
Identification of the Kinase-Substrate Recognition Interface between MYPT1 and Rho-Kinase


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Chemicals
2.2. Protein Preparation
2.3. Pull-Down Assay
2.4. Analysis of Phosphorylation of GFP-MYPT1 in COS7 Cells
2.5. In Vitro Kinase Assay
2.6. Immunocytochemistry
2.7. Statistics
3. Results
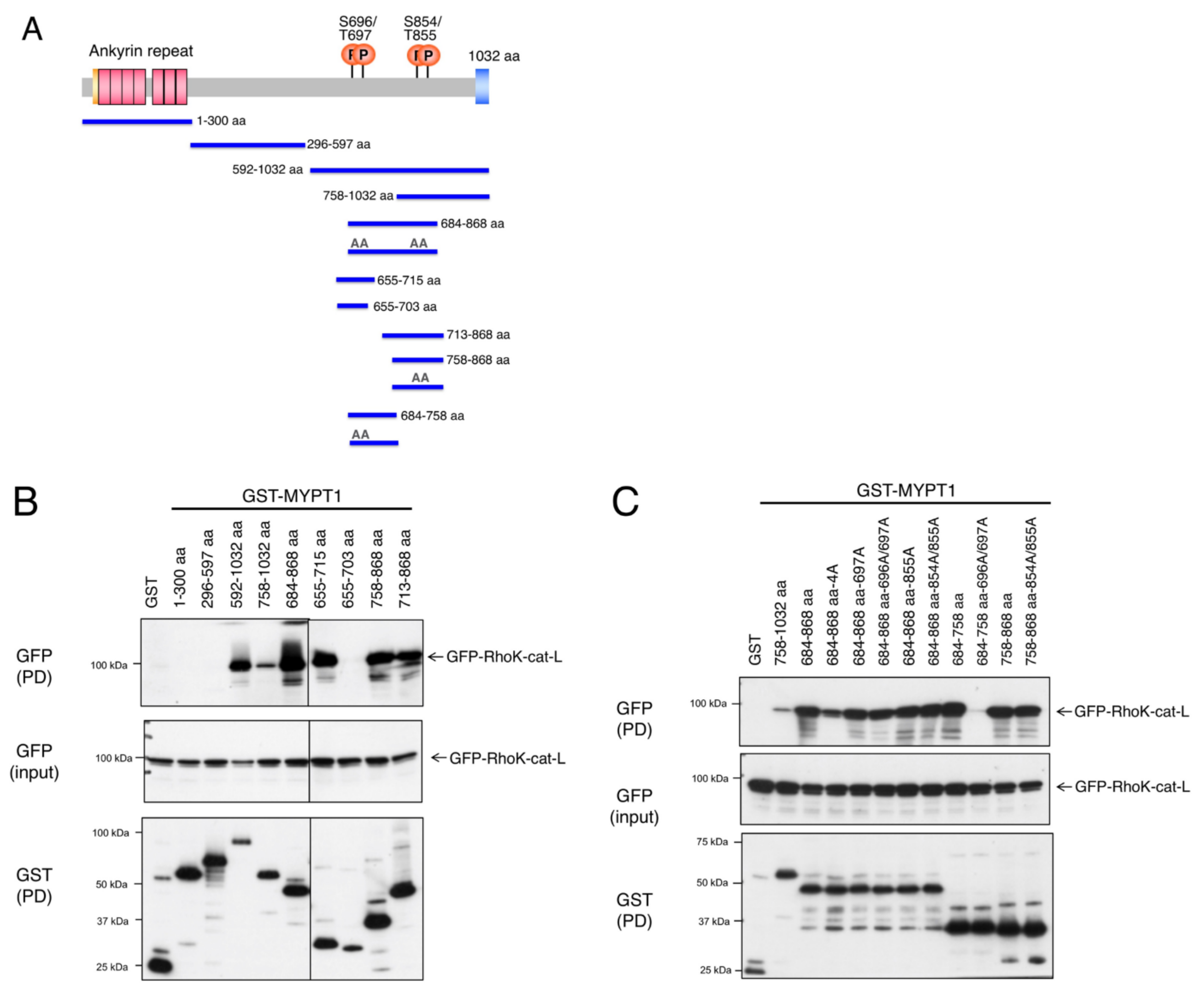
3.1. Interaction of MYPT1 with Rho-Kinase
3.2. Identification of the Docking Motif of MYPT1
3.3. Inhibition of Rho-Kinase by the Inhibitory Fragment/Peptide of MYPT1
3.4. Effects of Docking Motifs on the Phosphorylation of MYPT1
3.5. Kinase-Substrate Interaction of Rho-Kinase
4. Discussion
4.1. Rho-Kinase-MYPT1 Interaction
4.2. Docking Motifs of MYPT1
4.3. Inhibitors of Rho-Kinase Derived from Docking Motifs
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ubersax, J.A.; Ferrell, J.E., Jr. Mechanisms of specificity in protein phosphorylation. Nat. Rev. Mol. Cell Biol. 2007, 8, 530–541. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, P.S.L.; Ferraz, F.A.N.; Pena, D.A.; Pramio, D.T.; Morais, F.A.; Schechtman, D. Revisiting protein kinase–substrate interactions: Toward therapeutic development. Sci. Signal. 2016, 9, re3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amano, M.; Nishioka, T.; Tsuboi, D.; Kuroda, K.; Funahashi, Y.; Yamahashi, Y.; Kaibuchi, K. Comprehensive analysis of kinase-oriented phospho-signalling pathways. J. Biochem. 2019, 165, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Garai, A.; Zeke, A.; Gógl, G.; Törő, I.; Fördős, F.; Blankenburg, H.; Bárkai, T.; Varga, J.; Alexa, A.; Emig, D.; et al. Specificity of Linear Motifs That Bind to a Common Mitogen-Activated Protein Kinase Docking Groove. Sci. Signal. 2012, 5, ra74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peti, W.; Page, R. Molecular basis of MAP kinase regulation. Protein Sci. 2013, 22, 1698–1710. [Google Scholar] [CrossRef] [PubMed]
- Kaidanovich-Beilin, O.; Eldar-Finkelman, H. Peptides Targeting Protein Kinases: Strategies and Implications. Physiology 2006, 21, 411–418. [Google Scholar] [CrossRef]
- Amano, M.; Hamaguchi, T.; Shohag, M.H.; Kozawa, K.; Kato, K.; Zhang, X.; Yura, Y.; Matsuura, Y.; Kataoka, C.; Nishioka, T.; et al. Kinase-interacting substrate screening is a novel method to identify kinase substrates. J. Cell Biol. 2015, 209, 895–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yura, Y.; Amano, M.; Takefuji, M.; Bando, T.; Suzuki, K.; Kato, K.; Hamaguchi, T.; Shohag, H.; Takano, T.; Funahashi, Y.; et al. Focused proteomics revealed a novel Rho-kinase signaling pathway in the heart. Cell Struct. Funct. 2016, 41, 105–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amano, M.; Nishioka, T.; Yura, Y.; Kaibuchi, K. Identification of Protein Kinase Substrates by the Kinase-Interacting Substrate Screening (KISS) Approach. Curr. Protoc. Cell Biol. 2016, 72, 14.16.1–14.16.12. [Google Scholar] [CrossRef] [PubMed]
- Narumiya, S.; Thumkeo, D. Rho signaling research: History, current status and future directions. FEBS Lett. 2018, 592, 1763–1776. [Google Scholar] [CrossRef] [Green Version]
- Shahbazi, R.; Baradaran, B.; Khordadmehr, M.; Safaei, S.; Baghbanzadeh, A.; Jigari, F.; Ezzati, H. Targeting ROCK signaling in health, malignant and non-malignant diseases. Immunol. Lett. 2020, 219, 15–26. [Google Scholar] [CrossRef]
- Shimokawa, H. Reactive oxygen species in cardiovascular health and disease: Special references to nitric oxide, hydrogen peroxide, and Rho-kinase. J. Clin. Biochem. Nutr. 2020, 66, 83–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.B.; Lograsso, P.V.; Defert, O.; Li, R.S. Rho Kinase (ROCK) Inhibitors and Their Therapeutic Potential. J. Med. Chem. 2015, 59, 2269–2300. [Google Scholar] [CrossRef]
- Riento, K.; Ridley, A.J. ROCKs: Multifunctional kinases in cell behaviour. Nat. Rev. Mol. Cell Biol. 2003, 4, 446–456. [Google Scholar] [CrossRef]
- Ito, M.; Nakano, T.; Erdödi, F.; Hartshorne, D.J. Myosin phosphatase: Structure, regulation and function. Mol. Cell. Biochem. 2004, 259, 197–209. [Google Scholar] [CrossRef]
- Amano, M.; Nakayama, M.; Kaibuchi, K. Rho-kinase/ROCK: A key regulator of the cytoskeleton and cell polarity. Cytoskeleton 2010, 67, 545–554. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zheng, X.R.; Riddick, N.; Bryden, M.; Baur, W.; Zhang, X.; Surks, H.K. ROCK Isoform Regulation of Myosin Phosphatase and Contractility in Vascular Smooth Muscle Cells. Circ. Res. 2009, 104, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, S.; Nagata, S. pEF-BOS, a powerful mammalian expression vector. Nucleic Acids Res. 1990, 18, 5322. [Google Scholar] [CrossRef] [PubMed]
- Amano, M.; Chihara, K.; Nakamura, N.; Kaneko, T.; Matsuura, Y.; Kaibuchi, K. The COOH Terminus of Rho-kinase Negatively Regulates Rho-kinase Activity. J. Biol. Chem. 1999, 274, 32418–32424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Khromov, A.; Choudhury, N.; Stevenson, A.S.; Somlyo, A.V.; Eto, M. Phosphorylation-dependent Autoinhibition of Myosin Light Chain Phosphatase Accounts for Ca2+ Sensitization Force of Smooth Muscle Contraction. J. Biol. Chem. 2009, 284, 21569–21579. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, H.; Kasa, M.; Amano, M.; Kaibuchi, K.; Hakoshima, T. Molecular Mechanism for the Regulation of Rho-Kinase by Dimerization and Its Inhibition by Fasudil. Structure 2006, 14, 589–600. [Google Scholar] [CrossRef] [Green Version]
- Gold, M.G.; Barford, D.; Komander, D. Lining the pockets of kinases and phosphatases. Curr. Opin. Struct. Biol. 2006, 16, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, D.; Glossip, D.; Xing, H.; Muslin, A.J.; Kornfeld, K. Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK MAP kinase. Genes Dev. 1999, 13, 163–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, S.; Iwaoka, R.; Eto, M.; Ohki, S.-Y. Solution structure of the inhibitory phosphorylation domain of myosin phosphatase targeting subunit 1. Proteins Struct. Funct. Bioinform. 2009, 77, 732–735. [Google Scholar] [CrossRef] [Green Version]
- Qvit, N.; Kornfeld, O.S.; Mochly-Rosen, D. Engineered Substrate-Specific Delta PKC Antagonists to Enhance Cardiac Therapeutics. Angew. Chem. Int. Ed. 2016, 55, 15672–15679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Imanishi, A.; Komatsu, N.; Terai, K.; Amano, M.; Kaibuchi, K.; Matsuda, M. A FRET Biosensor for ROCK Based on a Consensus Substrate Sequence Identified by KISS Technology. Cell Struct. Funct. 2017, 42, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Amino Acid | Sequence |
|---|---|---|
| DM1 | 704–715 aa | DLQEAEKTIGRS |
| PS1 | 692–703 aa | QSRRAAQGVTLT |
| PS1 + DM1 | 692–715 aa | QSRRAAQGVTLTDLQEAEKTIGRS |
| DM2 | 836–849 aa | KSQPKSIRERRRPR |
| PS2 | 850–860 aa | EKRRAAGVSFW |
| PS2 + DM2 | 836–860 aa | KSQPKSIRERRRPREKRRAAGVSFW |
| DM3 | 683–691 aa | RKARSRQAR |
| PS1 + DM3 | 683–703 aa | RKARSRQARQSRRAAQGVTLT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amano, M.; Kanazawa, Y.; Kozawa, K.; Kaibuchi, K. Identification of the Kinase-Substrate Recognition Interface between MYPT1 and Rho-Kinase. Biomolecules 2022, 12, 159. https://doi.org/10.3390/biom12020159
Amano M, Kanazawa Y, Kozawa K, Kaibuchi K. Identification of the Kinase-Substrate Recognition Interface between MYPT1 and Rho-Kinase. Biomolecules. 2022; 12(2):159. https://doi.org/10.3390/biom12020159
Chicago/Turabian StyleAmano, Mutsuki, Yoko Kanazawa, Kei Kozawa, and Kozo Kaibuchi. 2022. "Identification of the Kinase-Substrate Recognition Interface between MYPT1 and Rho-Kinase" Biomolecules 12, no. 2: 159. https://doi.org/10.3390/biom12020159