
Prediction and Modeling of Protein–Protein Interactions Using “Spotted” Peptides with a Template-Based Approach



Abstract
:1. Introduction
2. Methodology
2.1. Required Inputs
2.2. Multiple Peptides Fragments Generation and Scoring by Alignment Matrices
2.3. Template Based 3D Modeling of Protein–Peptide Complexes
2.4. Scoring and Ranking of Protein–Peptide Complexes with 3D Scoring Functions
2.5. Benchmark Dataset
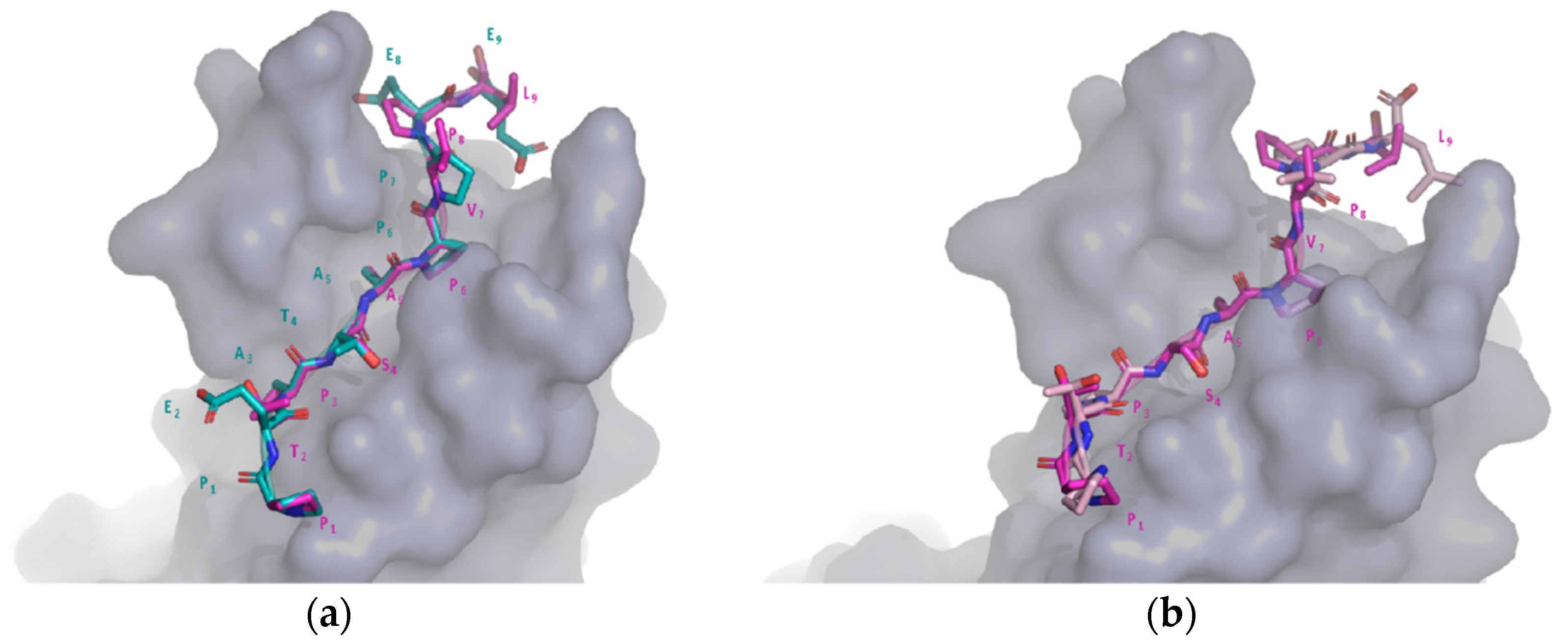
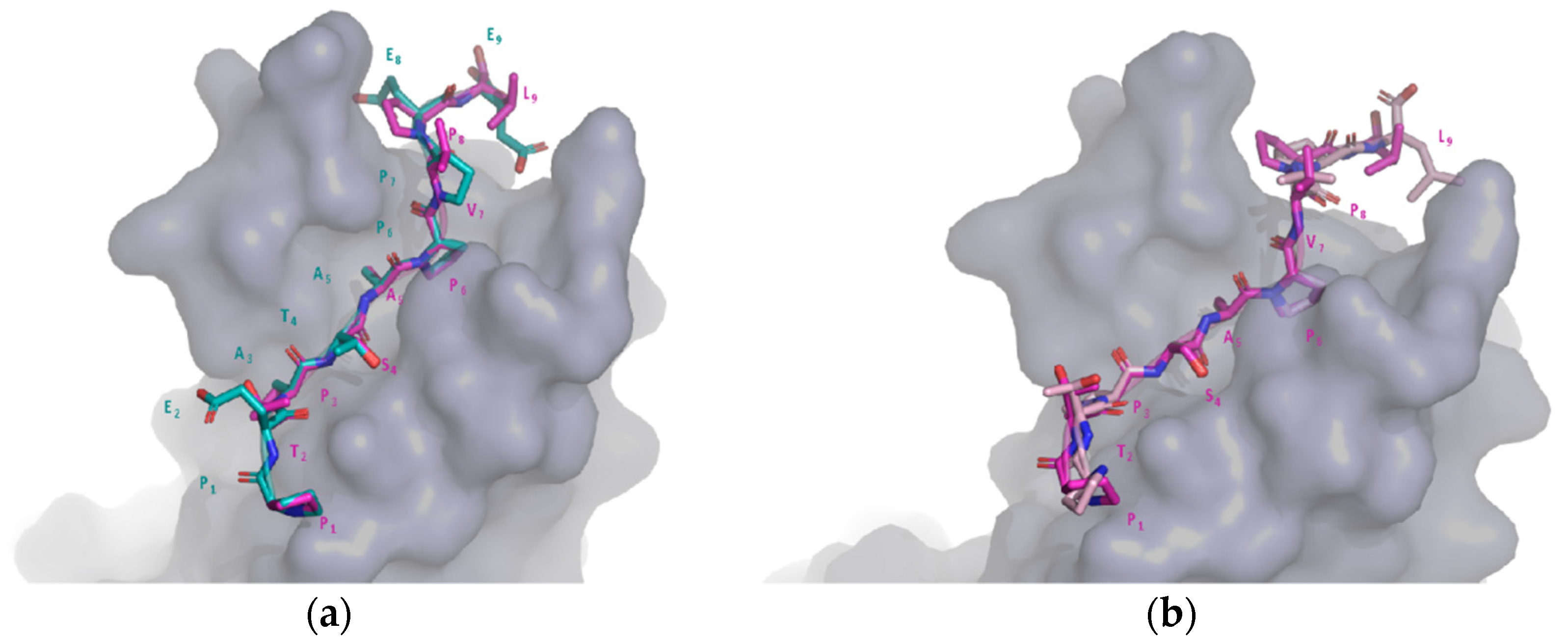
2.6. Comparison of Target and Template Peptide–Receptor Complexes
3. Results and Discussion
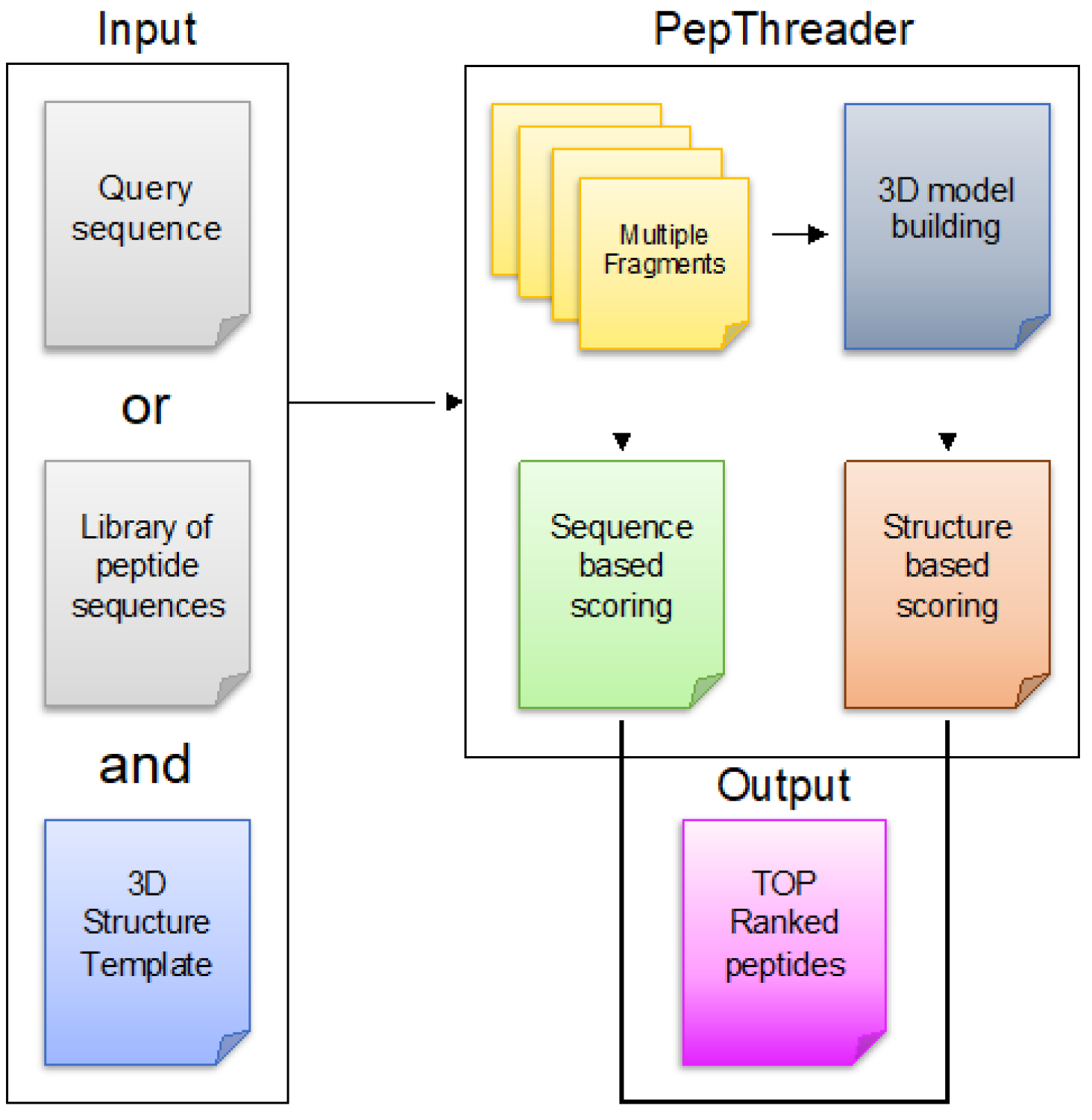
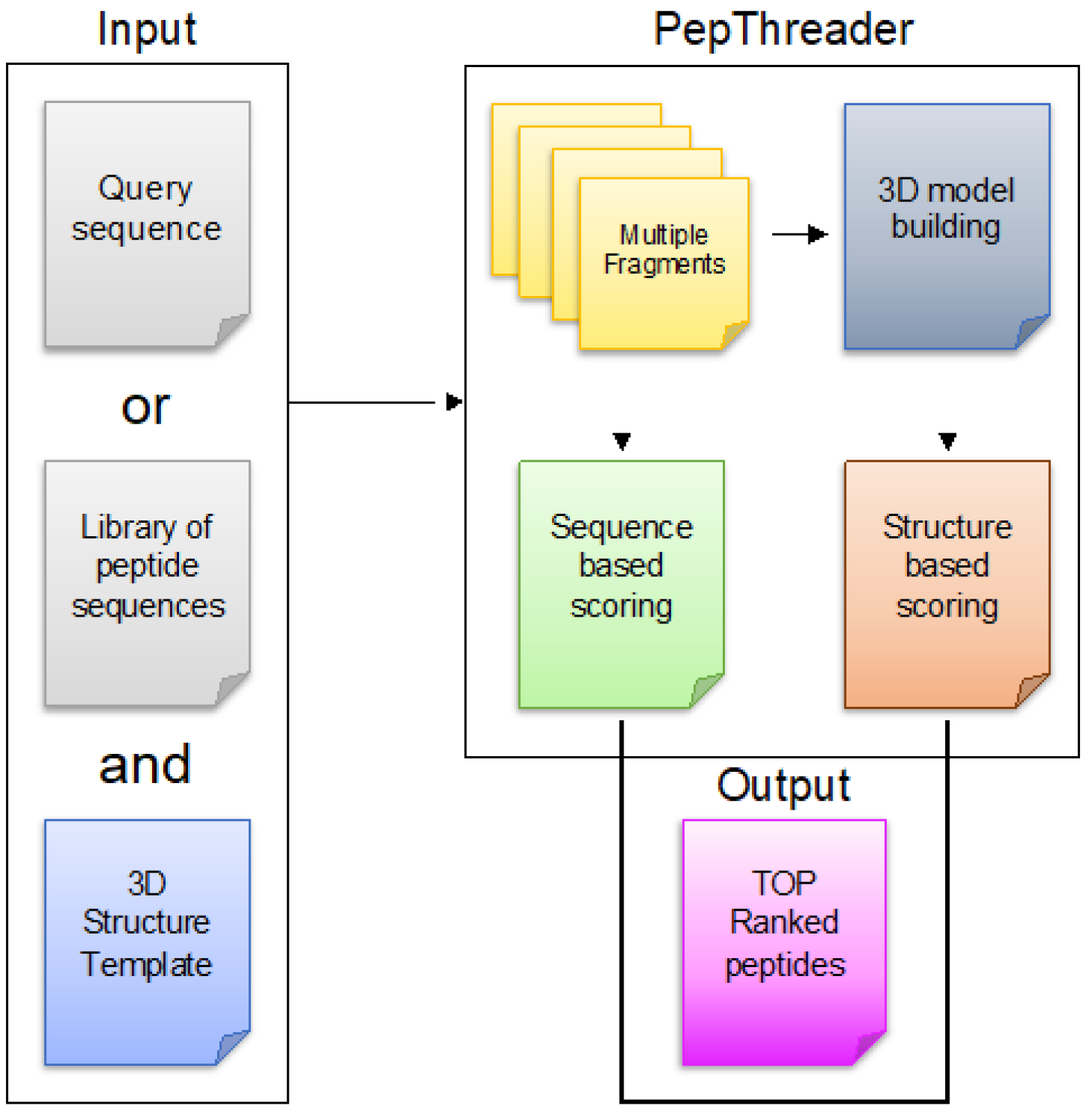
3.1. Overview of PepThreader
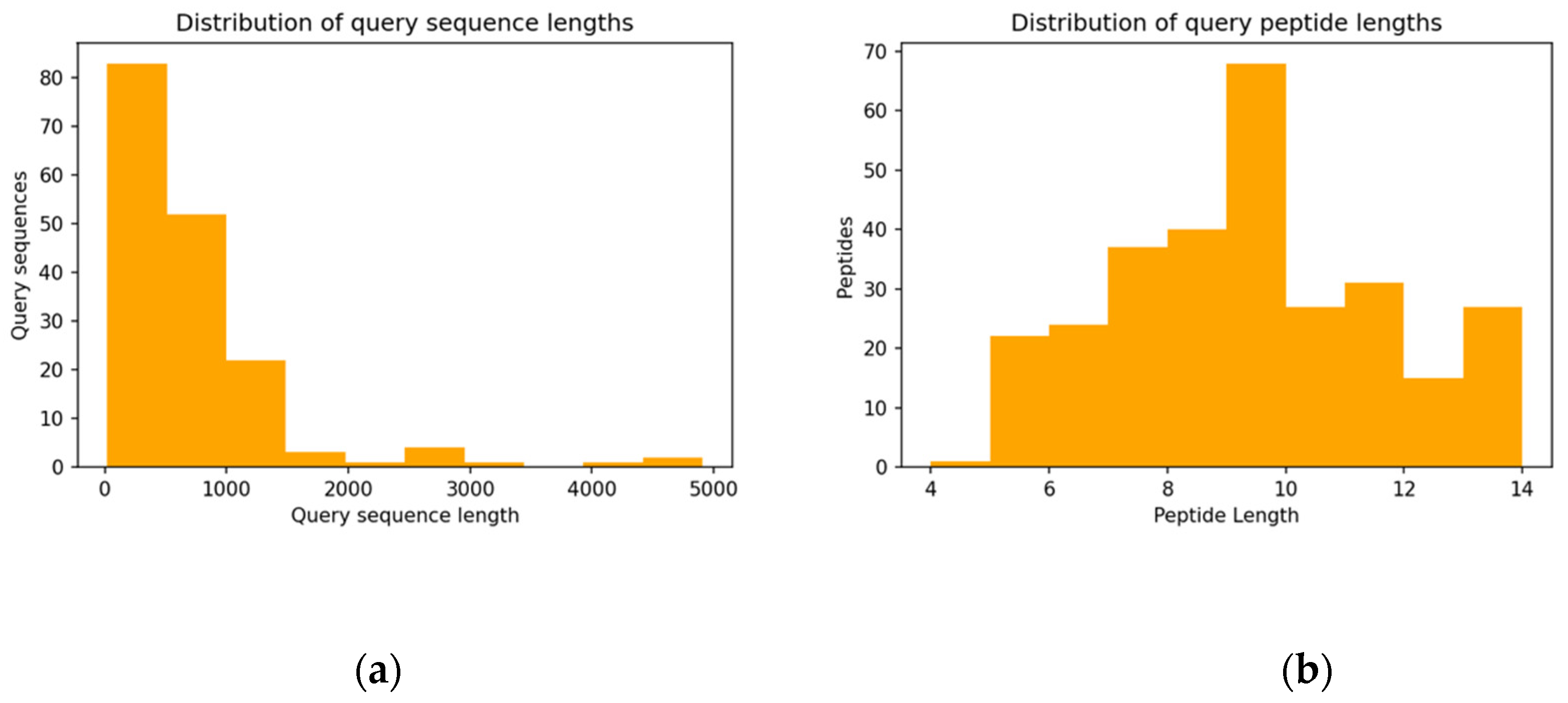
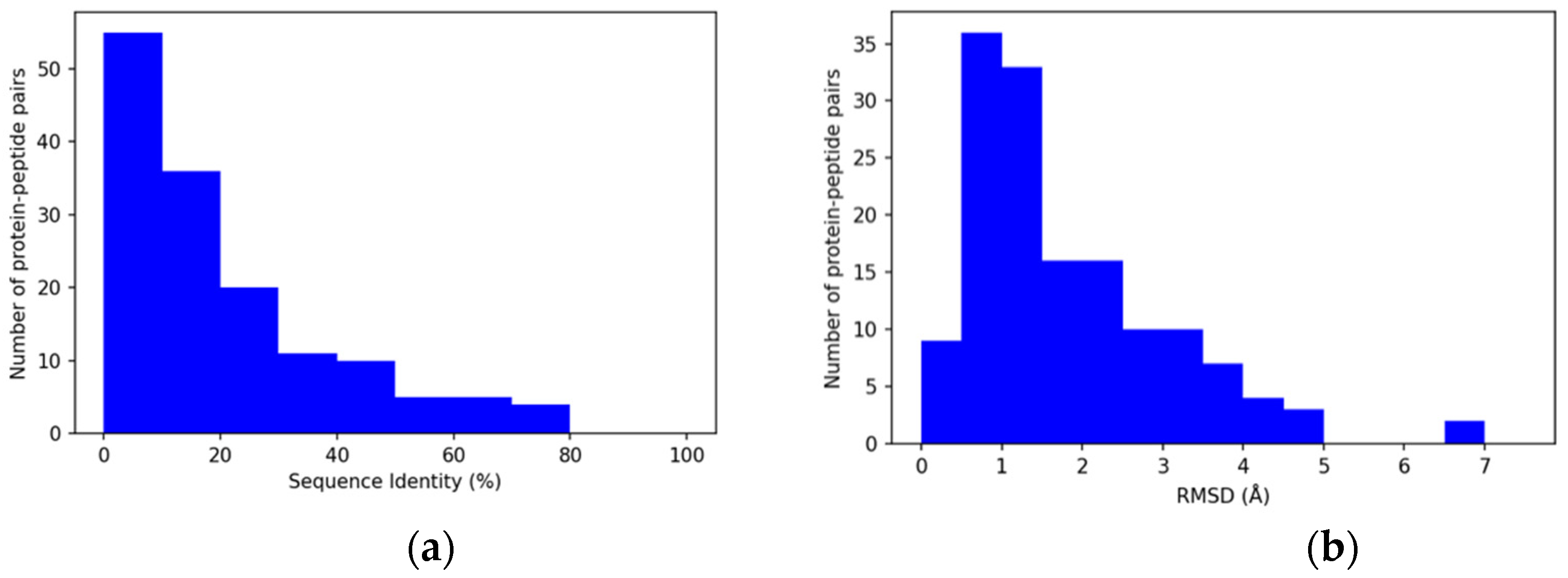
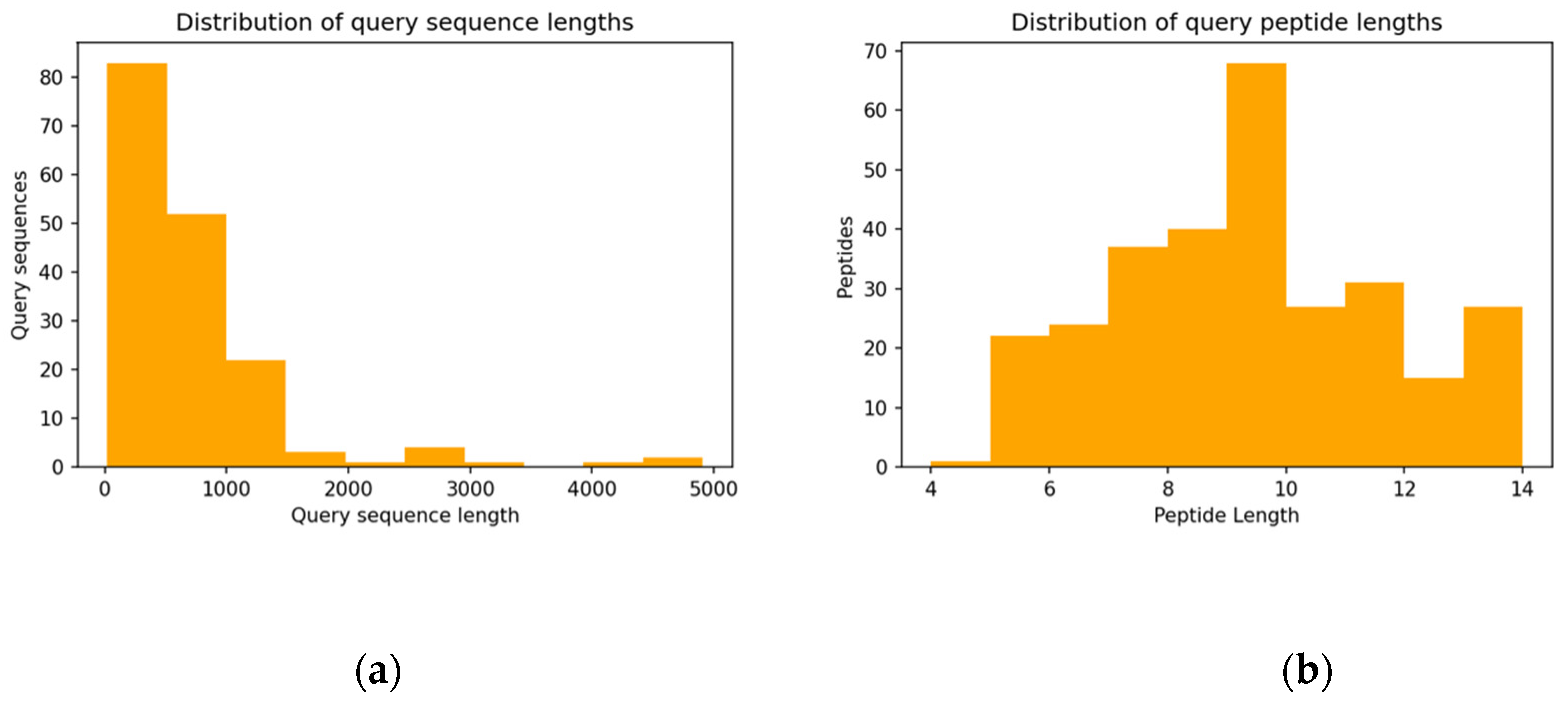
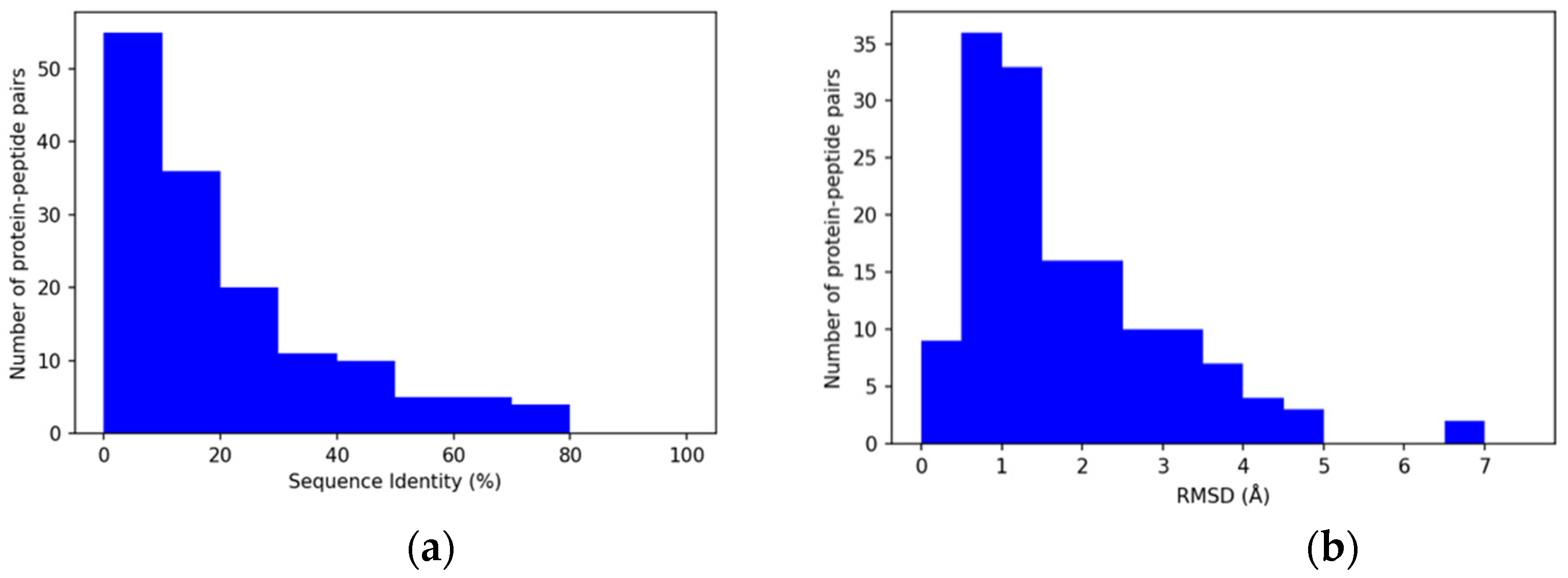
3.2. Properties of Query Sequences and Target–Template Pairs
3.3. Sequence Similarity-Based Score
3.4. Structure-Based Score
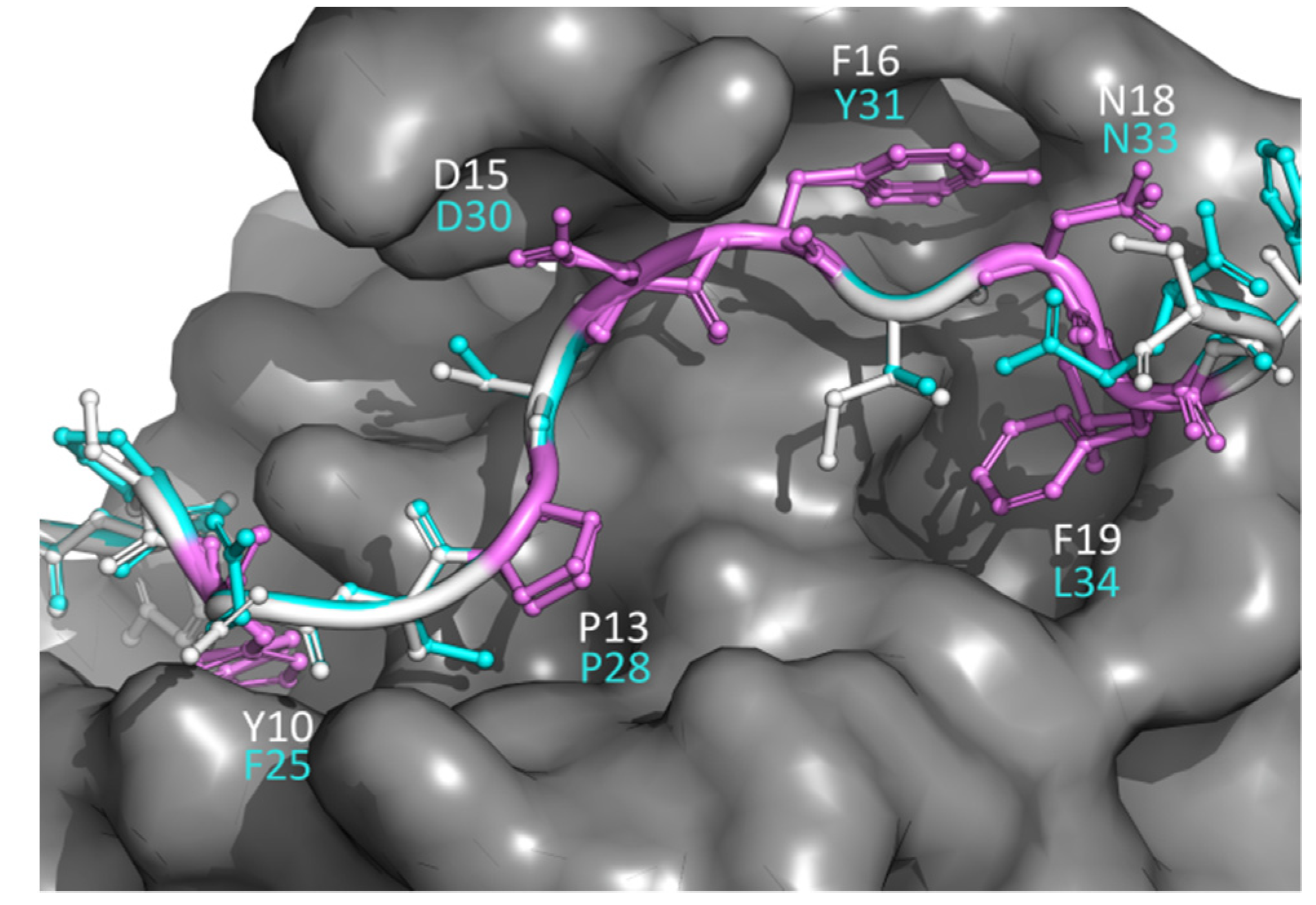
3.5. An Application of PepThreader: The ESCRT-I Interacting Peptides
3.6. An Application of PepThreader: Identifying the Critical Site of Bora to Bind and Activate Aurora-A
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carbonell, P.; Nussinov, R.; Del Sol, A. Energetic determinants of protein binding specificity: Insights into protein interaction networks. Proteomics 2009, 9, 1744–1753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilofer, C.; Sukhwal, A.; Mohanapriya, A.; Kangueane, P. Open access Volume 13(6) Hypothesis Protein-protein interfaces are vdW dominant with selective H-bonds and (or) electrostatics towards broad functional specificity Open access. Bioinformation 2017, 13, 164–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawyer, N.; Watkins, A.M.; Arora, P.S. Protein Domain Mimics as Modulators of Protein-Protein Interactions. Acc. Chem. Res. 2017, 50, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Seychell, B.C.; Beck, T. Molecular basis for protein-protein interactions. Beilstein J. Org. Chem. 2021, 17, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Powell, H.R. From then till now: Changing data collection methods in single crystal X-ray crystallography since 1912. Crystallogr. Rev. 2019, 25, 264–294. [Google Scholar] [CrossRef]
- McPherson, A.; Cudney, B. Optimization of crystallization conditions for biological macromolecules. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2014, 70, 1445–1467. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, M.; Pinto, C.; Houben, K.; Baldus, M. Nuclear magnetic resonance (NMR) applied to membrane–protein complexes. Q. Rev. Biophys. 2016, 49, e15. [Google Scholar] [CrossRef]
- Frank, J. Advances in the field of single-particle cryo-electron microscopy over the last decade. Nat. Protoc. 2017, 12, 209–212. [Google Scholar] [CrossRef]
- Rappsilber, J.; Siniossoglou, S.; Hurt, E.C.; Mann, M. A generic strategy to analyze the spatial organization of multi-protein complexes by cross-linking and mass spectrometry. Anal. Chem. 2000, 72, 267–275. [Google Scholar] [CrossRef]
- Masson, G.R.; Burke, J.E.; Ahn, N.G.; Anand, G.S.; Borchers, C.; Brier, S.; Bou-Assaf, G.M.; Engen, J.R.; Englander, S.W.; Faber, J.; et al. Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (HDX-MS) experiments. Nat. Methods 2019, 16, 595–602. [Google Scholar] [CrossRef] [Green Version]
- Myszka, D.G.; Rich, R.L. Implementing surface plasmon resonance biosensors in drug discovery. Pharm. Sci. Technol. Today 2000, 3, 310–317. [Google Scholar] [CrossRef]
- Velazquez-Campoy, A.; Leavitt, S.A.; Freire, E. Characterization of Protein-Protein Interactions by Isothermal Titration Calorimetry. In Protein-Protein Interactions: Methods and Applications: Second Edition; Humana Press: New York, NY, USA, 2015; pp. 183–204. [Google Scholar]
- Zong, H.; Wang, X.; Mu, X.; Wang, J.; Sun, M. Plasmon-Enhanced Fluorescence Resonance Energy Transfer. Chem. Rec. 2019, 19, 818–842. [Google Scholar] [CrossRef]
- Jerabek-Willemsen, M.; André, T.; Wanner, R.; Roth, H.M.; Duhr, S.; Baaske, P.; Breitsprecher, D. MicroScale Thermophoresis: Interaction analysis and beyond. J. Mol. Struct. 2014, 1077, 101–113. [Google Scholar] [CrossRef] [Green Version]
- Benyamini, H.; Friedler, A. Using peptides to study protein-protein interactions. Future Med. Chem. 2010, 2, 989–1003. [Google Scholar] [CrossRef] [Green Version]
- Merrifield, R.B. Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- Frank, R. Spot-synthesis: An easy technique for the positionally addressable, parallel chemical synthesis on a membrane support. Tetrahedron 1992, 48, 9217–9232. [Google Scholar] [CrossRef]
- Cretich, M.; Damin, F.; Pirri, G.; Chiari, M. Protein and peptide arrays: Recent trends and new directions. Biomol. Eng. 2006, 23, 77–88. [Google Scholar] [CrossRef]
- Cunningham, A.D.; Qvit, N.; Mochly-Rosen, D. Peptides and peptidomimetics as regulators of protein–protein interactions. Curr. Opin. Struct. Biol. 2017, 44, 59–66. [Google Scholar] [CrossRef]
- Li, H.; Lu, L.; Chen, R.; Quan, L.; Xia, X.; Lü, Q. PaFlexPepDock: Parallel Ab-initio docking of peptides onto their receptors with full flexibility based on Rosetta. PLoS ONE 2014, 9, e94769. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Yan, C.; Zou, X. MDockPeP: An ab-initio protein–peptide docking server. J. Comput. Chem. 2018, 39, 2409–2413. [Google Scholar] [CrossRef]
- Lamiable, A.; Thévenet, P.; Rey, J.; Vavrusa, M.; Derreumaux, P.; Tufféry, P. PEP-FOLD3: Faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016, 44, 1731–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- London, N.; Raveh, B.; Cohen, E.; Fathi, G.; Schueler-Furman, O. Rosetta FlexPepDock web server—High resolution modeling of peptide-protein interactions. Nucleic Acids Res. 2011, 39, 249–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez, C.; Boelens, R.; Bonvin, A.M.J.J. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 2409–2413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; Van Dijk, M.; De Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Heo, L.; Lee, M.S.; Seok, C. GalaxyPepDock: A protein-peptide docking tool based on interaction similarity and energy optimization. Nucleic Acids Res. 2015, 43, W431–W435. [Google Scholar] [CrossRef] [Green Version]
- Burley, S.K.; Berman, H.M.; Kleywegt, G.J.; Markley, J.L.; Nakamura, H.; Velankar, S. Protein Data Bank (PDB): The single global macromolecular structure archive. Methods Mol. Biol. 2017, 1607, 627–641. [Google Scholar] [CrossRef] [Green Version]
- Janin, J.; Henrick, K.; Moult, J.; Eyck, L.T.; Sternberg, M.J.E.; Vajda, S.; Vakser, I.; Wodak, S.J. CAPRI: A critical assessment of PRedicted interactions. Proteins Struct. Funct. Genet. 2003, 52, 2–9. [Google Scholar] [CrossRef]
- Xue, L.C.; Jordan, R.A.; Yasser, E.M.; Dobbs, D.; Honavar, V. DockRank: Ranking docked conformations using partner-specific sequence homology-based protein interface prediction. Proteins Struct. Funct. Bioinform. 2014, 82, 250–267. [Google Scholar] [CrossRef] [Green Version]
- Xue, L.C.; Rodrigues, J.P.G.L.M.; Dobbs, D.; Honavar, V.; Bonvin, A.M.J.J. Template-based protein-protein docking exploiting pairwise interfacial residue restraints. Brief. Bioinform. 2017, 18, 458–466. [Google Scholar] [CrossRef] [Green Version]
- Sinha, R.; Kundrotas, P.J.; Vakser, I.A. Docking by structural similarity at protein-protein interfaces. Proteins Struct. Funct. Bioinform. 2010, 78, 3235–3241. [Google Scholar] [CrossRef]
- London, N.; Movshovitz-Attias, D.; Schueler-Furman, O. The Structural Basis of Peptide-Protein Binding Strategies. Structure 2010, 18, 188–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekmekci, B.; McAnany, C.E.; Mura, C. An Introduction to Programming for Bioscientists: A Python-Based Primer. PLoS Comput. Biol. 2016, 12, e1004867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef] [PubMed]
- McKinney, W. Data Structures for Statistical Computing in Python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28 June–3 July 2010. [Google Scholar]
- Eddy, S.R. Where did the BLOSUM62 alignment score matrix come from? Nat. Biotechnol. 2004, 22, 1035–1036. [Google Scholar] [CrossRef]
- Qu, C.X.; Lai, L.H.; Xu, X.J.; Tang, Y.Q. Phyletic relationships of protein structures based on spatial preference of residues. J. Mol. Evol. 1993, 36, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinform. 2016, 15, 5–6. [Google Scholar] [CrossRef] [Green Version]
- Dong, G.Q.; Fan, H.; Schneidman-Duhovny, D.; Webb, B.; Sali, A.; Tramontano, A. Optimized atomic statistical potentials: Assessment of protein interfaces and loops. Bioinformatics 2013, 29, 3158–3166. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [Green Version]
- Olechnovič, K.; Venclovas, Č. VoroMQA: Assessment of protein structure quality using interatomic contact areas. Proteins Struct. Funct. Bioinform. 2017, 85, 1131–1145. [Google Scholar] [CrossRef]
- Frappier, V.; Duran, M.; Keating, A.E. PixelDB: Protein–peptide complexes annotated with structural conservation of the peptide binding mode. Protein Sci. 2018, 27, 276–285. [Google Scholar] [CrossRef] [Green Version]
- Hauser, A.S.; Windshügel, B. LEADS-PEP: A Benchmark Data Set for Assessment of Peptide Docking Performance. J. Chem. Inf. Model. 2016, 56, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zou, X. PepPro: A Nonredundant Structure Data Set for Benchmarking Peptide–Protein Computational Docking. J. Comput. Chem. 2020, 41, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Zou, X. Predicting peptide binding sites on protein surfaces by clustering chemical interactions. J. Comput. Chem. 2014, 36, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Im, Y.J.; Kuo, L.; Ren, X.; Burgos, P.V.; Zhao, X.Z.; Liu, F.; Burke, T.R.; Bonifacino, J.S.; Freed, E.O.; Hurley, J.H. Crystallographic and Functional Analysis of the ESCRT-I/HIV-1 Gag PTAP Interaction. Structure 2010, 18, 1536–1547. [Google Scholar] [CrossRef] [Green Version]
- Demirov, D.G.; Ono, A.; Orenstein, J.M.; Freed, E.O. Overexpression of the N-terminal domain of TSG101 inhibits HIV-1 budding by blocking late domain function. Proc. Natl. Acad. Sci. USA 2002, 99, 955–960. [Google Scholar] [CrossRef] [Green Version]
- Tavernier, N.; Thomas, Y.; Vigneron, S.; Maisonneuve, P.; Orlicky, S.; Mader, P.; Regmi, S.G.; Van Hove, L.; Levinson, N.M.; Gasmi-Seabrook, G.; et al. Bora phosphorylation substitutes in trans for T-loop phosphorylation in Aurora A to promote mitotic entry. Nat. Commun. 2021, 12, 1–22. [Google Scholar] [CrossRef]
- Bayliss, R.; Sardon, T.; Vernos, I.; Conti, E. Structural basis of Aurora-A activation by TPX2 at the mitotic spindle. Mol. Cell 2003, 12, 851–862. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Matrix | Top 1 | Top 10 | Top 25 | Top 50 | Top 75 |
|---|---|---|---|---|---|
| BLOSUM62 | 57/292 (19.5%) | 143/292 (49.0%) | 165/292 (56.5%) | 194/292 (66.4%) | 209/292 (71.6%) |
| BLOSUM62-M | 70/292 (24.0%) | 143/292 (49.0%) | 181/292 (62.0%) | 204/292 (69.9%) | 209/292 (71.6%) |
| BLOSUM62 GALAXY | 69/292 (23.6%) | 143/292 (49.0%) | 185/292 (63.3%) | 200/292 (69.8%) | 217/292 (74.3%) |
| QU3 | 48/292 (16.4%) | 117/292 (40.0%) | 154/292 (52.7%) | 183/292 (62.7%) | 211/292 (72.2%) |
| QU3-M | 65/292 (22.2%) | 138/292 (47.3%) | 177/292 (60.6%) | 200/292 (68.4%) | 214/292 (73.3%) |
| QU3 GALAXY | 61/292 (20.9%) | 139/292 (47.6%) | 178/292 (61.0%) | 201/292 (68.8%) | 214/292 (73.3%) |
| Matrix | Top 10 | Top 25 | Top 50 | Top 75 |
|---|---|---|---|---|
| Length 0–500 | 69/138 (50.0%) | 92/138 (66.6%) | 101/138 (73.2%) | 106/138 (76.8%) |
| Length 500–1000 | 30/79 (38.0%) | 38/79 (48.1%) | 45/79 (57.0%) | 49/79 (62.0%) |
| Length 1000–2000 | 32/58 (55.2%) | 39/58 (67.2%) | 42/58 (72.4%) | 44/58 (75.9%) |
| Length 2000–5000 | 12/17 (70.6%) | 12/17 (70.6%) | 16/17 (94.1%) | 16/17 (94.1%) |
| Matrix | Top 1 | Top 10 | Top 25 | Top 50 |
|---|---|---|---|---|
| SeqId 0–25 | 37/162 (22.8%) | 56/162 (34.6%) | 75/162 (46.3%) | 86/162 (53.1%) |
| SeqId 25–50 | 79/102 (77.4%) | 98/102 (96.1%) | 101/102 (99.0%) | 101/102 (99.0%) |
| SeqId 50–75 | 18/19 (94.7%) | 18/19 (94.7%) | 19/19 (100%) | 19/19 (100%) |
| SeqId 75–100 | 9/9 (100%) | 9/9 (100%) | 9/9 (100%) | 9/9 (100%) |
| Scoring Function | Number of Improved | Average Rank Change | Top 10 after Reranking | Top 25 after Reranking |
|---|---|---|---|---|
| DOPE | 42/127 (33.1%) | −5.9 (13) | 42/127 (33.1%) | 87/127 (68.5%) |
| SOAP-PPI | 68/127 (53.5%) | 0.5 (13) | 71/127 (55.9%) | 102/127 (80.0%) |
| SOAP-PEP | 62/127 (48.8%) | −0.1 (12) | 69/127 (54.3%) | 102/127 (80.0%) |
| voromqa | 51/127 (40.2%) | −1.6 (15) | 57/127 (44.8%) | 96/127 (75.6%) |
| voromqa-int | 50/127 (39.4%) | −2.2 (13) | 56/127 (44.1%) | 97/127 (76.4%) |
| Scoring Function | N. Improved | Average SeqId | Average Rank Change | Top 10 after Reranking | Top 25 after Reranking |
|---|---|---|---|---|---|
| DOPE (L) | 23/77 (29.9%) | 0.23 | −6.5 (13.4) | 42/127 (33.1%) | 87/127 (68.5%) |
| DOPE (M) | 11/36 (30.6%) | 0.16 | −5.9 (16.6) | 71/127 (55.9%) | 102/127 (80.0%) |
| DOPE (H) | 8/14 (57.1%) | 0.13 | −1.6 (8.2) | 69/127 (54.3%) | 102/127 (80.0%) |
| SOAP-PPI (L) | 43/77 (55.8%) | 0.27 | 0.13 (9.9) | 57/127 (44.8%) | 96/127 (75.6%) |
| SOAP-PPI (M) | 17/36 (47.2%) | 0.22 | 1.1 (15.5) | 56/127 (44.1%) | 97/127 (76.4%) |
| SOAP-PPI (H) | 8/14 (57.1%) | 0.09 | 1.1 (9.0) | 7/14 (50.0%) | 11/14 (78.6%) |
| SOAP-PEP (L) | 36/77 (46.8%) | 0.27 | −0.8 (10.5) | 44/77 (57.1%) | 62/77 (80.5%) |
| SOAP-PEP (M) | 18/36 (50.0%) | 0.20 | 1.0 (15.2) | 16/36 (44.4%) | 29/36 (80.6%) |
| SOAP-PEP (H) | 8/14 (57.1%) | 0.10 | 1.0 (8.4) | 9/14 (64.3%) | 11/14 (78.6%) |
| voromqa (L) | 31/77 (40.3%) | 0.22 | −1.7 (12.6) | 39/77 (50.6%) | 61/77 (79.2%) |
| voromqa (M) | 13/36 (36.1%) | 0.19 | −2.3 (18.2) | 13/36 (36.1%) | 24/36 (66.7%) |
| voromqa (H) | 7/14 (50.0%) | 0.11 | 0.71 (13.6) | 5/14 (35.7%) | 11/14 (78.6%) |
| voromqa-int (L) | 28/77 (36.4%) | 0.22 | −2.2 (13.6) | 37/77 (48.1%) | 60/77 (77.9%) |
| voromqa-int (M) | 15/36 (41.6%) | 0.19 | −3.2 (13.8) | 12/36 (33.3%) | 25/36 (69.4%) |
| voromqa-int (H) | 7/14 (50.0%) | 0.11 | 2.92 (16.1) | 7/14 (50.0%) | 12/14 (85.7%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gasbarri, C.; Rosignoli, S.; Janson, G.; Boi, D.; Paiardini, A. Prediction and Modeling of Protein–Protein Interactions Using “Spotted” Peptides with a Template-Based Approach. Biomolecules 2022, 12, 201. https://doi.org/10.3390/biom12020201
Gasbarri C, Rosignoli S, Janson G, Boi D, Paiardini A. Prediction and Modeling of Protein–Protein Interactions Using “Spotted” Peptides with a Template-Based Approach. Biomolecules. 2022; 12(2):201. https://doi.org/10.3390/biom12020201
Chicago/Turabian StyleGasbarri, Chiara, Serena Rosignoli, Giacomo Janson, Dalila Boi, and Alessandro Paiardini. 2022. "Prediction and Modeling of Protein–Protein Interactions Using “Spotted” Peptides with a Template-Based Approach" Biomolecules 12, no. 2: 201. https://doi.org/10.3390/biom12020201
APA StyleGasbarri, C., Rosignoli, S., Janson, G., Boi, D., & Paiardini, A. (2022). Prediction and Modeling of Protein–Protein Interactions Using “Spotted” Peptides with a Template-Based Approach. Biomolecules, 12(2), 201. https://doi.org/10.3390/biom12020201