
An Introduction to Bacterial Biofilms and Their Proteases, and Their Roles in Host Infection and Immune Evasion


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
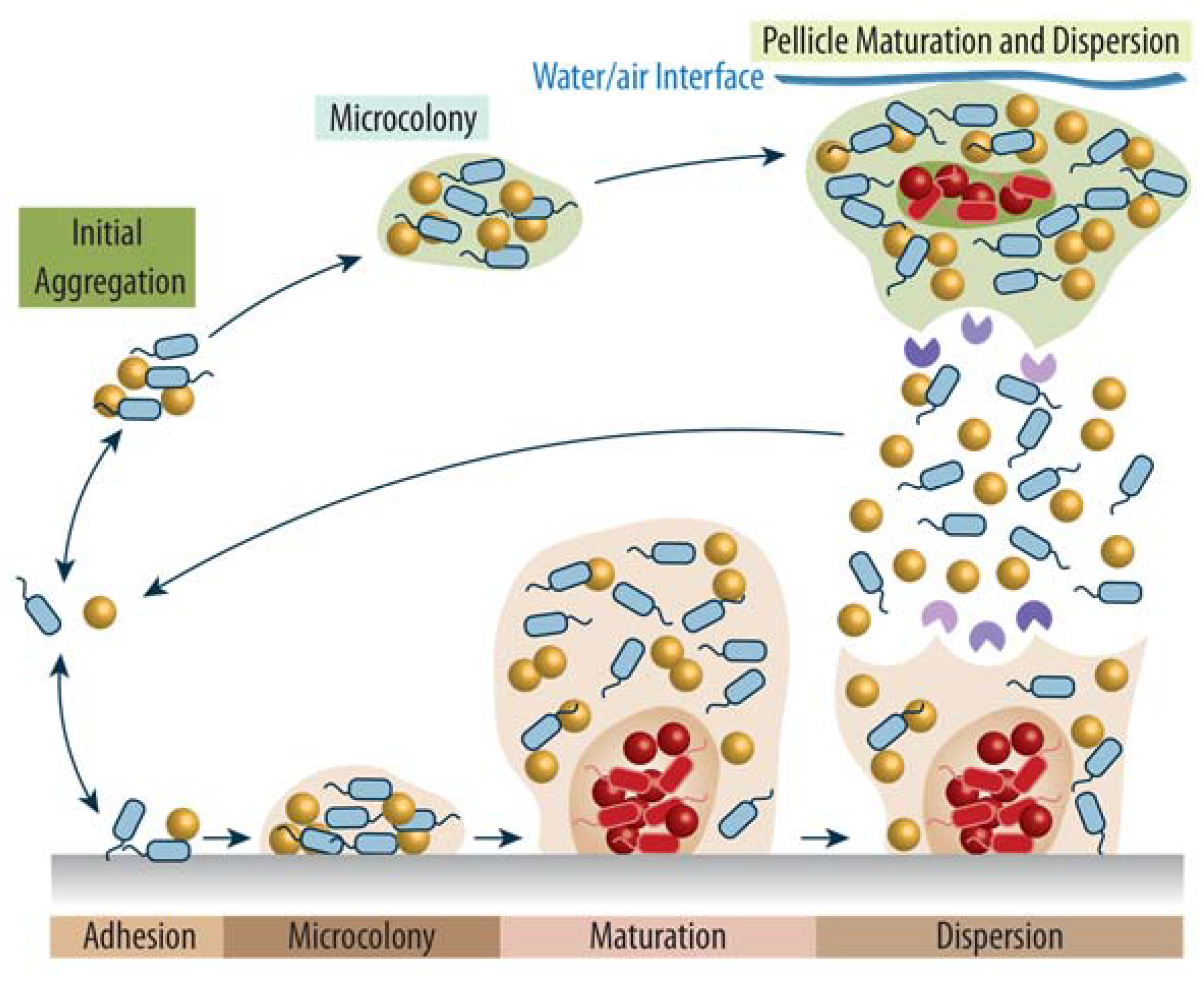
:1. The Complexity of Bacterial Biofilms
2. Proteolytic Enzymes—How They Work and What They Do
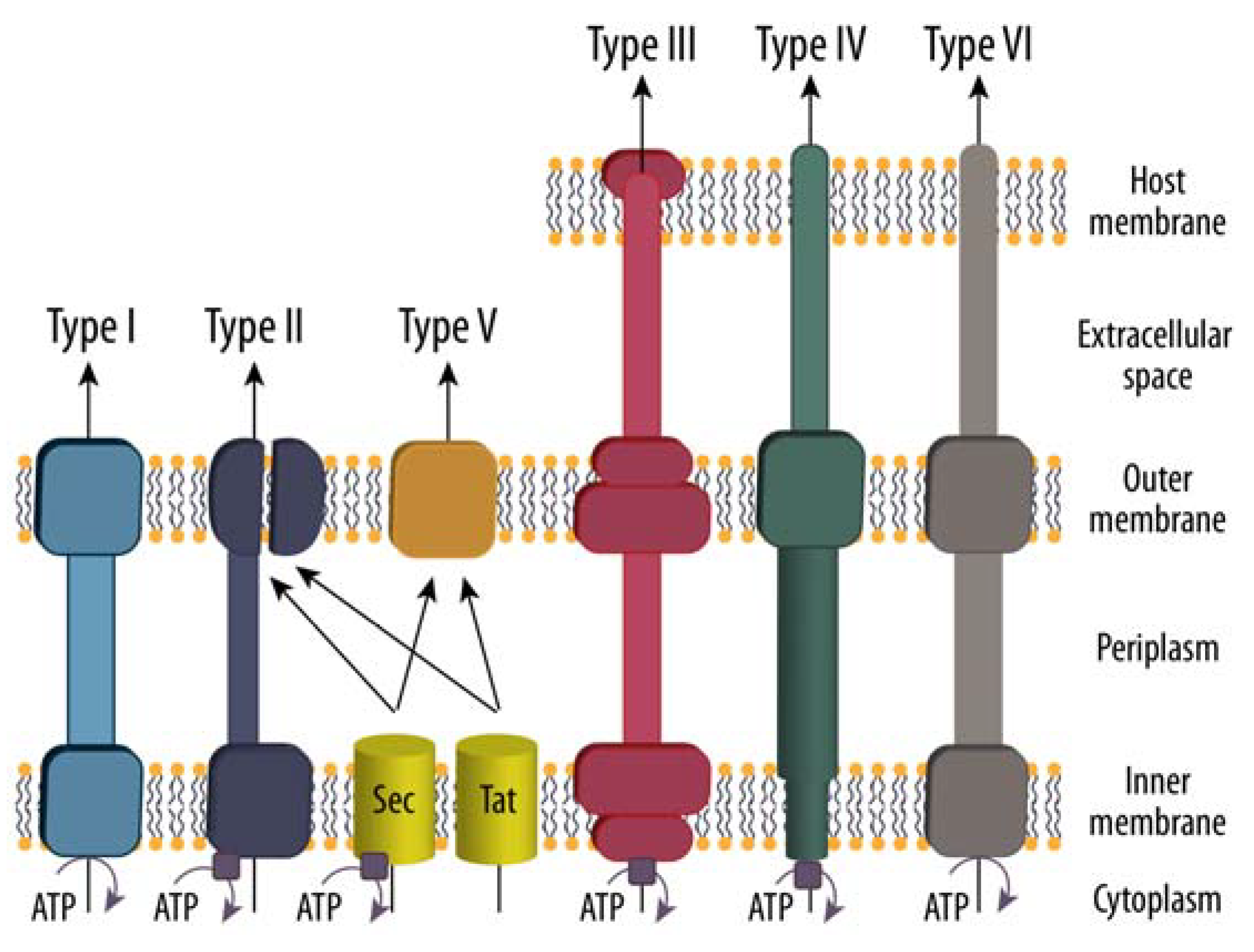
3. Protein Secretion to the Extracellular Space
4. The Immune System and How Pathogenic Bacteria Evade to Establish Infection
5. Biofilm Remodeling and the Role of Proteases in Virulence
6. Biomedical and Biotechnological Applications of Bacterial Proteases
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stoodley, P.; Sauer, K.; Davies, D.G.; Costerton, J.W. Biofilms as Complex Differentiated Communities. Annu. Rev. Microbiol. 2002, 56, 187–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, A.L.S.D.; Galdino, A.C.M.; de Mello, T.P.; Ramos, L.d.S.; Branquinha, M.H.; Bolognese, A.M.; Columbano Neto, J.; Roudbary, M. What Are the Advantages of Living in a Community? A Microbial Biofilm Perspective! Mem. Inst. Oswaldo Cruz 2018, 113, e180212. [Google Scholar] [CrossRef] [Green Version]
- Flemming, H.-C.; Wingender, J.; Szewzyk, U.; Steinberg, P.; Rice, S.A.; Kjelleberg, S. Biofilms: An Emergent Form of Bacterial Life. Nat. Rev. Microbiol. 2016, 14, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Høiby, N.; Ciofu, O.; Johansen, H.K.; Song, Z.; Moser, C.; Jensen, P.Ø.; Molin, S.; Givskov, M.; Tolker-Nielsen, T.; Bjarnsholt, T. The Clinical Impact of Bacterial Biofilms. Int. J. Oral Sci. 2011, 3, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Maali, Y.; Journo, C.; Mahieux, R.; Dutartre, H. Microbial Biofilms: Human T-Cell Leukemia Virus Type 1 First in Line for Viral Biofilm but Far Behind Bacterial Biofilms. Front. Microbiol. 2020, 11, 2041. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.-S.; Henderson, J.P. Emerging Concepts of Biofilms in Infectious Diseases. Mo. Med. 2009, 106, 292–296. [Google Scholar]
- Arnaouteli, S.; Bamford, N.C.; Stanley-Wall, N.R.; Kovács, Á.T. Bacillus Subtilis Biofilm Formation and Social Interactions. Nat. Rev. Microbiol. 2021, 19, 600–614. [Google Scholar] [CrossRef]
- Eckhard, U.; Bandukwala, H.; Mansfield, M.J.; Marino, G.; Cheng, J.; Wallace, I.; Holyoak, T.; Charles, T.C.; Austin, J.; Overall, C.M.; et al. Discovery of a Proteolytic Flagellin Family in Diverse Bacterial Phyla That Assembles Enzymatically Active Flagella. Nat. Commun. 2017, 8, 521. [Google Scholar] [CrossRef]
- Eckhard, U.; Blöchl, C.; Jenkins, B.G.L.; Mansfield, M.J.; Huber, C.G.; Doxey, A.C.; Brandstetter, H. Identification and Characterization of the Proteolytic Flagellin from the Common Freshwater Bacterium Hylemonella Gracilis. Sci. Rep. 2020, 10, 19052. [Google Scholar] [CrossRef]
- Domka, J.; Lee, J.; Bansal, T.; Wood, T.K. Temporal Gene-Expression in Escherichia Coli K-12 Biofilms. Environ. Microbiol. 2007, 9, 332–346. [Google Scholar] [CrossRef]
- Serra, D.O.; Richter, A.M.; Klauck, G.; Mika, F.; Hengge, R. Microanatomy at Cellular Resolution and Spatial Order of Physiological Differentiation in a Bacterial Biofilm. mBio 2013, 4, e00103–e00113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghafoor, A.; Hay, I.D.; Rehm, B.H.A. Role of Exopolysaccharides in Pseudomonas Aeruginosa Biofilm Formation and Architecture. Appl. Environ. Microbiol. 2011, 77, 5238–5246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, P.; Joshi, Y.; Rawat, K.; Bisht, R. Biofilms: Architecture, Resistance, Quorum Sensing and Control Mechanisms. Indian J. Microbiol. 2019, 59, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Koul, S.; Prakash, J.; Mishra, A.; Kalia, V.C. Potential Emergence of Multi-Quorum Sensing Inhibitor Resistant (MQSIR) Bacteria. Indian J. Microbiol. 2016, 56, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Jamal, M.; Ahmad, W.; Andleeb, S.; Jalil, F.; Imran, M.; Nawaz, M.A.; Hussain, T.; Ali, M.; Rafiq, M.; Kamil, M.A. Bacterial Biofilm and Associated Infections. J. Chin. Med. Assoc. JCMA 2018, 81, 7–11. [Google Scholar] [CrossRef]
- Sauer, K.; Cullen, M.C.; Rickard, A.H.; Zeef, L.A.H.; Davies, D.G.; Gilbert, P. Characterization of Nutrient-Induced Dispersion in Pseudomonas Aeruginosa PAO1 Biofilm. J. Bacteriol. 2004, 186, 7312–7326. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, J.B.; Velliyagounder, K.; Ragunath, C.; Rohde, H.; Mack, D.; Knobloch, J.K.-M.; Ramasubbu, N. Genes Involved in the Synthesis and Degradation of Matrix Polysaccharide in Actinobacillus Actinomycetemcomitans and Actinobacillus Pleuropneumoniae Biofilms. J. Bacteriol. 2004, 186, 8213–8220. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, J.B. Biofilm Dispersal. J. Dent. Res. 2010, 89, 205–218. [Google Scholar] [CrossRef] [Green Version]
- Pisithkul, T.; Schroeder, J.W.; Trujillo, E.A.; Yeesin, P.; Stevenson, D.M.; Chaiamarit, T.; Coon, J.J.; Wang, J.D.; Amador-Noguez, D. Metabolic Remodeling during Biofilm Development of Bacillus Subtilis. mBio 2019, 10, e00623-19. [Google Scholar] [CrossRef] [Green Version]
- Wood, T.K.; González Barrios, A.F.; Herzberg, M.; Lee, J. Motility Influences Biofilm Architecture in Escherichia Coli. Appl. Microbiol. Biotechnol. 2006, 72, 361–367. [Google Scholar] [CrossRef]
- Harris, L.G.; Murray, S.; Pascoe, B.; Bray, J.; Meric, G.; Mageiros, L.; Wilkinson, T.S.; Jeeves, R.; Rohde, H.; Schwarz, S.; et al. Biofilm Morphotypes and Population Structure among Staphylococcus Epidermidis from Commensal and Clinical Samples. PloS ONE 2016, 11, e0151240. [Google Scholar] [CrossRef]
- Flemming, H.-C.; Wingender, J. The Biofilm Matrix. Nat. Rev. Microbiol. 2010, 8, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Flemming, H.-C. EPS—Then, and Now. Microorganisms 2016, 4, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, O.Y.A.; Raaijmakers, J.M.; Kuramae, E.E. Microbial Extracellular Polymeric Substances: Ecological Function and Impact on Soil Aggregation. Front. Microbiol. 2018, 9, 1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limoli, D.H.; Jones, C.J.; Wozniak, D.J. Bacterial Extracellular Polysaccharides in Biofilm Formation and Function. Microbiol. Spectr. 2015, 3, 3. [Google Scholar] [CrossRef] [Green Version]
- Montanaro, L.; Poggi, A.; Visai, L.; Ravaioli, S.; Campoccia, D.; Speziale, P.; Arciola, C.R. Extracellular DNA in Biofilms. Int. J. Artif. Organs 2011, 34, 824–831. [Google Scholar] [CrossRef]
- Whitchurch, C.B.; Tolker-Nielsen, T.; Ragas, P.C.; Mattick, J.S. Extracellular DNA Required for Bacterial Biofilm Formation. Science 2002, 295, 1487. [Google Scholar] [CrossRef]
- Tielker, D.; Hacker, S.; Loris, R.; Strathmann, M.; Wingender, J.; Wilhelm, S.; Rosenau, F.; Jaeger, K.-E. Pseudomonas Aeruginosa Lectin LecB Is Located in the Outer Membrane and Is Involved in Biofilm Formation. Microbiol. Read. Engl. 2005, 151, 1313–1323. [Google Scholar] [CrossRef]
- Lasa, I.; Penadés, J.R. Bap: A Family of Surface Proteins Involved in Biofilm Formation. Res. Microbiol. 2006, 157, 99–107. [Google Scholar] [CrossRef]
- Taglialegna, A.; Lasa, I.; Valle, J. Amyloid Structures as Biofilm Matrix Scaffolds. J. Bacteriol. 2016, 198, 2579–2588. [Google Scholar] [CrossRef] [Green Version]
- Barnhart, M.M.; Chapman, M.R. Curli Biogenesis and Function. Annu. Rev. Microbiol. 2006, 60, 131–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passos da Silva, D.; Matwichuk, M.L.; Townsend, D.O.; Reichhardt, C.; Lamba, D.; Wozniak, D.J.; Parsek, M.R. The Pseudomonas Aeruginosa Lectin LecB Binds to the Exopolysaccharide Psl and Stabilizes the Biofilm Matrix. Nat. Commun. 2019, 10, 2183. [Google Scholar] [CrossRef] [PubMed]
- Potempa, M.; Potempa, J. Protease-Dependent Mechanisms of Complement Evasion by Bacterial Pathogens. Biol. Chem. 2012, 393, 873–888. [Google Scholar] [CrossRef] [Green Version]
- Laarman, A.J.; Ruyken, M.; Malone, C.L.; van Strijp, J.A.G.; Horswill, A.R.; Rooijakkers, S.H.M. Staphylococcus Aureus Metalloprotease Aureolysin Cleaves Complement C3 to Mediate Immune Evasion. J. Immunol. 2011, 186, 6445–6453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Pawel-Rammingen, U.; Johansson, B.P.; Björck, L. IdeS, a Novel Streptococcal Cysteine Proteinase with Unique Specificity for Immunoglobulin G. EMBO J. 2002, 21, 1607–1615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naqvi, N.; Liu, K.; Graham, R.M.; Husain, A. Molecular Basis of Exopeptidase Activity in the C-Terminal Domain of Human Angiotensin I-Converting Enzyme: Insights into the Origins of Its Exopeptidase Activity. J. Biol. Chem. 2005, 280, 6669–6675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooper, N.M. Proteases: A Primer. Essays Biochem. 2002, 38, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, T.; Eckhard, U.; Dufour, A.; Solis, N.; Overall, C.M. Proteolytic Cleavage-Mechanisms, Function, and “Omic” Approaches for a Near-Ubiquitous Posttranslational Modification. Chem. Rev. 2018, 118, 1137–1168. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS Database of Proteolytic Enzymes, Their Substrates and Inhibitors in 2017 and a Comparison with Peptidases in the PANTHER Database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef]
- Martínez-García, S.; Rodríguez-Martínez, S.; Cancino-Diaz, M.E.; Cancino-Diaz, J.C. Extracellular Proteases of Staphylococcus Epidermidis: Roles as Virulence Factors and Their Participation in Biofilm. APMIS Acta Pathol. Microbiol. Immunol. Scand. 2018, 126, 177–185. [Google Scholar] [CrossRef]
- Chen, C.; Krishnan, V.; Macon, K.; Manne, K.; Narayana, S.V.L.; Schneewind, O. Secreted Proteases Control Autolysin-Mediated Biofilm Growth of Staphylococcus Aureus. J. Biol. Chem. 2013, 288, 29440–29452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Banbula, A.; Potempa, J.; Travis, J.; Fernandez-Catalán, C.; Mann, K.; Huber, R.; Bode, W.; Medrano, F. Amino-Acid Sequence and Three-Dimensional Structure of the Staphylococcus Aureus Metalloproteinase at 1.72 A Resolution. Struct. Lond. Engl. 1998, 6, 1185–1193. [Google Scholar] [CrossRef] [Green Version]
- DeLano, W.L. The Case for Open-Source Software in Drug Discovery. Drug Discov. Today 2005, 10, 213–217. [Google Scholar] [CrossRef]
- Hooper, N.M. Families of Zinc Metalloproteases. FEBS Lett. 1994, 354, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Cerdà-Costa, N.; Gomis-Rüth, F.X. Architecture and Function of Metallopeptidase Catalytic Domains. Protein Sci. Publ. Protein Soc. 2014, 23, 123–144. [Google Scholar] [CrossRef] [Green Version]
- Kataoka, Y.; Takada, K.; Oyama, H.; Tsunemi, M.; James, M.N.G.; Oda, K. Catalytic Residues and Substrate Specificity of Scytalidoglutamic Peptidase, the First Member of the Eqolisin in Family (G1) of Peptidases. FEBS Lett. 2005, 579, 2991–2994. [Google Scholar] [CrossRef] [Green Version]
- Blundell, T.L.; Guruprasad, K.; Albert, A.; Williams, M.; Sibanda, B.L.; Dhanaraj, V. The Aspartic Proteinases. An Historical Overview. Adv. Exp. Med. Biol. 1998, 436, 1–13. [Google Scholar]
- Davies, D.R. The Structure and Function of the Aspartic Proteinases. Annu. Rev. Biophys. Biophys. Chem. 1990, 19, 189–215. [Google Scholar] [CrossRef]
- Jensen, K.; Østergaard, P.R.; Wilting, R.; Lassen, S.F. Identification and Characterization of a Bacterial Glutamic Peptidase. BMC Biochem. 2010, 11, 47. [Google Scholar] [CrossRef] [Green Version]
- Hedstrom, L. Serine Protease Mechanism and Specificity. Chem. Rev. 2002, 102, 4501–4524. [Google Scholar] [CrossRef] [PubMed]
- Polgár, L. The Catalytic Triad of Serine Peptidases. Cell. Mol. Life Sci. CMLS 2005, 62, 2161–2172. [Google Scholar] [CrossRef] [PubMed]
- Rzychon, M.; Chmiel, D.; Stec-Niemczyk, J. Modes of Inhibition of Cysteine Proteases. Acta Biochim. Pol. 2004, 51, 861–873. [Google Scholar] [PubMed]
- Vicik, R.; Busemann, M.; Baumann, K.; Schirmeister, T. Inhibitors of Cysteine Proteases. Curr. Top. Med. Chem. 2006, 6, 331–353. [Google Scholar] [CrossRef] [PubMed]
- Brannigan, J.A.; Dodson, G.; Duggleby, H.J.; Moody, P.C.; Smith, J.L.; Tomchick, D.R.; Murzin, A.G. A Protein Catalytic Framework with an N-Terminal Nucleophile Is Capable of Self-Activation. Nature 1995, 378, 416–419. [Google Scholar] [CrossRef]
- Dodson, G.; Wlodawer, A. Catalytic Triads and Their Relatives. Trends Biochem. Sci. 1998, 23, 347–352. [Google Scholar] [CrossRef]
- Murphy, G. The ADAMs: Signalling Scissors in the Tumour Microenvironment. Nat. Rev. Cancer 2008, 8, 929–941. [Google Scholar] [CrossRef]
- Nguyen, T.T.H.; Myrold, D.D.; Mueller, R.S. Distributions of Extracellular Peptidases Across Prokaryotic Genomes Reflect Phylogeny and Habitat. Front. Microbiol. 2019, 10, 413. [Google Scholar] [CrossRef] [Green Version]
- Fernández, L.; Breidenstein, E.B.M.; Song, D.; Hancock, R.E.W. Role of Intracellular Proteases in the Antibiotic Resistance, Motility, and Biofilm Formation of Pseudomonas Aeruginosa. Antimicrob. Agents Chemother. 2012, 56, 1128–1132. [Google Scholar] [CrossRef] [Green Version]
- Soltes, G.R.; Martin, N.R.; Park, E.; Sutterlin, H.A.; Silhavy, T.J. Distinctive Roles for Periplasmic Proteases in the Maintenance of Essential Outer Membrane Protein Assembly. J. Bacteriol. 2017, 199, e00418-17. [Google Scholar] [CrossRef] [Green Version]
- Frees, D.; Brøndsted, L.; Ingmer, H. Bacterial Proteases and Virulence. Subcell. Biochem. 2013, 66, 161–192. [Google Scholar] [CrossRef] [PubMed]
- Green, E.R.; Mecsas, J. Bacterial Secretion Systems: An Overview. Microbiol. Spectr. 2016, 4, 4–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natale, P.; Brüser, T.; Driessen, A.J.M. Sec- and Tat-Mediated Protein Secretion across the Bacterial Cytoplasmic Membrane--Distinct Translocases and Mechanisms. Biochim. Biophys. Acta 2008, 1778, 1735–1756. [Google Scholar] [CrossRef] [Green Version]
- Müller, M. Twin-Arginine-Specific Protein Export in Escherichia Coli. Res. Microbiol. 2005, 156, 131–136. [Google Scholar] [CrossRef]
- Goosens, V.J.; Monteferrante, C.G.; van Dijl, J.M. The Tat System of Gram-Positive Bacteria. Biochim. Biophys. Acta 2014, 1843, 1698–1706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anné, J.; Economou, A.; Bernaerts, K. Protein Secretion in Gram-Positive Bacteria: From Multiple Pathways to Biotechnology. Curr. Top. Microbiol. Immunol. 2017, 404, 267–308. [Google Scholar] [CrossRef] [Green Version]
- Bottai, D.; Gröschel, M.I.; Brosch, R. Type VII Secretion Systems in Gram-Positive Bacteria. Curr. Top. Microbiol. Immunol. 2017, 404, 235–265. [Google Scholar] [CrossRef]
- Depluverez, S.; Devos, S.; Devreese, B. The Role of Bacterial Secretion Systems in the Virulence of Gram-Negative Airway Pathogens Associated with Cystic Fibrosis. Front. Microbiol. 2016, 7, 1336. [Google Scholar] [CrossRef] [Green Version]
- Tseng, T.-T.; Tyler, B.M.; Setubal, J.C. Protein Secretion Systems in Bacterial-Host Associations, and Their Description in the Gene Ontology. BMC Microbiol. 2009, 9, S2. [Google Scholar] [CrossRef] [Green Version]
- Costa, T.R.D.; Felisberto-Rodrigues, C.; Meir, A.; Prevost, M.S.; Redzej, A.; Trokter, M.; Waksman, G. Secretion Systems in Gram-Negative Bacteria: Structural and Mechanistic Insights. Nat. Rev. Microbiol. 2015, 13, 343–359. [Google Scholar] [CrossRef]
- Nicholson, L.B. The Immune System. Essays Biochem. 2016, 60, 275–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woof, J.M.; Burton, D.R. Human Antibody-Fc Receptor Interactions Illuminated by Crystal Structures. Nat. Rev. Immunol. 2004, 4, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Sarma, J.V.; Ward, P.A. The Complement System. Cell Tissue Res. 2011, 343, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Dunkelberger, J.R.; Song, W.-C. Complement and Its Role in Innate and Adaptive Immune Responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef] [Green Version]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part I - Molecular Mechanisms of Activation and Regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef] [Green Version]
- Cole, J.N.; Nizet, V. Bacterial Evasion of Host Antimicrobial Peptide Defenses. Microbiol. Spectr. 2016, 4, 4–11. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Haney, E.F.; Vogel, H.J. The Expanding Scope of Antimicrobial Peptide Structures and Their Modes of Action. Trends Biotechnol. 2011, 29, 464–472. [Google Scholar] [CrossRef]
- Oren, Z.; Shai, Y. Mode of Action of Linear Amphipathic Alpha-Helical Antimicrobial Peptides. Biopolymers 1998, 47, 451–463. [Google Scholar] [CrossRef]
- Park, S.-C.; Park, Y.; Hahm, K.-S. The Role of Antimicrobial Peptides in Preventing Multidrug-Resistant Bacterial Infections and Biofilm Formation. Int. J. Mol. Sci. 2011, 12, 5971–5992. [Google Scholar] [CrossRef] [Green Version]
- de Vor, L.; Rooijakkers, S.H.M.; van Strijp, J.A.G. Staphylococci Evade the Innate Immune Response by Disarming Neutrophils and Forming Biofilms. FEBS Lett. 2020, 594, 2556–2569. [Google Scholar] [CrossRef] [Green Version]
- von Pawel-Rammingen, U.; Björck, L. IdeS and SpeB: Immunoglobulin-Degrading Cysteine Proteinases of Streptococcus Pyogenes. Curr. Opin. Microbiol. 2003, 6, 50–55. [Google Scholar] [CrossRef]
- Vindebro, R.; Spoerry, C.; von Pawel-Rammingen, U. Rapid IgG Heavy Chain Cleavage by the Streptococcal IgG Endopeptidase IdeS Is Mediated by IdeS Monomers and Is Not Due to Enzyme Dimerization. FEBS Lett. 2013, 587, 1818–1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenig, K.; Chatwell, L.; von Pawel-Rammingen, U.; Björck, L.; Huber, R.; Sondermann, P. Structure of the Streptococcal Endopeptidase IdeS, a Cysteine Proteinase with Strict Specificity for IgG. Proc. Natl. Acad. Sci. USA 2004, 101, 17371–17376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brezski, R.J.; Vafa, O.; Petrone, D.; Tam, S.H.; Powers, G.; Ryan, M.H.; Luongo, J.L.; Oberholtzer, A.; Knight, D.M.; Jordan, R.E. Tumor-Associated and Microbial Proteases Compromise Host IgG Effector Functions by a Single Cleavage Proximal to the Hinge. Proc. Natl. Acad. Sci. USA 2009, 106, 17864–17869. [Google Scholar] [CrossRef] [Green Version]
- Blöchl, C.; Holzner, C.; Luciano, M.; Bauer, R.; Horejs-Hoeck, J.; Eckhard, U.; Brandstetter, H.; Huber, C.G. Proteolytic Profiling of Streptococcal Pyrogenic Exotoxin B (SpeB) by Complementary HPLC-MS Approaches. Int. J. Mol. Sci. 2022, 23, 412. [Google Scholar] [CrossRef]
- Collin, M.; Olsén, A. Effect of SpeB and EndoS from Streptococcus Pyogenes on Human Immunoglobulins. Infect. Immun. 2001, 69, 7187–7189. [Google Scholar] [CrossRef] [Green Version]
- Woehl, J.L.; Kitamura, S.; Dillon, N.; Han, Z.; Edgar, L.J.; Nizet, V.; Wolan, D.W. An Irreversible Inhibitor to Probe the Role of Streptococcus Pyogenes Cysteine Protease SpeB in Evasion of Host Complement Defenses. ACS Chem. Biol. 2020, 15, 2060–2069. [Google Scholar] [CrossRef]
- McKenna, S.; Huse, K.K.; Giblin, S.; Pearson, M.; Majid Al Shibar, M.S.; Sriskandan, S.; Matthews, S.; Pease, J.E. The Role of Streptococcal Cell-Envelope Proteases in Bacterial Evasion of the Innate Immune System. J. Innate Immun. 2021, 14, 69–88. [Google Scholar] [CrossRef]
- Edwards, R.J.; Taylor, G.W.; Ferguson, M.; Murray, S.; Rendell, N.; Wrigley, A.; Bai, Z.; Boyle, J.; Finney, S.J.; Jones, A.; et al. Specific C-Terminal Cleavage and Inactivation of Interleukin-8 by Invasive Disease Isolates of Streptococcus Pyogenes. J. Infect. Dis. 2005, 192, 783–790. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.; McLandsborough, L.; Kondagunta, A.; Cleary, P.P. C5a Peptidase Alters Clearance and Trafficking of Group A Streptococci by Infected Mice. Infect. Immun. 1996, 64, 503–510. [Google Scholar] [CrossRef] [Green Version]
- Zinkernagel, A.S.; Timmer, A.M.; Pence, M.A.; Locke, J.B.; Buchanan, J.T.; Turner, C.E.; Mishalian, I.; Sriskandan, S.; Hanski, E.; Nizet, V. The IL-8 Protease SpyCEP/ScpC of Group A Streptococcus Promotes Resistance to Neutrophil Killing. Cell Host Microbe 2008, 4, 170–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abreu, A.G.; Fraga, T.R.; Granados Martínez, A.P.; Kondo, M.Y.; Juliano, M.A.; Juliano, L.; Navarro-Garcia, F.; Isaac, L.; Barbosa, A.S.; Elias, W.P. The Serine Protease Pic From Enteroaggregative Escherichia Coli Mediates Immune Evasion by the Direct Cleavage of Complement Proteins. J. Infect. Dis. 2015, 212, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Orth, D.; Ehrlenbach, S.; Brockmeyer, J.; Khan, A.B.; Huber, G.; Karch, H.; Sarg, B.; Lindner, H.; Würzner, R. EspP, a Serine Protease of Enterohemorrhagic Escherichia Coli, Impairs Complement Activation by Cleaving Complement Factors C3/C3b and C5. Infect. Immun. 2010, 78, 4294–4301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandeputte-Rutten, L.; Kramer, R.A.; Kroon, J.; Dekker, N.; Egmond, M.R.; Gros, P. Crystal Structure of the Outer Membrane Protease OmpT from Escherichia Coli Suggests a Novel Catalytic Site. EMBO J. 2001, 20, 5033–5039. [Google Scholar] [CrossRef] [Green Version]
- Stumpe, S.; Schmid, R.; Stephens, D.L.; Georgiou, G.; Bakker, E.P. Identification of OmpT as the Protease That Hydrolyzes the Antimicrobial Peptide Protamine before It Enters Growing Cells of Escherichia Coli. J. Bacteriol. 1998, 180, 4002–4006. [Google Scholar] [CrossRef] [Green Version]
- Brannon, J.R.; Thomassin, J.-L.; Desloges, I.; Gruenheid, S.; Le Moual, H. Role of Uropathogenic Escherichia Coli OmpT in the Resistance against Human Cathelicidin LL-37. FEMS Microbiol. Lett. 2013, 345, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Desloges, I.; Taylor, J.A.; Leclerc, J.-M.; Brannon, J.R.; Portt, A.; Spencer, J.D.; Dewar, K.; Marczynski, G.T.; Manges, A.; Gruenheid, S.; et al. Identification and Characterization of OmpT-like Proteases in Uropathogenic Escherichia Coli Clinical Isolates. MicrobiologyOpen 2019, 8, e915. [Google Scholar] [CrossRef] [Green Version]
- Guina, T.; Yi, E.C.; Wang, H.; Hackett, M.; Miller, S.I. A PhoP-Regulated Outer Membrane Protease of Salmonella Enterica Serovar Typhimurium Promotes Resistance to Alpha-Helical Antimicrobial Peptides. J. Bacteriol. 2000, 182, 4077–4086. [Google Scholar] [CrossRef] [Green Version]
- Ramu, P.; Tanskanen, R.; Holmberg, M.; Lähteenmäki, K.; Korhonen, T.K.; Meri, S. The Surface Protease PgtE of Salmonella Enterica Affects Complement Activity by Proteolytically Cleaving C3b, C4b and C5. FEBS Lett. 2007, 581, 1716–1720. [Google Scholar] [CrossRef] [Green Version]
- Overhage, J.; Campisano, A.; Bains, M.; Torfs, E.C.W.; Rehm, B.H.A.; Hancock, R.E.W. Human Host Defense Peptide LL-37 Prevents Bacterial Biofilm Formation. Infect. Immun. 2008, 76, 4176–4182. [Google Scholar] [CrossRef] [Green Version]
- Schmidtchen, A.; Frick, I.-M.; Andersson, E.; Tapper, H.; Björck, L. Proteinases of Common Pathogenic Bacteria Degrade and Inactivate the Antibacterial Peptide LL-37. Mol. Microbiol. 2002, 46, 157–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Ge, X.; Xie, D.; Wang, S.; Chen, F.; Pan, S. Clinical Strains of Pseudomonas Aeruginosa Secrete LasB Elastase to Induce Hemorrhagic Diffuse Alveolar Damage in Mice. J. Inflamm. Res. 2021, 14, 3767–3780. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.Q.; Ghebrehiwet, B. Effect of Pseudomonas Aeruginosa Elastase and Alkaline Protease on Serum Complement and Isolated Components C1q and C3. Clin. Immunol. Immunopathol. 1992, 62, 133–138. [Google Scholar] [CrossRef]
- Holder, I.A.; Wheeler, R. Experimental Studies of the Pathogenesis of Infections Owing to Pseudomonas Aeruginosa: Elastase, an IgG Protease. Can. J. Microbiol. 1984, 30, 1118–1124. [Google Scholar] [CrossRef]
- Heck, L.W.; Alarcon, P.G.; Kulhavy, R.M.; Morihara, K.; Russell, M.W.; Mestecky, J.F. Degradation of IgA Proteins by Pseudomonas Aeruginosa Elastase. J. Immunol. 1990, 144, 2253–2257. [Google Scholar]
- Spoerry, C.; Karlsson, J.; Aschtgen, M.-S.; Loh, E. Neisseria Meningitidis IgA1-Specific Serine Protease Exhibits Novel Cleavage Activity against IgG3. Virulence 2021, 12, 389–403. [Google Scholar] [CrossRef]
- Plaut, A.G.; Gilbert, J.V.; Artenstein, M.S.; Capra, J.D. Neisseria Gonorrhoeae and Neisseria Meningitidis: Extracellular Enzyme Cleaves Human Immunoglobulin A. Science 1975, 190, 1103–1105. [Google Scholar] [CrossRef]
- Coureuil, M.; Join-Lambert, O.; Lécuyer, H.; Bourdoulous, S.; Marullo, S.; Nassif, X. Pathogenesis of Meningococcemia. Cold Spring Harb. Perspect. Med. 2013, 3, a012393. [Google Scholar] [CrossRef]
- Schulze, A.; Mitterer, F.; Pombo, J.P.; Schild, S. Biofilms by Bacterial Human Pathogens: Clinical Relevance—Development, Composition and Regulation—Therapeutical Strategies. Microb. Cell Graz Austria 2021, 8, 28–56. [Google Scholar] [CrossRef]
- Matsushita, O.; Okabe, A. Clostridial Hydrolytic Enzymes Degrading Extracellular Components. Toxicon Off. J. Int. Soc. Toxinology 2001, 39, 1769–1780. [Google Scholar] [CrossRef]
- Eckhard, U.; Schönauer, E.; Nüss, D.; Brandstetter, H. Structure of Collagenase G Reveals a Chew-and-Digest Mechanism of Bacterial Collagenolysis. Nat. Struct. Mol. Biol. 2011, 18, 1109–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckhard, U.; Schönauer, E.; Brandstetter, H. Structural Basis for Activity Regulation and Substrate Preference of Clostridial Collagenases G, H, and T. J. Biol. Chem. 2013, 288, 20184–20194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrington, S.M.; Sheikh, J.; Henderson, I.R.; Ruiz-Perez, F.; Cohen, P.S.; Nataro, J.P. The Pic Protease of Enteroaggregative Escherichia Coli Promotes Intestinal Colonization and Growth in the Presence of Mucin. Infect. Immun. 2009, 77, 2465–2473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Saitz-Rojas, W.; Smith, R.; Gonyar, L.; In, J.G.; Kovbasnjuk, O.; Zachos, N.C.; Donowitz, M.; Nataro, J.P.; Ruiz-Perez, F. Mucus Layer Modeling of Human Colonoids during Infection with Enteroaggragative E. Coli. Sci. Rep. 2020, 10, 10533. [Google Scholar] [CrossRef] [PubMed]
- Goulas, T.; Arolas, J.L.; Gomis-Rüth, F.X. Structure, Function and Latency Regulation of a Bacterial Enterotoxin Potentially Derived from a Mammalian Adamalysin/ADAM Xenolog. Proc. Natl. Acad. Sci. USA 2011, 108, 1856–1861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Tassell, R.L.; Lyerly, D.M.; Wilkins, T.D. Purification and Characterization of an Enterotoxin from Bacteroides Fragilis. Infect. Immun. 1992, 60, 1343–1350. [Google Scholar] [CrossRef] [Green Version]
- Obiso, R.J.; Azghani, A.O.; Wilkins, T.D. The Bacteroides Fragilis Toxin Fragilysin Disrupts the Paracellular Barrier of Epithelial Cells. Infect. Immun. 1997, 65, 1431–1439. [Google Scholar] [CrossRef] [Green Version]
- Devaux, C.A.; Mezouar, S.; Mege, J.-L. The E-Cadherin Cleavage Associated to Pathogenic Bacteria Infections Can Favor Bacterial Invasion and Transmigration, Dysregulation of the Immune Response and Cancer Induction in Humans. Front. Microbiol. 2019, 10, 2598. [Google Scholar] [CrossRef]
- Pierce, J.V.; Bernstein, H.D. Genomic Diversity of Enterotoxigenic Strains of Bacteroides Fragilis. PloS ONE 2016, 11, e0158171. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-R.; Rhee, K.-J.; Eom, Y.-B. Anti-Biofilm and Antimicrobial Effects of Zerumbone against Bacteroides Fragilis. Anaerobe 2019, 57, 99–106. [Google Scholar] [CrossRef]
- Rhee, K.-J.; Wu, S.; Wu, X.; Huso, D.L.; Karim, B.; Franco, A.A.; Rabizadeh, S.; Golub, J.E.; Mathews, L.E.; Shin, J.; et al. Induction of Persistent Colitis by a Human Commensal, Enterotoxigenic Bacteroides Fragilis, in Wild-Type C57BL/6 Mice. Infect. Immun. 2009, 77, 1708–1718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, W.T.; Kantilal, H.K.; Davamani, F. The Mechanism of Bacteroides Fragilis Toxin Contributes to Colon Cancer Formation. Malays. J. Med. Sci. MJMS 2020, 27, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Hoy, B.; Löwer, M.; Weydig, C.; Carra, G.; Tegtmeyer, N.; Geppert, T.; Schröder, P.; Sewald, N.; Backert, S.; Schneider, G.; et al. Helicobacter Pylori HtrA Is a New Secreted Virulence Factor That Cleaves E-Cadherin to Disrupt Intercellular Adhesion. EMBO Rep. 2010, 11, 798–804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrer, A.; Boehm, M.; Backert, S.; Tegtmeyer, N. Overexpression of Serine Protease HtrA Enhances Disruption of Adherens Junctions, Paracellular Transmigration and Type IV Secretion of CagA by Helicobacter Pylori. Gut Pathog. 2017, 9, 40. [Google Scholar] [CrossRef]
- Zarzecka, U.; Modrak-Wójcik, A.; Figaj, D.; Apanowicz, M.; Lesner, A.; Bzowska, A.; Lipinska, B.; Zawilak-Pawlik, A.; Backert, S.; Skorko-Glonek, J. Properties of the HtrA Protease From Bacterium Helicobacter Pylori Whose Activity Is Indispensable for Growth Under Stress Conditions. Front. Microbiol. 2019, 10, 961. [Google Scholar] [CrossRef] [PubMed]
- Zarzecka, U.; Harrer, A.; Zawilak-Pawlik, A.; Skorko-Glonek, J.; Backert, S. Chaperone Activity of Serine Protease HtrA of Helicobacter Pylori as a Crucial Survival Factor under Stress Conditions. Cell Commun. Signal. CCS 2019, 17, 161. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.; Biswas, I. Role of HtrA in Surface Protein Expression and Biofilm Formation by Streptococcus Mutans. Infect. Immun. 2005, 73, 6923–6934. [Google Scholar] [CrossRef] [Green Version]
- Shaw, L.; Golonka, E.; Potempa, J.; Foster, S.J. The Role and Regulation of the Extracellular Proteases of Staphylococcus Aureus. Microbiol. Read. Engl. 2004, 150, 217–228. [Google Scholar] [CrossRef] [Green Version]
- Fortelny, N.; Cox, J.H.; Kappelhoff, R.; Starr, A.E.; Lange, P.F.; Pavlidis, P.; Overall, C.M. Network Analyses Reveal Pervasive Functional Regulation between Proteases in the Human Protease Web. PLoS Biol. 2014, 12, e1001869. [Google Scholar] [CrossRef]
- Valle, J.; Latasa, C.; Gil, C.; Toledo-Arana, A.; Solano, C.; Penadés, J.R.; Lasa, I. Bap, a Biofilm Matrix Protein of Staphylococcus Aureus Prevents Cellular Internalization through Binding to GP96 Host Receptor. PLoS Pathog. 2012, 8, e1002843. [Google Scholar] [CrossRef] [Green Version]
- Pietrocola, G.; Nobile, G.; Rindi, S.; Speziale, P. Staphylococcus Aureus Manipulates Innate Immunity through Own and Host-Expressed Proteases. Front. Cell. Infect. Microbiol. 2017, 7, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendes, S.R.; Eckhard, U.; Rodríguez-Banqueri, A.; Guevara, T.; Czermak, P.; Marcos, E.; Vilcinskas, A.; Xavier Gomis-Rüth, F. An Engineered Protein-Based Submicromolar Competitive Inhibitor of the Staphylococcus Aureus Virulence Factor Aureolysin. Comput. Struct. Biotechnol. J. 2022, 20, 534–544. [Google Scholar] [CrossRef]
- Corrigan, R.M.; Rigby, D.; Handley, P.; Foster, T.J. The Role of Staphylococcus Aureus Surface Protein SasG in Adherence and Biofilm Formation. Microbiol. Read. Engl. 2007, 153, 2435–2446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.F.; Gao, L. Mutational Analysis of the C-Terminal Anchoring Domains of Streptococcus Mutans P1 Antigen: Role of the LPXTGX Motif in P1 Association with the Cell Wall. Can. J. Microbiol. 2000, 46, 584–592. [Google Scholar] [CrossRef]
- Lister, J.L.; Horswill, A.R. Staphylococcus Aureus Biofilms: Recent Developments in Biofilm Dispersal. Front. Cell. Infect. Microbiol. 2014, 4, 178. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.F.; Boran, T.L. Roles of Sortase in Surface Expression of the Major Protein Adhesin P1, Saliva-Induced Aggregation and Adherence, and Cariogenicity of Streptococcus Mutans. Infect. Immun. 2003, 71, 676–681. [Google Scholar] [CrossRef] [Green Version]
- Paterson, G.K.; Mitchell, T.J. The Biology of Gram-Positive Sortase Enzymes. Trends Microbiol. 2004, 12, 89–95. [Google Scholar] [CrossRef]
- Chen, S.; Paterson, G.K.; Tong, H.H.; Mitchell, T.J.; DeMaria, T.F. Sortase A Contributes to Pneumococcal Nasopharyngeal Colonization in the Chinchilla Model. FEMS Microbiol. Lett. 2005, 253, 151–154. [Google Scholar] [CrossRef] [Green Version]
- Qin, Z.; Ou, Y.; Yang, L.; Zhu, Y.; Tolker-Nielsen, T.; Molin, S.; Qu, D. Role of Autolysin-Mediated DNA Release in Biofilm Formation of Staphylococcus Epidermidis. Microbiol. Read. Engl. 2007, 153, 2083–2092. [Google Scholar] [CrossRef] [Green Version]
- Raju, R.M.; Goldberg, A.L.; Rubin, E.J. Bacterial Proteolytic Complexes as Therapeutic Targets. Nat. Rev. Drug Discov. 2012, 11, 777–789. [Google Scholar] [CrossRef]
- Razzaq, A.; Shamsi, S.; Ali, A.; Ali, Q.; Sajjad, M.; Malik, A.; Ashraf, M. Microbial Proteases Applications. Front. Bioeng. Biotechnol. 2019, 7, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nigam, P.K.; Nigam, A. BOTULINUM TOXIN. Indian J. Dermatol. 2010, 55, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Craik, C.S.; Page, M.J.; Madison, E.L. Proteases as Therapeutics. Biochem. J. 2011, 435, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doft, M.A.; Hardy, K.L.; Ascherman, J.A. Treatment of Hyperhidrosis with Botulinum Toxin. Aesthet. Surg. J. 2012, 32, 238–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, A. History and Uses of BOTOX (Botulinum Toxin Type A). Lippincotts Case Manag. Manag. Process Patient Care 2004, 9, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Dressler, D.; Eleopra, R. Clinical Use of Non-A Botulinum Toxins: Botulinum Toxin Type B. Neurotox. Res. 2006, 9, 121–125. [Google Scholar] [CrossRef]
- Hurst, L.C.; Badalamente, M.A.; Hentz, V.R.; Hotchkiss, R.N.; Kaplan, F.T.D.; Meals, R.A.; Smith, T.M.; Rodzvilla, J. CORD I Study Group Injectable Collagenase Clostridium Histolyticum for Dupuytren’s Contracture. N. Engl. J. Med. 2009, 361, 968–979. [Google Scholar] [CrossRef] [Green Version]
- Cocci, A.; Russo, G.I.; Salamanca, J.I.M.; Ralph, D.; Palmieri, A.; Mondaini, N. The End of an Era: Withdrawal of Xiapex (Clostridium Histolyticum Collagenase) from the European Market. Eur. Urol. 2020, 77, 660–661. [Google Scholar] [CrossRef]
- Lonze, B.E.; Tatapudi, V.S.; Weldon, E.P.; Min, E.S.; Ali, N.M.; Deterville, C.L.; Gelb, B.E.; Benstein, J.A.; Dagher, N.N.; Wu, M.; et al. IdeS (Imlifidase): A Novel Agent That Cleaves Human IgG and Permits Successful Kidney Transplantation Across High-Strength Donor-Specific Antibody. Ann. Surg. 2018, 268, 488–496. [Google Scholar] [CrossRef]
- Lamm, M.E.; Emancipator, S.N.; Robinson, J.K.; Yamashita, M.; Fujioka, H.; Qiu, J.; Plaut, A.G. Microbial IgA Protease Removes IgA Immune Complexes from Mouse Glomeruli in Vivo: Potential Therapy for IgA Nephropathy. Am. J. Pathol. 2008, 172, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Wall, R.J.; Powell, A.M.; Paape, M.J.; Kerr, D.E.; Bannerman, D.D.; Pursel, V.G.; Wells, K.D.; Talbot, N.; Hawk, H.W. Genetically Enhanced Cows Resist Intramammary Staphylococcus Aureus Infection. Nat. Biotechnol. 2005, 23, 445–451. [Google Scholar] [CrossRef]
- Cabanillas, B.; Pedrosa, M.M.; Rodríguez, J.; Muzquiz, M.; Maleki, S.J.; Cuadrado, C.; Burbano, C.; Crespo, J.F. Influence of Enzymatic Hydrolysis on the Allergenicity of Roasted Peanut Protein Extract. Int. Arch. Allergy Immunol. 2012, 157, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Banik, R.M.; Prakash, M. Laundry Detergent Compatibility of the Alkaline Protease from Bacillus Cereus. Microbiol. Res. 2004, 159, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, C.; Regl, C.; Huber, C.G.; Winter, P.; Weiss, R.; Wohlschlager, T. Towards Middle-up Analysis of Polyclonal Antibodies: Subclass-Specific N-Glycosylation Profiling of Murine Immunoglobulin G (IgG) by Means of HPLC-MS. Sci. Rep. 2020, 10, 18080. [Google Scholar] [CrossRef] [PubMed]
- Duivelshof, B.L.; Denorme, S.; Sandra, K.; Liu, X.; Beck, A.; Lauber, M.A.; Guillarme, D.; D’Atri, V. Quantitative N-Glycan Profiling of Therapeutic Monoclonal Antibodies Performed by Middle-Up Level HILIC-HRMS Analysis. Pharmaceutics 2021, 13, 1744. [Google Scholar] [CrossRef]
- Lu, R.-M.; Hwang, Y.-C.; Liu, I.-J.; Lee, C.-C.; Tsai, H.-Z.; Li, H.-J.; Wu, H.-C. Development of Therapeutic Antibodies for the Treatment of Diseases. J. Biomed. Sci. 2020, 27, 1. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramírez-Larrota, J.S.; Eckhard, U. An Introduction to Bacterial Biofilms and Their Proteases, and Their Roles in Host Infection and Immune Evasion. Biomolecules 2022, 12, 306. https://doi.org/10.3390/biom12020306
Ramírez-Larrota JS, Eckhard U. An Introduction to Bacterial Biofilms and Their Proteases, and Their Roles in Host Infection and Immune Evasion. Biomolecules. 2022; 12(2):306. https://doi.org/10.3390/biom12020306
Chicago/Turabian StyleRamírez-Larrota, Juan Sebastián, and Ulrich Eckhard. 2022. "An Introduction to Bacterial Biofilms and Their Proteases, and Their Roles in Host Infection and Immune Evasion" Biomolecules 12, no. 2: 306. https://doi.org/10.3390/biom12020306