
microRNA-Mediated Encoding and Decoding of Time-Dependent Signals in Tumorigenesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Feedforward Loops and Feedback Loops Interpret Temporally Encoded Patterns
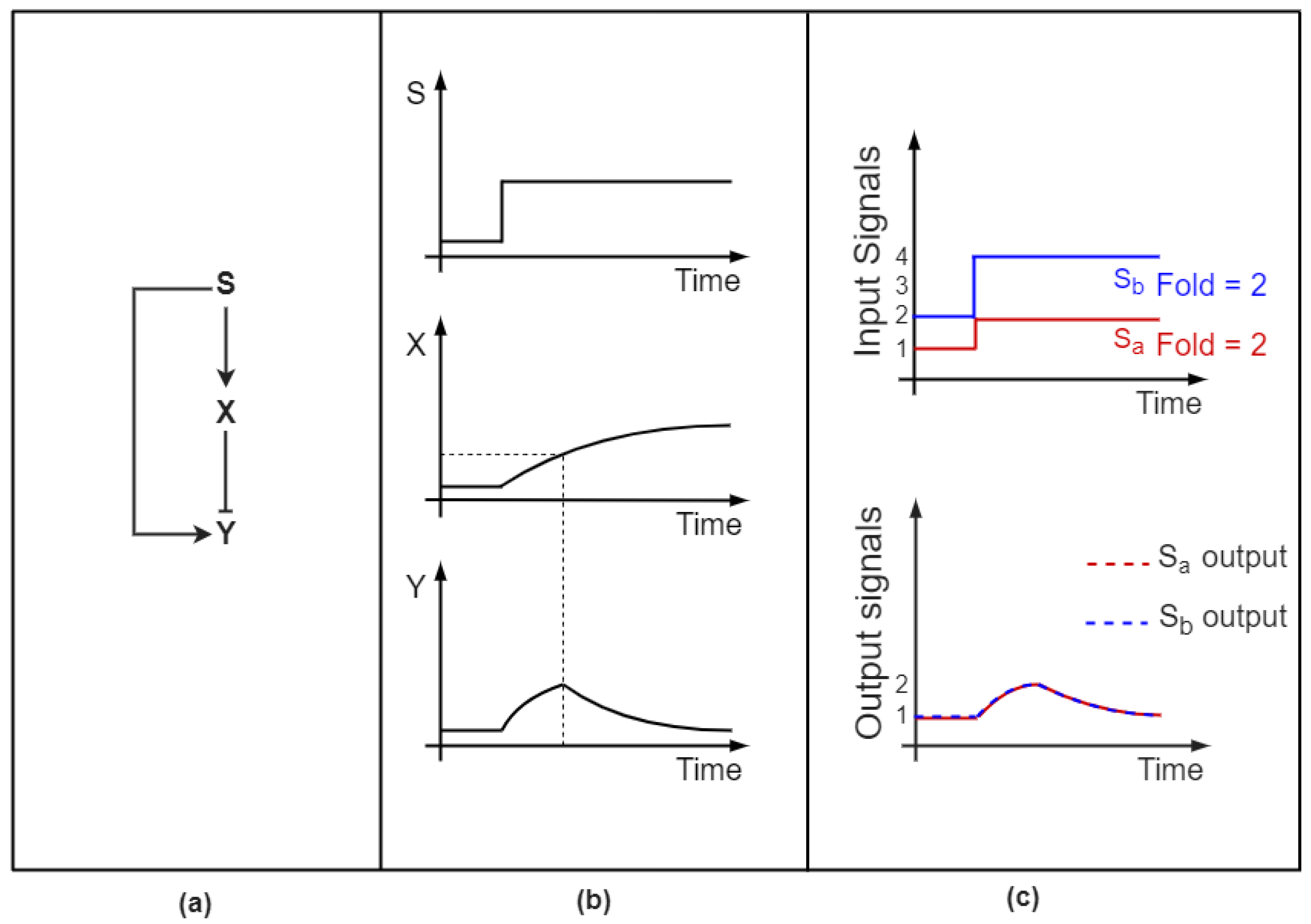
2.1. Feedforward Loops Differentiate between Pulsatile and Transient Signals
2.2. Feedback Loops: From Memory Effects to Oscillating Behaviors
3. microRNA-Mediated Gene Regulatory Networks and Temporal Dynamics
3.1. microRNAs and p53: Cooperation to Guard the Genome
3.2. microRNAs and Myc: Mediating Cell Proliferation and Differentiation
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lytle, N.K.; Barber, A.G.; Reya, T. Stem cell fate in cancer growth, progression and therapy resistance. Nat. Rev. Cancer 2018, 18, 669–680. [Google Scholar] [CrossRef]
- Waddington, C.H. The Strategy of the Genes; Routledge: London, UK, 2014. [Google Scholar]
- Wang, J.; Zhang, K.; Xu, L.; Wang, E. Quantifying the Waddington landscape and biological paths for development and differentiation. Proc. Natl. Acad. Sci. USA 2011, 108, 8257–8262. [Google Scholar] [CrossRef] [Green Version]
- Ferrell, J.E., Jr. Bistability, bifurcations, and Waddington’s epigenetic landscape. Curr. Biol. 2012, 22, R458–R466. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Oltvai, Z.N.; Bayır, H.; Silverman, G.A.; Pak, S.C.; Perlmutter, D.H.; Bahar, I. Quantitative assessment of cell fate decision between autophagy and apoptosis. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Frum, T.; Ralston, A. Cell signaling and transcription factors regulating cell fate during formation of the mouse blastocyst. Trends Genet. 2015, 31, 402–410. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, J.A.; Hsieh, Y.W.; Chen, S.; Pirri, J.K.; Alkema, M.J.; Li, W.H.; Chang, C.; Chuang, C.F. Intercellular calcium signaling in a gap junction-coupled cell network establishes asymmetric neuronal fates in C. elegans. Development 2012, 139, 4191–4201. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Ernberg, I.; Kauffman, S. Cancer attractors: A systems view of tumors from a gene network dynamics and developmental perspective. Semin. Cell Dev. Biol. 2009, 20, 869–876. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.Y.; Muller, W.J. Oncogenes and tumor suppressor genes. Cold Spring Harb. Perspect. Biol. 2010, 2, a003236. [Google Scholar] [CrossRef] [Green Version]
- Sternlicht, M.D.; Lochter, A.; Sympson, C.J.; Huey, B.; Rougier, J.P.; Gray, J.W.; Pinkel, D.; Bissell, M.J.; Werb, Z. The stromal proteinase MMP3/stromelysin-1 promotes mammary carcinogenesis. Cell 1999, 98, 137–146. [Google Scholar] [CrossRef] [Green Version]
- Raj, A.; Van Oudenaarden, A. Nature, nurture, or chance: Stochastic gene expression and its consequences. Cell 2008, 135, 216–226. [Google Scholar] [CrossRef] [Green Version]
- Purvis, J.E.; Lahav, G. Encoding and decoding cellular information through signaling dynamics. Cell 2013, 152, 945–956. [Google Scholar] [CrossRef] [Green Version]
- Friedman, R.C.; Farh, K.K.H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [Green Version]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- Jo, M.H.; Shin, S.; Jung, S.R.; Kim, E.; Song, J.J.; Hohng, S. Human Argonaute 2 has diverse reaction pathways on target RNAs. Mol. Cell 2015, 59, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Shalgi, R.; Lieber, D.; Oren, M.; Pilpel, Y. Global and local architecture of the mammalian microRNA–transcription factor regulatory network. PLoS Comput. Biol. 2007, 3, e131. [Google Scholar] [CrossRef]
- Schmitz, U.; Lai, X.; Winter, F.; Wolkenhauer, O.; Vera, J.; Gupta, S.K. Cooperative gene regulation by microRNA pairs and their identification using a computational workflow. Nucleic Acids Res. 2014, 42, 7539–7552. [Google Scholar] [CrossRef] [Green Version]
- Lai, X.; Schmitz, U.; Gupta, S.K.; Bhattacharya, A.; Kunz, M.; Wolkenhauer, O.; Vera, J. Computational analysis of target hub gene repression regulated by multiple and cooperative miRNAs. Nucleic Acids Res. 2012, 40, 8818–8834. [Google Scholar] [CrossRef] [Green Version]
- Mukherji, S.; Ebert, M.S.; Zheng, G.X.; Tsang, J.S.; Sharp, P.A.; van Oudenaarden, A. microRNAs can generate thresholds in target gene expression. Nat. Genet. 2011, 43, 854–859. [Google Scholar] [CrossRef] [Green Version]
- Bosia, C.; Sgrò, F.; Conti, L.; Baldassi, C.; Brusa, D.; Cavallo, F.; Di Cunto, F.; Turco, E.; Pagnani, A.; Zecchina, R. RNAs competing for microRNAs mutually influence their fluctuations in a highly non-linear microRNA-dependent manner in single cells. Genome Biol. 2017, 18, 1–14. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Sun, G.; Li, S.; Shi, Y. A feedback regulatory loop involving microRNA-9 and nuclear receptor TLX in neural stem cell fate determination. Nat. Struct. Mol. Biol. 2009, 16, 365–371. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.D.; Esquela-Kerscher, A.; Stefani, G.; Byrom, M.; Kelnar, K.; Ovcharenko, D.; Wilson, M.; Wang, X.; Shelton, J.; Shingara, J.; et al. The let-7 microRNA represses cell proliferation pathways in human cells. Cancer Res. 2007, 67, 7713–7722. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Liu, M.; Stribinskis, V.; Klinge, C.; Ramos, K.; Colburn, N.; Li, Y. microRNA-21 promotes cell transformation by targeting the programmed cell death 4 gene. Oncogene 2008, 27, 4373–4379. [Google Scholar] [CrossRef] [Green Version]
- Bracken, C.P.; Scott, H.S.; Goodall, G.J. A network-biology perspective of microRNA function and dysfunction in cancer. Nat. Rev. Genet. 2016, 17, 719–732. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, P.; Wang, F.; Zhang, H.; Yang, Y.; Shi, C.; Xia, Y.; Peng, J.; Liu, W.; Yang, Z.; et al. Elevated oncofoetal miR-17-5p expression regulates colorectal cancer progression by repressing its target gene P130. Nat. Commun. 2012, 3, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Korpal, M.; Ell, B.J.; Buffa, F.M.; Ibrahim, T.; Blanco, M.A.; Celià-Terrassa, T.; Mercatali, L.; Khan, Z.; Goodarzi, H.; Hua, Y.; et al. Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nat. Med. 2011, 17, 1101–1108. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, W.T.; Tzeng, K.R.; Ciou, J.S.; Tsai, J.J.; Kurubanjerdjit, N.; Huang, C.H.; Ng, K.L. Transcription factor and microRNA-regulated network motifs for cancer and signal transduction networks. BMC Syst. Biol. 2015, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Gong, B.; Yue, Y.; Wang, R.; Zhang, Y.; Jin, Q.; Zhou, X. Overexpression of microRNA-194 suppresses the epithelial–mesenchymal transition in targeting stem cell transcription factor Sox3 in endometrial carcinoma stem cells. Tumor Biol. 2017, 39, 1010428317706217. [Google Scholar] [CrossRef] [Green Version]
- Re, A.; Caselle, M.; Bussolino, F. microRNA-mediated regulatory circuits: Outlook and perspectives. Phys. Biol. 2017, 14, 045001. [Google Scholar]
- Ferro, E.; Enrico Bena, C.; Grigolon, S.; Bosia, C. From endogenous to synthetic microRNA-mediated regulatory circuits: An overview. Cells 2019, 8, 1540. [Google Scholar] [CrossRef] [Green Version]
- Levine, J.H.; Lin, Y.; Elowitz, M.B. Functional roles of pulsing in genetic circuits. Science 2013, 342, 1193–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippi, S.; Barnes, C.P.; Kirk, P.D.; Kudo, T.; Kunida, K.; McMahon, S.S.; Tsuchiya, T.; Wada, T.; Kuroda, S.; Stumpf, M.P. Robustness of MEK-ERK dynamics and origins of cell-to-cell variability in MAPK signaling. Cell Rep. 2016, 15, 2524–2535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bootman, M.D.; Bultynck, G. Fundamentals of cellular calcium signaling: A primer. Cold Spring Harb. Perspect. Biol. 2020, 12, a038802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, D.M.; Purvis, J.E. Computational analysis of signaling patterns in single cells. Semin. Cell Dev. Biol. 2015, 37, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Kwon, Y.K.; Cho, K.H. Coupled positive and negative feedback circuits form an essential building block of cellular signaling pathways. BioEssays 2007, 29, 85–90. [Google Scholar] [CrossRef]
- Mangan, S.; Alon, U. Structure and function of the feed-forward loop network motif. Proc. Natl. Acad. Sci. USA 2003, 100, 11980–11985. [Google Scholar] [CrossRef] [Green Version]
- Shen-Orr, S.S.; Milo, R.; Mangan, S.; Alon, U. Network motifs in the transcriptional regulation network of Escherichia coli. Nat. Genet. 2002, 31, 64–68. [Google Scholar] [CrossRef]
- Tsang, J.; Zhu, J.; van Oudenaarden, A. microRNA-mediated feedback and feedforward loops are recurrent network motifs in mammals. Mol. Cell 2007, 26, 753–767. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.I.; Rinaldi, N.J.; Robert, F.; Odom, D.T.; Bar-Joseph, Z.; Gerber, G.K.; Hannett, N.M.; Harbison, C.T.; Thompson, C.M.; Simon, I.; et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 2002, 298, 799–804. [Google Scholar] [CrossRef] [Green Version]
- Boyer, L.A.; Lee, T.I.; Cole, M.F.; Johnstone, S.E.; Levine, S.S.; Zucker, J.P.; Guenther, M.G.; Kumar, R.M.; Murray, H.L.; Jenner, R.G.; et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 2005, 122, 947–956. [Google Scholar] [CrossRef] [Green Version]
- Chevrier, N.; Mertins, P.; Artyomov, M.N.; Shalek, A.K.; Iannacone, M.; Ciaccio, M.F.; Gat-Viks, I.; Tonti, E.; DeGrace, M.M.; Clauser, K.R.; et al. Systematic discovery of TLR signaling components delineates viral-sensing circuits. Cell 2011, 147, 853–867. [Google Scholar] [CrossRef] [Green Version]
- Johnston, R.J., Jr.; Otake, Y.; Sood, P.; Vogt, N.; Behnia, R.; Vasiliauskas, D.; McDonald, E.; Xie, B.; Koenig, S.; Wolf, R.; et al. Interlocked feedforward loops control cell-type-specific Rhodopsin expression in the Drosophila eye. Cell 2011, 145, 956–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, S.; Mehreja, R.; Thiberge, S.; Chen, M.T.; Weiss, R. Spatiotemporal control of gene expression with pulse-generating networks. Proc. Natl. Acad. Sci. USA 2004, 101, 6355–6360. [Google Scholar] [CrossRef] [Green Version]
- Matthews, H.R.; Reisert, J. Calcium, the two-faced messenger of olfactory transduction and adaptation. Curr. Opin. Neurobiol. 2003, 13, 469–475. [Google Scholar] [CrossRef]
- Kollmann, M.; Løvdok, L.; Bartholomé, K.; Timmer, J.; Sourjik, V. Design principles of a bacterial signalling network. Nature 2005, 438, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Trusina, A.; El-Samad, H.; Lim, W.A.; Tang, C. Defining network topologies that can achieve biochemical adaptation. Cell 2009, 138, 760–773. [Google Scholar] [CrossRef] [Green Version]
- Strovas, T.J.; Rosenberg, A.B.; Kuypers, B.E.; Muscat, R.A.; Seelig, G. microRNA-based single-gene circuits buffer protein synthesis rates against perturbations. ACS Synth. Biol. 2014, 3, 324–331. [Google Scholar] [CrossRef]
- Guo, S.; Murray, R.M. Construction of incoherent feedforward loop circuits in a cell-free system and in cells. ACS Synth. Biol. 2019, 8, 606–610. [Google Scholar] [CrossRef]
- Gasch, A.P.; Spellman, P.T.; Kao, C.M.; Carmel-Harel, O.; Eisen, M.B.; Storz, G.; Botstein, D.; Brown, P.O. Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell 2000, 11, 4241–4257. [Google Scholar] [CrossRef]
- Braun, E.; Brenner, N. Transient responses and adaptation to steady state in a eukaryotic gene regulation system. Phys. Biol. 2004, 1, 67. [Google Scholar] [CrossRef]
- Busch, H.; Camacho-Trullio, D.; Rogon, Z.; Breuhahn, K.; Angel, P.; Eils, R.; Szabowski, A. Gene network dynamics controlling keratinocyte migration. Mol. Syst. Biol. 2008, 4, 199. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, E.; Mock, C.S.; Bhan, I.; Loewer, A.; Lahav, G. Recurrent initiation: A mechanism for triggering p53 pulses in response to DNA damage. Mol. Cell 2008, 30, 277–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chechik, G.; Koller, D. Timing of gene expression responses to environmental changes. J. Comput. Biol. 2009, 16, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Goentoro, L.; Shoval, O.; Kirschner, M.W.; Alon, U. The incoherent feedforward loop can provide fold-change detection in gene regulation. Mol. Cell 2009, 36, 894–899. [Google Scholar] [CrossRef] [Green Version]
- Goentoro, L.; Kirschner, M.W. Evidence that fold-change, and not absolute level, of β-catenin dictates Wnt signaling. Mol. Cell 2009, 36, 872–884. [Google Scholar] [CrossRef] [Green Version]
- Cohen-Saidon, C.; Cohen, A.A.; Sigal, A.; Liron, Y.; Alon, U. Dynamics and variability of ERK2 response to EGF in individual living cells. Mol. Cell 2009, 36, 885–893. [Google Scholar] [CrossRef]
- Bosia, C.; Osella, M.; El Baroudi, M.; Corà, D.; Caselle, M. Gene autoregulation via intronic microRNAs and its functions. BMC Syst. Biol. 2012, 6, 1–16. [Google Scholar] [CrossRef]
- Cerone, L.; Neufeld, Z. Differential gene expression regulated by oscillatory transcription factors. PLoS ONE 2012, 7, e30283. [Google Scholar] [CrossRef]
- Shalgi, R.; Brosh, R.; Oren, M.; Pilpel, Y.; Rotter, V. Coupling transcriptional and post-transcriptional miRNA regulation in the control of cell fate. Aging 2009, 1, 762. [Google Scholar] [CrossRef] [Green Version]
- Cournac, A.; Sepulchre, J.A. Simple molecular networks that respond optimally to time-periodic stimulation. BMC Syst. Biol. 2009, 3, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Osella, M.; Bosia, C.; Corá, D.; Caselle, M. The Role of Incoherent microRNA-Mediated Feedforward Loops in Noise Buffering. PLoS Comput. Biol. 2011, 7, e1001101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyun Kim, D.; Grün, D.; van Oudenaarden, A. Dampening of expression oscillations by synchronous regulation of a microRNA and its target. Nat. Genet. 2013, 45, 1337–1344. [Google Scholar]
- Alon, U. Network motifs: Theory and experimental approaches. Nat. Rev. Genet. 2007, 8, 450–461. [Google Scholar] [CrossRef] [PubMed]
- Mutzel, V.; Okamoto, I.; Dunkel, I.; Saitou, M.; Giorgetti, L.; Heard, E.; Schulz, E.G. A symmetric toggle switch explains the onset of random X inactivation in different mammals. Nat. Struct. Mol. Biol. 2019, 26, 350–360. [Google Scholar] [CrossRef]
- Palani, S.; Sarkar, C.A. Transient noise amplification and gene expression synchronization in a bistable mammalian cell-fate switch. Cell Rep. 2012, 1, 215–224. [Google Scholar] [CrossRef] [Green Version]
- Xiong, W.; Ferrell, J.E. A positive-feedback-based bistable ‘memory module’ that governs a cell fate decision. Nature 2003, 426, 460–465. [Google Scholar] [CrossRef]
- Davidson, E.H.; Rast, J.P.; Oliveri, P.; Ransick, A.; Calestani, C.; Yuh, C.H.; Minokawa, T.; Amore, G.; Hinman, V.; Arenas-Mena, C.; et al. A genomic regulatory network for development. Science 2002, 295, 1669–1678. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Khankan, R.R.; Caneda, C.; Godoy, M.I.; Haney, M.S.; Krawczyk, M.C.; Bassik, M.C.; Sloan, S.A.; Zhang, Y. Astrocyte-to-astrocyte contact and a positive feedback loop of growth factor signaling regulate astrocyte maturation. Glia 2019, 67, 1571–1597. [Google Scholar] [CrossRef]
- Osella, M.; Riba, A.; Testori, A.; Corà, D.; Caselle, M. Interplay of microRNA and epigenetic regulation in the human regulatory network. Front. Genet. 2014, 5, 345. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Lv, X.; Huang, C.; Hua, G.; Ma, B.; Chen, X.; Angeletti, P.C.; Dong, J.; Zhou, J.; Wang, Z.; et al. YAP 1-LATS 2 feedback loop dictates senescent or malignant cell fate to maintain tissue homeostasis. EMBO Rep. 2019, 20, e44948. [Google Scholar] [CrossRef]
- Ptacek, J.; Devgan, G.; Michaud, G.; Zhu, H.; Zhu, X.; Fasolo, J.; Guo, H.; Jona, G.; Breitkreutz, A.; Sopko, R.; et al. Global analysis of protein phosphorylation in yeast. Nature 2005, 438, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Afshar, A.S.; Xu, J.; Goutsias, J. Identifying deregulated TF/miRNA negative and double-negative feedback loops in prostate cancer. In Proceedings of the 2013 IEEE Global Conference on Signal and Information Processing, Austin, TX, USA, 3–5 December 2013; pp. 85–88. [Google Scholar] [CrossRef]
- Melling, G.E.; Flannery, S.E.; Abidin, S.A.; Clemmens, H.; Prajapati, P.; Hinsley, E.E.; Hunt, S.; Catto, J.W.; Coletta, R.D.; Mellone, M.; et al. A miRNA-145/TGF-β1 negative feedback loop regulates the cancer-associated fibroblast phenotype. Carcinogenesis 2018, 39, 798–807. [Google Scholar] [CrossRef] [PubMed]
- Hausser, J.; Syed, A.P.; Selevsek, N.; Van Nimwegen, E.; Jaskiewicz, L.; Aebersold, R.; Zavolan, M. Timescales and bottlenecks in miRNA-dependent gene regulation. Mol. Syst. Biol. 2013, 9, 711. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.R.; Yang, H.T.; Liu, W.C.; Hwang, M.J. The role of microRNA in the delayed negative feedback regulation of gene expression. Biochem. Biophys. Res. Commun. 2007, 358, 722–726. [Google Scholar] [CrossRef]
- Pigolotti, S.; Krishna, S.; Jensen, M.H. Oscillation patterns in negative feedback loops. Proc. Natl. Acad. Sci. USA 2007, 104, 6533–6537. [Google Scholar] [CrossRef] [Green Version]
- Nelson, D.; Ihekwaba, A.; Elliott, M.; Johnson, J.; Gibney, C.; Foreman, B.; Nelson, G.; See, V.; Horton, C.; Spiller, D.; et al. Oscillations in NF-κB signaling control the dynamics of gene expression. Science 2004, 306, 704–708. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; D’Alessandro, M.; Lee, C. miRNAs are required for generating a time delay critical for the circadian oscillator. Curr. Biol. 2013, 23, 1959–1968. [Google Scholar] [CrossRef] [Green Version]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef]
- Vennstrom, B.; Sheiness, D.; Zabielski, J.; Bishop, J. Isolation and characterization of c-myc, a cellular homolog of the oncogene (v-myc) of avian myelocytomatosis virus strain 29. J. Virol. 1982, 42, 773–779. [Google Scholar] [CrossRef] [Green Version]
- Lakin, N.D.; Jackson, S.P. Regulation of p53 in response to DNA damage. Oncogene 1999, 18, 7644–7655. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, K.D.; Galbraith, M.D.; Andrysik, Z.; Espinosa, J.M. Mechanisms of transcriptional regulation by p53. Cell Death Differ. 2018, 25, 133–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lourenco, C.; Resetca, D.; Redel, C.; Lin, P.; MacDonald, A.S.; Ciaccio, R.; Kenney, T.M.; Wei, Y.; Andrews, D.W.; Sunnerhagen, M.; et al. MYC protein interactors in gene transcription and cancer. Nat. Rev. Cancer 2021, 21, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.K.; Sharp, P.A. microRNAs: A safeguard against turmoil? Cell 2007, 130, 581–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, A.K.; Sharp, P.A. microRNA functions in stress responses. Mol. Cell 2010, 40, 205–215. [Google Scholar] [CrossRef] [Green Version]
- Sionov, R.V.; Haupt, Y. The cellular response to p53: The decision between life and death. Oncogene 1999, 18, 6145–6157. [Google Scholar] [CrossRef] [Green Version]
- Lukin, D.J.; Carvajal, L.A.; Liu, W.j.; Resnick-Silverman, L.; Manfredi, J.J. p53 Promotes cell survival due to the reversibility of its cell-cycle checkpoints. Mol. Cancer Res. 2015, 13, 16–28. [Google Scholar] [CrossRef] [Green Version]
- Dashzeveg, N.; Yoshida, K. Cell death decision by p53 via control of the mitochondrial membrane. Cancer Lett. 2015, 367, 108–112. [Google Scholar] [CrossRef]
- Kim, Y.Y.; Um, J.H.; Shin, D.J.; Jeong, D.J.; Hong, Y.B.; Yun, J. p53-mediated regulation of mitochondrial dynamics plays a pivotal role in the senescence of various normal cells as well as cancer cells. FASEB J. 2021, 35, e21319. [Google Scholar] [CrossRef]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef]
- Karakostis, K.; Ponnuswamy, A.; Fusée, L.T.; Bailly, X.; Laguerre, L.; Worall, E.; Vojtesek, B.; Nylander, K.; Fåhraeus, R. p53 mRNA and p53 protein structures have evolved independently to interact with MDM2. Mol. Biol. Evol. 2016, 33, 1280–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Deiry, W.S. The role of p53 in chemosensitivity and radiosensitivity. Oncogene 2003, 22, 7486–7495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiewe, T. The p53 family in differentiation and tumorigenesis. Nat. Rev. Cancer 2007, 7, 165–167. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [Green Version]
- Stracquadanio, G.; Wang, X.; Wallace, M.D.; Grawenda, A.M.; Zhang, P.; Hewitt, J.; Zeron-Medina, J.; Castro-Giner, F.; Tomlinson, I.P.; Goding, C.R.; et al. The importance of p53 pathway genetics in inherited and somatic cancer genomes. Nat. Rev. Cancer 2016, 16, 251–265. [Google Scholar] [CrossRef]
- Lahav, G.; Rosenfeld, N.; Sigal, A.; Geva-Zatorsky, N.; Levine, A.J.; Elowitz, M.B.; Alon, U. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat. Genet. 2004, 36, 147–150. [Google Scholar] [CrossRef]
- Batchelor, E.; Loewer, A.; Mock, C.; Lahav, G. Stimulus-dependent dynamics of p53 in single cells. Mol. Syst. Biol. 2011, 7, 488. [Google Scholar] [CrossRef]
- Purvis, J.E.; Karhohs, K.W.; Mock, C.; Batchelor, E.; Loewer, A.; Lahav, G. p53 dynamics control cell fate. Science 2012, 336, 1440–1444. [Google Scholar] [CrossRef] [Green Version]
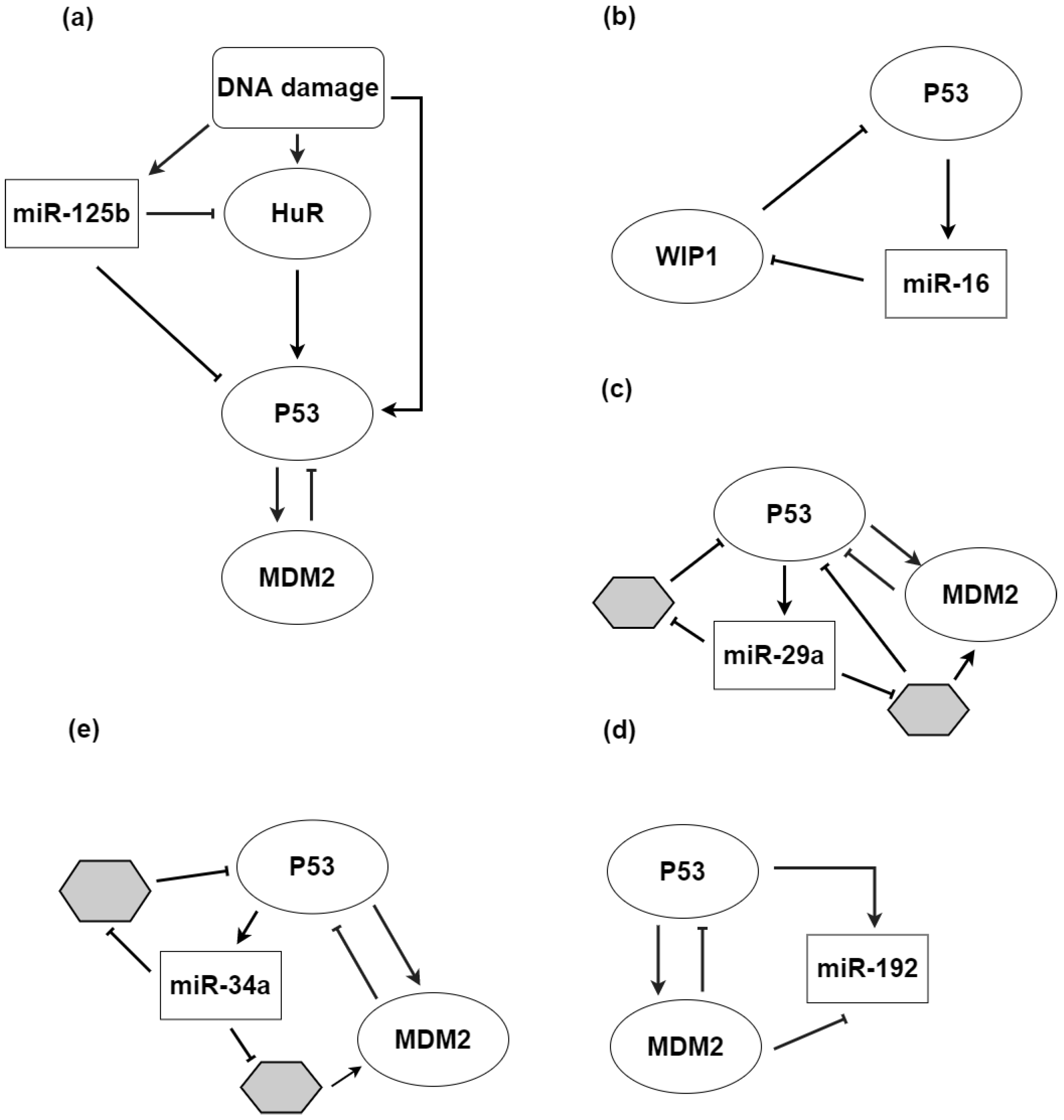
- Guha, A.; Ahuja, D.; Mandal, S.D.; Parasar, B.; Deyasi, K.; Roy, D.; Sharma, V.; Willard, B.; Ghosh, A.; Ray, P.S. Integrated regulation of HuR by translation repression and protein degradation determines pulsatile expression of p53 Under DNA damage. Iscience 2019, 15, 342–359. [Google Scholar] [CrossRef] [Green Version]
- Mazan-Mamczarz, K.; Galban, S.; de Silanes, I.; Martindale, J.; Atasoy, U.; Keene, J.; Gorospe, M. RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proc. Natl. Acad. Sci. USA 2003, 100, 8354–8359. [Google Scholar] [CrossRef] [Green Version]
- Ahuja, D.; Goyal, A.; Ray, P.S. Interplay between RNA-binding protein HuR and microRNA-125b regulates p53 mRNA translation in response to genotoxic stress. RNA Biol. 2016, 13, 1152–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Novere, N. Quantitative and logic modelling of molecular and gene networks. Nat. Rev. Genet. 2015, 16, 146–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abou-Jaoude, W.; Traynard, P.; Monteiro, P.; Saez-Rodriguez, J.; Helikar, T.; Thieffry, D.; Chaouiya, C. Logical Modeling and Dynamical Analysis of Cellular Networks. Front. Genet. 2016, 7, 94. [Google Scholar] [CrossRef] [PubMed]
- Issler, M.V.C.; Mombach, J.C.M. microRNA-16 feedback loop with p53 and Wip1 can regulate cell fate determination between apoptosis and senescence in DNA damage response. PLoS ONE 2017, 12, e0185794. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.I.; Yamagata, K.; Sugimoto, K.; Iwamoto, T.; Kato, S.; Miyazono, K. Modulation of microRNA processing by p53. Nature 2009, 460, 529–533. [Google Scholar] [CrossRef]
- Moore, R.; Ooi, H.K.; Kang, T.; Bleris, L.; Ma, L. MiR-192-Mediated Positive Feedback Loop Controls the Robustness of Stress-Induced p53 Oscillations in Breast Cancer Cells. PLoS Comput. Biol. 2015, 11, e1004653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Wu, W.S.; Heinrichs, S.; Xu, D.; Garrison, S.P.; Zambetti, G.P.; Adams, J.M.; Look, A.T. Slug antagonizes p53-mediated apoptosis of hematopoietic progenitors by repressing puma. Cell 2005, 123, 641–653. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Bhatt, D.; Oltvai, Z.N.; Greenberger, J.S.; Bahar, I. Significance of p53 dynamics in regulating apoptosis in response to ionizing radiation and polypharmacological strategies. Sci. Rep. 2014, 4, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Raver-Shapira, N.; Marciano, E.; Meiri, E.; Spector, Y.; Rosenfeld, N.; Moskovits, N.; Bentwich, Z.; Oren, M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol. Cell 2007, 26, 731–743. [Google Scholar] [CrossRef]
- Le, M.T.; Teh, C.; Shyh-Chang, N.; Xie, H.; Zhou, B.; Korzh, V.; Lodish, H.F.; Lim, B. microRNA-125b is a novel negative regulator of p53. Genes Dev. 2009, 23, 862–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leemans, C.R.; Braakhuis, B.J.M.; Brakenhoff, R.H. The molecular biology of head and neck cancer. Nat. Rev. Cancer 2011, 11, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.H.; Tsai, K.L.; Chen, Y.W.; Yu, C.C.; Chang, K.W.; Chiou, S.H.; Ku, H.H.; Chu, P.Y.; Tseng, L.M.; Huang, P.I.; et al. microRNA as a novel modulator in head and neck squamous carcinoma. J. Oncol. 2010, 2010, 135632. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sun, Q.; Zhang, Z.; Ge, S.; Han, Z.; Chen, W. Loss of microRNA-143/145 disturbs cellular growth and apoptosis of human epithelial cancers by impairing the MDM2-p53 feedback loop. Oncogene 2013, 32, 61–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Liu, J.; Tan, C.; Yue, X.; Zhao, Y.; Peng, J.; Wang, X.; Laddha, S.V.; Chan, C.S.; Zheng, S.; et al. microRNA-1827 represses MDM2 to positively regulate tumor suppressor p53 and suppress tumorigenesis. Oncotarget 2016, 7, 8783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La, T.; Liu, G.Z.; Farrelly, M.; Cole, N.; Feng, Y.C.; Zhang, Y.Y.; Sherwin, S.K.; Yari, H.; Tabatabaee, H.; Yan, X.G.; et al. A p53-responsive miRNA network promotes cancer cell quiescence. Cancer Res. 2018, 78, 6666–6679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adhikary, S.; Eilers, M. Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell Biol. 2005, 6, 635–645. [Google Scholar] [CrossRef]
- Lemaitre, J.M.; Buckle, R.S.; Méchali, M. c-Myc in the Controlm of Cell Proliferation and Embryonic Development. Adv. Cancer Res. 1996, 70, 95–144. [Google Scholar]
- Gandarillas, A.; Watt, F.M. c-Myc promotes differentiation of human epidermal stem cells. Genes Dev. 1997, 11, 2869–2882. [Google Scholar] [CrossRef] [Green Version]
- Evan, G.I.; Wyllie, A.H.; Gilbert, C.S.; Littlewood, T.D.; Land, H.; Brooks, M.; Waters, C.M.; Penn, L.Z.; Hancock, D.C. Induction of apoptosis in fibroblasts by c-myc protein. Cell 1992, 69, 119–128. [Google Scholar] [CrossRef]
- Zhou, Z.Q.; Hurlin, P.J. The interplay between Mad and Myc in proliferation and differentiation. Trends Cell Biol. 2001, 11, S10–S14. [Google Scholar] [CrossRef]
- Pelengaris, S.; Khan, M.; Evan, G.I. Suppression of Myc-induced apoptosis in β cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell 2002, 109, 321–334. [Google Scholar] [CrossRef] [Green Version]
- Kalkat, M.; De Melo, J.; Hickman, K.A.; Lourenco, C.; Redel, C.; Resetca, D.; Tamachi, A.; Tu, W.B.; Penn, L.Z. MYC deregulation in primary human cancers. Genes 2017, 8, 151. [Google Scholar] [CrossRef] [Green Version]
- Kaczmarek, L.; Hyland, J.K.; Watt, R.; Rosenberg, M.; Baserga, R. Microinjected c-myc as a competence factor. Science 1985, 228, 1313–1315. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Dong, F. Gfi1 upregulates c-Myc expression and promotes c-Myc-driven cell proliferation. Sci. Rep. 2020, 10, 1–11. [Google Scholar]
- Mollaoglu, G.; Guthrie, M.R.; Böhm, S.; Brägelmann, J.; Can, I.; Ballieu, P.M.; Marx, A.; George, J.; Heinen, C.; Chalishazar, M.D.; et al. MYC drives progression of small cell lung cancer to a variant neuroendocrine subtype with vulnerability to aurora kinase inhibition. Cancer Cell 2017, 31, 270–285. [Google Scholar] [CrossRef] [Green Version]
- Sorrentino, V.; Drozdoff, V.; McKinney, M.D.; Zeitz, L.; Fleissner, E. Potentiation of growth factor activity by exogenous c-myc expression. Proc. Natl. Acad. Sci. USA 1986, 83, 8167–8171. [Google Scholar] [CrossRef] [Green Version]
- Gartel, A.L.; Ye, X.; Goufman, E.; Shianov, P.; Hay, N.; Najmabadi, F.; Tyner, A.L. Myc represses the p21 (WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc. Natl. Acad. Sci. USA 2001, 98, 4510–4515. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, J.W.; Zhao, X.; De Cecco, M.; Peterson, A.L.; Pagliaroli, L.; Manivannan, J.; Hubbard, G.B.; Ikeno, Y.; Zhang, Y.; Feng, B.; et al. Reduced expression of MYC increases longevity and enhances healthspan. Cell 2015, 160, 477–488. [Google Scholar] [CrossRef] [Green Version]
- Dangi, S.; Chen, F.; Shapiro, P. Activation of extracellular signal-regulated kinase (ERK) in G2 phase delays mitotic entry through p21CIP1. Cell Prolif. 2006, 39, 261–279. [Google Scholar] [CrossRef]
- Lee, T.; Yao, G.; Nevins, J.; You, L. Sensing and integration of Erk and PI3K signals by Myc. PLoS Comput. Biol. 2008, 4, e1000013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, C. Specificity of receptor tyrosine kinase signaling: Transient versus sustained extracellular signal-regulated kinase activation. Cell 1995, 80, 179–185. [Google Scholar] [CrossRef] [Green Version]
- Albeck, J.G.; Mills, G.B.; Brugge, J.S. Frequency-modulated pulses of ERK activity transmit quantitative proliferation signals. Mol. Cell 2013, 49, 249–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki, K.; Kumagai, Y.; Sakurai, A.; Komatsu, N.; Fujita, Y.; Shionyu, C.; Matsuda, M. Stochastic ERK activation induced by noise and cell-to-cell propagation regulates cell density-dependent proliferation. Mol. Cell 2013, 52, 529–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kholodenko, B.N. Negative feedback and ultrasensitivity can bring about oscillations in the mitogen-activated protein kinase cascades. Eur. J. Biochem. 2000, 267, 1583–1588. [Google Scholar] [CrossRef] [Green Version]
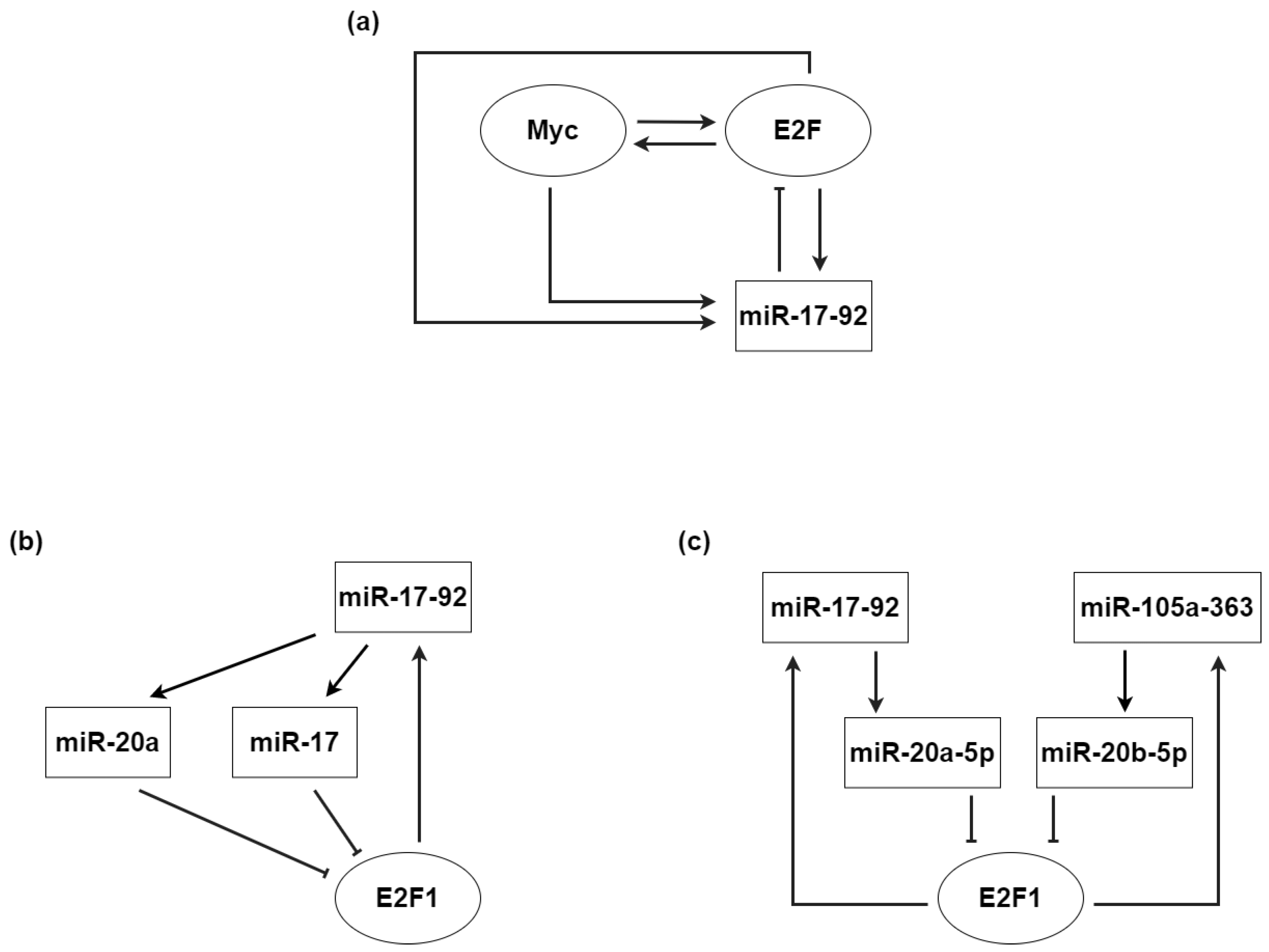
- O’Donnell, K.A.; Wentzel, E.A.; Zeller, K.I.; Dang, C.V.; Mendell, J.T. c-Myc-regulated microRNAs modulate E2F1 expression. Nature 2005, 435, 839–843. [Google Scholar] [CrossRef]
- He, P.; Billy, K.J.; Ma, H.; Jia, Y.; Yang, L. The dynamical roles of miR-17-92 on the E2F-related network during the G1/S transition. Nonlinear Dyn. 2019, 95, 259–271. [Google Scholar] [CrossRef]
- Zhang, Y.; Song, X.; Herrup, K. Context-dependent functions of E2F1: Cell cycle, cell death, and DNA damage repair in cortical neurons. Mol. Neurobiol. 2020, 57, 2377–2390. [Google Scholar] [CrossRef]
- Yao, G.; Lee, T.J.; Mori, S.; Nevins, J.R.; You, L. A bistable Rb–E2F switch underlies the restriction point. Nat. Cell Biol. 2008, 10, 476–482. [Google Scholar] [CrossRef]
- Gedeon, T.; Bokes, P. Delayed protein synthesis reduces the correlation between mRNA and protein fluctuations. Biophys. J. 2012, 103, 377–385. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Shi, B.; Chen, J.; Li, C.; Wang, S.; Wang, Z.; Zhu, G. An E2F1/MiR-17-92 negative feedback loop mediates proliferation of mouse palatal mesenchymal cells. Sci. Rep. 2017, 7, 1–9. [Google Scholar]
- Luo, W.; Li, G.; Yi, Z.; Nie, Q.; Zhang, X. E2F1-miR-20a-5p/20b-5p auto-regulatory feedback loop involved in myoblast proliferation and differentiation. Sci. Rep. 2016, 6, 1–13. [Google Scholar]
- Palmbos, P.L.; Wang, L.; Yang, H.; Wang, Y.; Leflein, J.; Ahmet, M.L.; Wilkinson, J.E.; Kumar-Sinha, C.; Ney, G.M.; Tomlins, S.A.; et al. ATDC/TRIM29 drives invasive bladder cancer formation through miRNA-mediated and epigenetic mechanisms. Cancer Res. 2015, 75, 5155–5166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Cao, Y.; Zhai, Y.; Ma, X.; An, X.; Zhang, S.; Li, Z. Micro RNA-29b regulates DNA methylation by targeting Dnmt3a/3b and Tet1/2/3 in porcine early embryo development. Dev. Growth Differ. 2018, 60, 197–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, H.F.; Liu, Y.Z.; Du, R.; Wang, B.; Chen, M.T.; Zhang, Y.Y.; Deng, Z.L.; Li, J. miR-377 induces senescence in human skin fibroblasts by targeting DNA methyltransferase 1. Cell Death Dis. 2017, 8, e2663. [Google Scholar] [CrossRef] [Green Version]
- Shao, L.; Chen, Z.; Peng, D.; Soutto, M.; Zhu, S.; Bates, A.; Zhang, S.; El-Rifai, W. Methylation of the HOXA10 promoter directs miR-196b-5p–dependent cell proliferation and invasion of gastric cancer cells. Mol. Cancer Res. 2018, 16, 696–706. [Google Scholar] [CrossRef] [Green Version]
- Pang, A.L.Y.; Rennert, O.M. Modulation of microRNA expression in human lung cancer cells by the G9a histone methyltransferase inhibitor BIX01294. Oncol. Lett. 2014, 7, 1819–1825. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Chen, T.Y.; Dhar, S.S.; Gu, B.; Chen, K.; Kim, Y.Z.; Li, W.; Lee, M.G. A feedback loop comprising PRMT7 and miR-24-2 interplays with Oct4, Nanog, Klf4 and c-Myc to regulate stemness. Nucleic Acids Res. 2016, 44, 10603–10618. [Google Scholar] [CrossRef]
- Chang, T.C.; Yu, D.; Lee, Y.S.; Wentzel, E.A.; Arking, D.E.; West, K.M.; Dang, C.V.; Thomas-Tikhonenko, A.; Mendell, J.T. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat. Genet. 2008, 40, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhao, X.; Shi, J.; Pan, Y.; Chen, Q.; Leng, P.; Wang, Y. miR-451 suppresses bladder cancer cell migration and invasion via directly targeting c-Myc. Oncol. Rep. 2016, 36, 2049–2058. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Zhang, G.; Li, J.; Liu, J.; Lv, P. The tumor-suppressive microRNA-135b targets c-myc in osteoscarcoma. PLoS ONE 2014, 9, e102621. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liang, Z.; Xu, X.; Li, J.; Zhu, Y.; Meng, S.; Li, S.; Wang, S.; Xie, B.; Ji, A.; et al. miR-148a-3p represses proliferation and EMT by establishing regulatory circuits between ERBB3/AKT2/c-myc and DNMT1 in bladder cancer. Cell Death Dis. 2016, 7, e2503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansouri, M.; Strittmatter, T.; Fussenegger, M. Light-controlled mammalian cells and their therapeutic applications in synthetic biology. Adv. Sci. 2019, 6, 1800952. [Google Scholar] [CrossRef] [PubMed]
- Muzzey, D.; van Oudenaarden, A. Quantitative time-lapse fluorescence microscopy in single cells. Annu. Rev. Cell Dev. 2009, 25, 301–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, F.; Bai, L. Using time-lapse fluorescence microscopy to study gene regulation. Methods 2019, 159, 138–145. [Google Scholar] [CrossRef]
- Turk, M.A.; Chung, C.Z.; Manni, E.; Zukowski, S.A.; Engineer, A.; Badakhshi, Y.; Bi, Y.; Heinemann, I.U. MiRAR—miRNA activity reporter for living cells. Genes 2018, 9, 305. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tealdi, S.; Ferro, E.; Campa, C.C.; Bosia, C. microRNA-Mediated Encoding and Decoding of Time-Dependent Signals in Tumorigenesis. Biomolecules 2022, 12, 213. https://doi.org/10.3390/biom12020213
Tealdi S, Ferro E, Campa CC, Bosia C. microRNA-Mediated Encoding and Decoding of Time-Dependent Signals in Tumorigenesis. Biomolecules. 2022; 12(2):213. https://doi.org/10.3390/biom12020213
Chicago/Turabian StyleTealdi, Simone, Elsi Ferro, Carlo Cosimo Campa, and Carla Bosia. 2022. "microRNA-Mediated Encoding and Decoding of Time-Dependent Signals in Tumorigenesis" Biomolecules 12, no. 2: 213. https://doi.org/10.3390/biom12020213