yasutomi, Y.; Aonuma, K.; Imanaka-Yoshida, K. Tenascin-C Aggravates Autoimmune Myocarditis via Dendritic Cell Activation and Th17 Cell Differentiation. J. Am. Hear. Assoc. 2014, 3, e001052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, T.; Tajiri, K.; Sato, A.; Sakai, S.; Wang, Z.; Yoshida, T.; Uede, T.; Hiroe, M.; Aonuma, K.; Ieda, M.; et al. Tenascin-C accelerates adverse ventricular remodelling after myocardial infarction by modulating macrophage polarization. Cardiovasc. Res. 2019, 115, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Maqbool, A.; Spary, E.J.; Manfield, I.W.; Ruhmann, M.; Zuliani-Alvarez, L.; Gamboa-Esteves, F.O.; Porter, K.E.; Drinkhill, M.J.; Midwood, K.S.; Turner, N.A.J. Tenascin C upregulates interleukin-6 expression in human cardiac myofibroblasts via toll-like receptor 4. World J. Cardiol. 2016, 8, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Piccinini, A.M.; Zuliani-Alvarez, L.; Lim, J.M.; Midwood, K.S. Distinct microenvironmental cues stimulate divergent TLR4-mediated signaling pathways in macrophages. Sci. Signal. 2016, 9, ra86. [Google Scholar] [CrossRef] [Green Version]
- Benbow, J.H.; Thompson, K.J.; Cope, H.L.; Brandon-Warner, E.; Culberson, C.R.; Bossi, K.L.; Li, T.; Russo, M.W.; Gersin, K.S.; McKillop, I.H.; et al. Diet-Induced Obesity Enhances Progression of Hepatocellular Carcinoma through Tenascin-C/Toll-Like Receptor 4 Signaling. Am. J. Pathol. 2016, 186, 145–158. [Google Scholar] [CrossRef] [Green Version]
- Kuriyama, N.; Duarte, S.; Hamada, T.; Busuttil, R.W.; Coito, A.J.J.H. Tenascin-C: A novel mediator of hepatic ischemia and reperfusion injury. Hepatology 2011, 54, 2125–2136. [Google Scholar] [CrossRef] [Green Version]
- Shimojo, N.; Hashizume, R.; Kanayama, K.; Hara, M.; Suzuki, Y.; Nishioka, T.; Hiroe, M.; Yoshida, T.; Imanaka-Yoshida, K. Tenascin-C May Accelerate Cardiac Fibrosis by Activating Macrophages via the Integrin alpha V beta 3/NuclearFactor-kappa B/Interleukin-6 Axis. Hypertension 2015, 66, 757–766. [Google Scholar] [CrossRef] [Green Version]
- Eming, S.A.; Krieg, T.; Davidson, J.M. Inflammation in wound repair: Molecular and cellular mechanisms. J. Investig. Dermatol. 2007, 127, 514–525. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wei, Q.; Han, L.; Cao, K.; Lan, T.; Xu, Z.; Wang, Y.; Gao, Y.; Xue, J.; Shan, F.; et al. Tenascin-c renders a proangiogenic phenotype in macrophage via annexin II. J. Cell. Mol. Med. 2018, 22, 429–438. [Google Scholar] [CrossRef]
- Murakami, T.; Kikuchi, H.; Ishimatsu, H.; Iino, I.; Hirotsu, A.; Matsumoto, T.; Ozaki, Y.; Kawabata, T.; Hiramatsu, Y.; Ohta, M.; et al. Tenascin C in colorectal cancer stroma is a predictive marker for liver metastasis and is a potent target of miR-198 as identified by microRNA analysis. Br. J. Cancer 2017, 117, 1360–1370. [Google Scholar] [CrossRef]
- Piccinini, A.M.; Midwood, K.S. Endogenous control of immunity against infection: Tenascin-C regulates TLR4-mediated inflammation via microRNA-155. Cell Rep. 2012, 2, 914–926. [Google Scholar] [CrossRef] [Green Version]
- Jetten, N.; Verbruggen, S.; Gijbels, M.J.; Post, M.J.; De Winther, M.P.; Donners, M.M. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis 2014, 17, 109–118. [Google Scholar] [CrossRef]
- Huang, Y.; He, B.; Wang, L.; Yuan, B.; Shu, H.; Zhang, F.; Sun, L. Bone marrow mesenchymal stem cell-derived exosomes promote rotator cuff tendon-bone healing by promoting angiogenesis and regulating M1 macrophages in rats. Stem Cell Res. Ther. 2020, 11, 496. [Google Scholar] [CrossRef]
- Hoeben, A.; Landuyt, B.; Highley, M.S.; Wildiers, H.; Van Oosterom, A.T.; De Bruijn, E.A. Vascular endothelial growth factor and angiogenesis. Pharmacol. Rev. 2004, 56, 549–580. [Google Scholar] [CrossRef] [Green Version]
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62. [Google Scholar] [CrossRef]
- Calvo, A.; Catena, R.; Noble, M.S.; Carbott, D.; Gil-Bazo, I.; Gonzalez-Moreno, O.; Huh, J.I.; Sharp, R.; Qiu, T.H.; Anver, M.R.; et al. Identification of VEGF-regulated genes associated with increased lung metastatic potential: Functional involvement of tenascin-C in tumor growth and lung metastasis. Oncogene 2008, 27, 5373–5384. [Google Scholar] [CrossRef] [Green Version]
- O’Connell, J.T.; Sugimoto, H.; Cooke, V.G.; MacDonald, B.A.; Mehta, A.I.; LeBleu, V.S.; Dewar, R.; Rocha, R.M.; Brentani, R.R.; Resnick, M.B.; et al. VEGF-A and Tenascin-C produced by S100A4+ stromal cells are important for metastatic colonization. Proc. Natl. Acad. Sci. USA 2011, 108, 16002–16007. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, K.; Kohno, R.I.; Mori, K.; Murakami, Y.; Nakao, S.; Akiyama, M.; Yoshida, S.; Sonoda, K.H. Increased expression of periostin and tenascin-C in eyes with neovascular glaucoma secondary to PDR. Graefe’s Arch. Clin. Exp. Ophthalmol. = Albrecht Von Graefes Arch. Fur Klin. Und Exp. Ophthalmol. 2020, 258, 621–628. [Google Scholar] [CrossRef]
- Bertolino, P.; Deckers, M.; Lebrin, F.; ten Dijke, P. Transforming growth factor-β signal transduction in angiogenesis and vascular disorders. Chest 2005, 128, 585S–590S. [Google Scholar] [CrossRef]
- Aubert, A.; Mercier-Gouy, P.; Aguero, S.; Berthier, L.; Liot, S.; Prigent, L.; Alcaraz, L.B.; Verrier, B.; Terreux, R.; Moali, C.; et al. Latent TGF-β Activation Is a Hallmark of the Tenascin Family. Front. Immunol. 2021, 12, 613438. [Google Scholar] [CrossRef]
- Sun, Z.; Velázquez-Quesada, I.; Murdamoothoo, D.; Ahowesso, C.; Yilmaz, A.; Spenlé, C.; Averous, G.; Erne, W.; Oberndorfer, F.; Oszwald, A.; et al. Tenascin-C increases lung metastasis by impacting blood vessel invasions. Matrix Biol. J. Int. Soc. Matrix Biol. 2019, 83, 26–47. [Google Scholar] [CrossRef]
- Margolis, R.; Budd, G.T.; Singh, A.D. Unusual macular degeneration following breast cancer. Acta Ophthalmol. Scand. 2007, 85, 686–687. [Google Scholar] [CrossRef]
- González, Á.F.-V. Vitelliform retinopathy associated with breast cancer. Arch. Soc. Esp. Oftalmol. 2018, 93, e53–e54. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Yoshida, S.; Zhou, Y.; Nakama, T.; Ishikawa, K.; Kubo, Y.; Arima, M.; Nakao, S.; Hisatomi, T.; Ikeda, Y.; et al. Tenascin-C secreted by transdifferentiated retinal pigment epithelial cells promotes choroidal neovascularization via integrin αV. Lab. Investig. 2016, 96, 1178–1188. [Google Scholar] [CrossRef] [Green Version]
- Maisonpierre, P.C.; Suri, C.; Jones, P.F.; Bartunkova, S.; Wiegand, S.J.; Radziejewski, C.; Compton, D.; McClain, J.; Aldrich, T.H.; Papadopoulos, N.; et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science 1997, 277, 55–60. [Google Scholar] [CrossRef]
- Park, Y.S.; Kim, N.H.; Jo, I. Hypoxia and vascular endothelial growth factor acutely up-regulate angiopoietin-1 and Tie2 mRNA in bovine retinal pericytes. Microvasc. Res. 2003, 65, 125–131. [Google Scholar] [CrossRef]
- Thygesen, K.; Alpert, J.S.; White, H.D. Universal definition of myocardial infarction. J. Am. Coll. Cardiol. 2007, 50, 2173–2195. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, I.F.; Acar, E.; Costantino, S.; Szabo, P.L.; Hamza, O.; Tretter, E.V.; Klein, K.U.; Trojanek, S.; Abraham, D.; Paneni, F.; et al. Epigenetic modulation of tenascin C in the heart: Implications on myocardial ischemia, hypertrophy and metabolism. J. Hypertens. 2019, 37, 1861–1870. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Mulholland, E.J.; Dunne, N.; McCarthy, H.O. MicroRNA as Therapeutic Targets for Chronic Wound Healing. Mol. Ther. Nucleic Acids 2017, 8, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.M.; Tao, J.; Chen, D.D.; Cai, J.J.; Irani, K.; Wang, Q.; Yuan, H.; Chen, A.F. MicroRNA miR-27b rescues bone marrow-derived angiogenic cell function and accelerates wound healing in type 2 diabetes mellitus. Arterioscler. Thromb Vasc. Biol. 2014, 34, 99–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, W.; Jiao, J.; Cheng, C.; Shao, J. MicroRNA-21 in the Pathogenesis of Traumatic Brain Injury. Neurochem. Res. 2018, 43, 1863–1868. [Google Scholar] [CrossRef] [PubMed]
- Hodges, W.M.; O’Brien, F.; Fulzele, S.; Hamrick, M.W. Function of microRNAs in the Osteogenic Differentiation and Therapeutic Application of Adipose-Derived Stem Cells (ASCs). Int. J. Mol. Sci. 2017, 18, 2597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moghaddam, A.S.; Afshari, J.T.; Esmaeili, S.A.; Saburi, E.; Joneidi, Z.; Momtazi-Borojeni, A.A. Cardioprotective microRNAs: Lessons from stem cell-derived exosomal microRNAs to treat cardiovascular disease. Atherosclerosis 2019, 285, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Dang, S.; Zhang, R.; Tian, S.; Hou, P.; Li, G.; Ji, M. MicroRNA-218 inhibits the malignant phenotypes of glioma by modulating the TNC/AKT/AP-1/TGFβ1 feedback signaling loop. Int. J. Mol. Med. 2021, 48, 205. [Google Scholar] [CrossRef]
- Chen, L.; Chen, X.R.; Zhang, R.; Li, P.; Liu, Y.; Yan, K.; Jiang, X.D. MicroRNA-107 inhibits glioma cell migration and invasion by modulating Notch2 expression. J. Neuro-Oncol. 2013, 112, 59–66. [Google Scholar] [CrossRef]
- Xi, L. Pigment Epithelium-Derived Factor as a Possible Treatment Agent for Choroidal Neovascularization. Oxidative Med. Cell. Longev. 2020, 2020, 8941057. [Google Scholar] [CrossRef]
- Yamagishi, S.I.; Matsui, T. Pigment Epithelium-Derived Factor: A Novel Therapeutic Target for Cardiometabolic Diseases and Related Complications. Curr. Med. Chem. 2018, 25, 1480–1500. [Google Scholar] [CrossRef]
- Rychli, K.; Huber, K.; Wojta, J. Pigment epithelium-derived factor (PEDF) as a therapeutic target in cardiovascular disease. Expert Opin. Ther. Targets 2009, 13, 1295–1302. [Google Scholar] [CrossRef]
- Falero-Perez, J.; Park, S.; Sorenson, C.M.; Sheibani, N. PEDF expression affects retinal endothelial cell proangiogenic properties through alterations in cell adhesive mechanisms. Am. J. Physiol. Cell Physiol. 2017, 313, C405–C420. [Google Scholar] [CrossRef]
- Shin, E.S.; Sorenson, C.M.; Sheibani, N. PEDF expression regulates the proangiogenic and proinflammatory phenotype of the lung endothelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L620–L634. [Google Scholar] [CrossRef] [Green Version]
- Ma, B.; Khazali, A.; Wells, A. CXCR3 in carcinoma progression. Histol. Histopathol. 2015, 30, 781–792. [Google Scholar] [CrossRef]
- Tokunaga, R.; Zhang, W.; Naseem, M.; Puccini, A.; Berger, M.D.; Soni, S.; McSkane, M.; Baba, H.; Lenz, H.J. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation—A target for novel cancer therapy. Cancer Treat. Rev. 2018, 63, 40–47. [Google Scholar] [CrossRef]
- Yates, C.C.; Rodrigues, M.; Nuschke, A.; Johnson, Z.I.; Whaley, D.; Stolz, D.; Newsome, J.; Wells, A. Multipotent stromal cells/mesenchymal stem cells and fibroblasts combine to minimize skin hypertrophic scarring. Stem Cell Res. Ther. 2017, 8, 193. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Mediators | Receptors/ Ligands | Functions |
|---|---|---|
| Pro-Angiogenesis Phage | ||
| VEGF | VEGFR-1, VEGFR-2 | Cell proliferation, migration, vascular permeability |
| TGF-β | TβR | Cell proliferation, differentiation; HIF expression |
| SDF-1 | CXCR4 | Cell survival, proliferation, chemotaxis |
| Ang | TIE2 | Cell adhesion, migration, and homing |
| miRNA | Cell movement, migration | |
| Anti-Angiogenesis Phage | ||
| PEDF | PEDFR | Cell adhesion, migration, differentiation |
| CXCR3 | CXCL9/10/11 | Cell growth, chemotaxis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Wang, G.; Liu, H. Tenascin-C: A Key Regulator in Angiogenesis during Wound Healing. Biomolecules 2022, 12, 1689. https://doi.org/10.3390/biom12111689
Wang Y, Wang G, Liu H. Tenascin-C: A Key Regulator in Angiogenesis during Wound Healing. Biomolecules. 2022; 12(11):1689. https://doi.org/10.3390/biom12111689
Chicago/Turabian StyleWang, Yucai, Guangfu Wang, and Hao Liu. 2022. "Tenascin-C: A Key Regulator in Angiogenesis during Wound Healing" Biomolecules 12, no. 11: 1689. https://doi.org/10.3390/biom12111689
APA StyleWang, Y., Wang, G., & Liu, H. (2022). Tenascin-C: A Key Regulator in Angiogenesis during Wound Healing. Biomolecules, 12(11), 1689. https://doi.org/10.3390/biom12111689