
PD-L1 Silencing in Liver Using siRNAs Enhances Efficacy of Therapeutic Vaccination for Chronic Hepatitis B



Abstract
:1. Introduction
2. Materials and Methods
3. Results
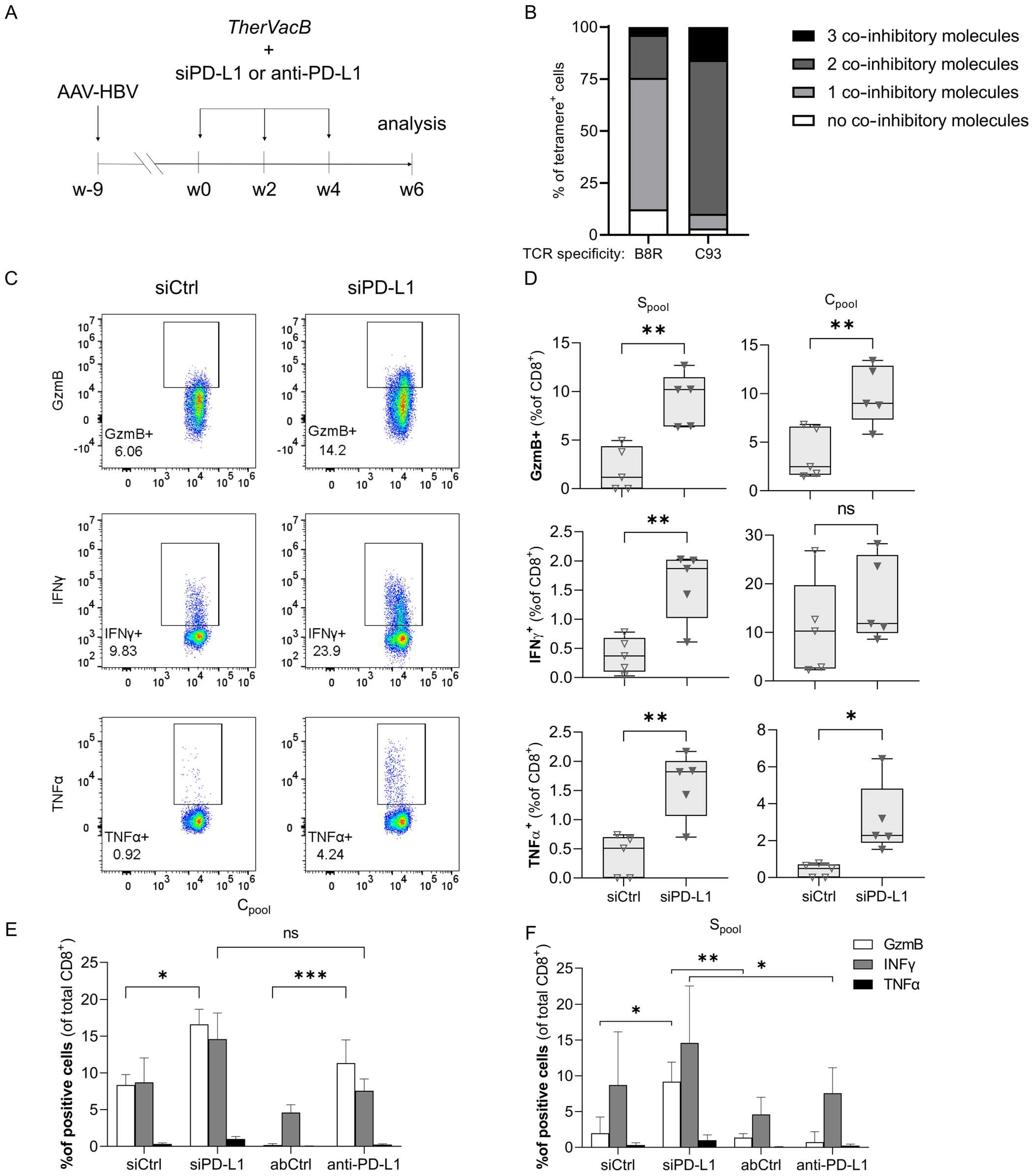
3.1. A Knock-Down of PD-L1 in the Liver by siRNA Improves T Cell Functionality
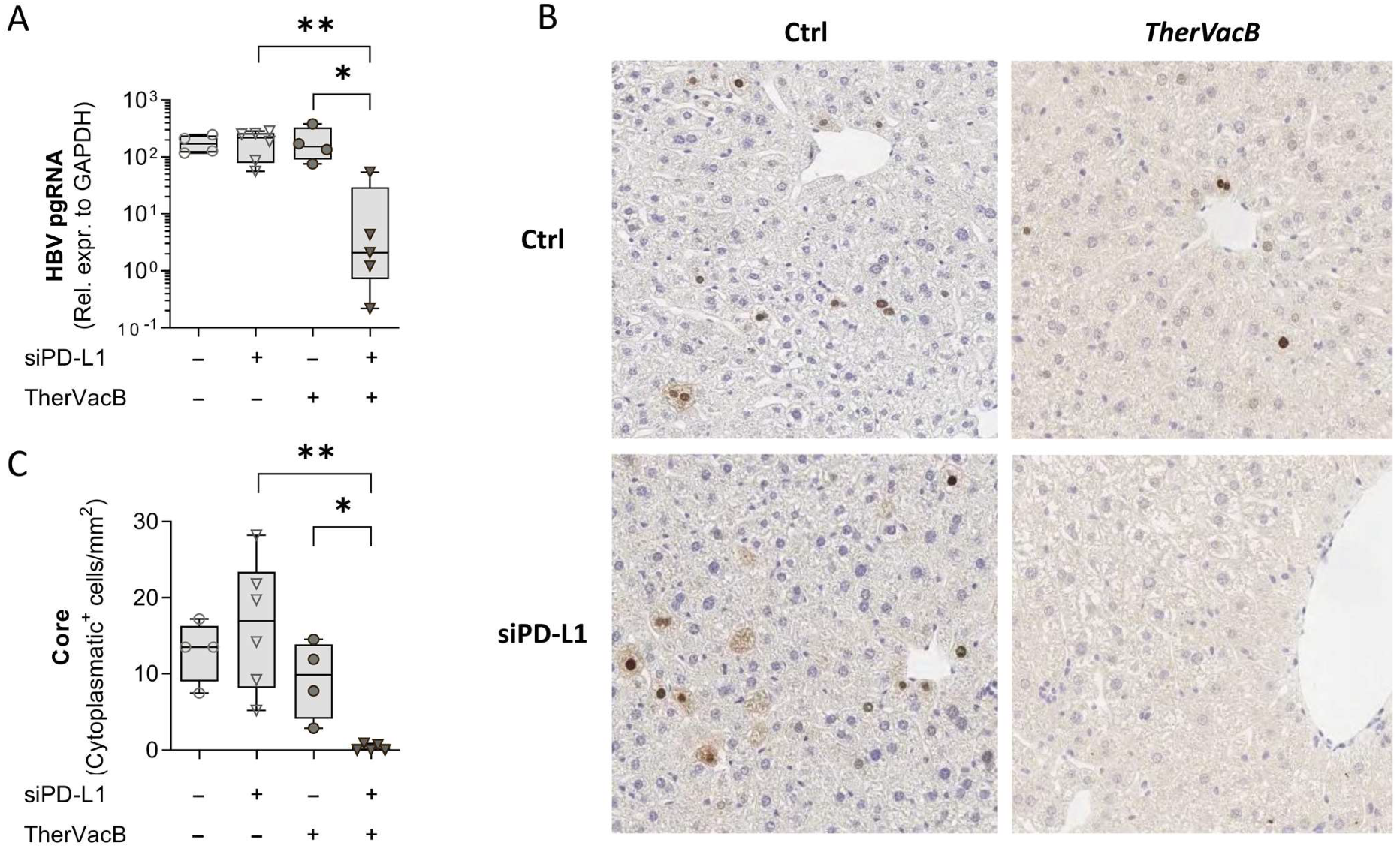
3.2. T Cells Activated by Therapeutic Vaccination Show an Improved Antiviral Effect after siRNA-Mediated Knock-Down of PD-L1
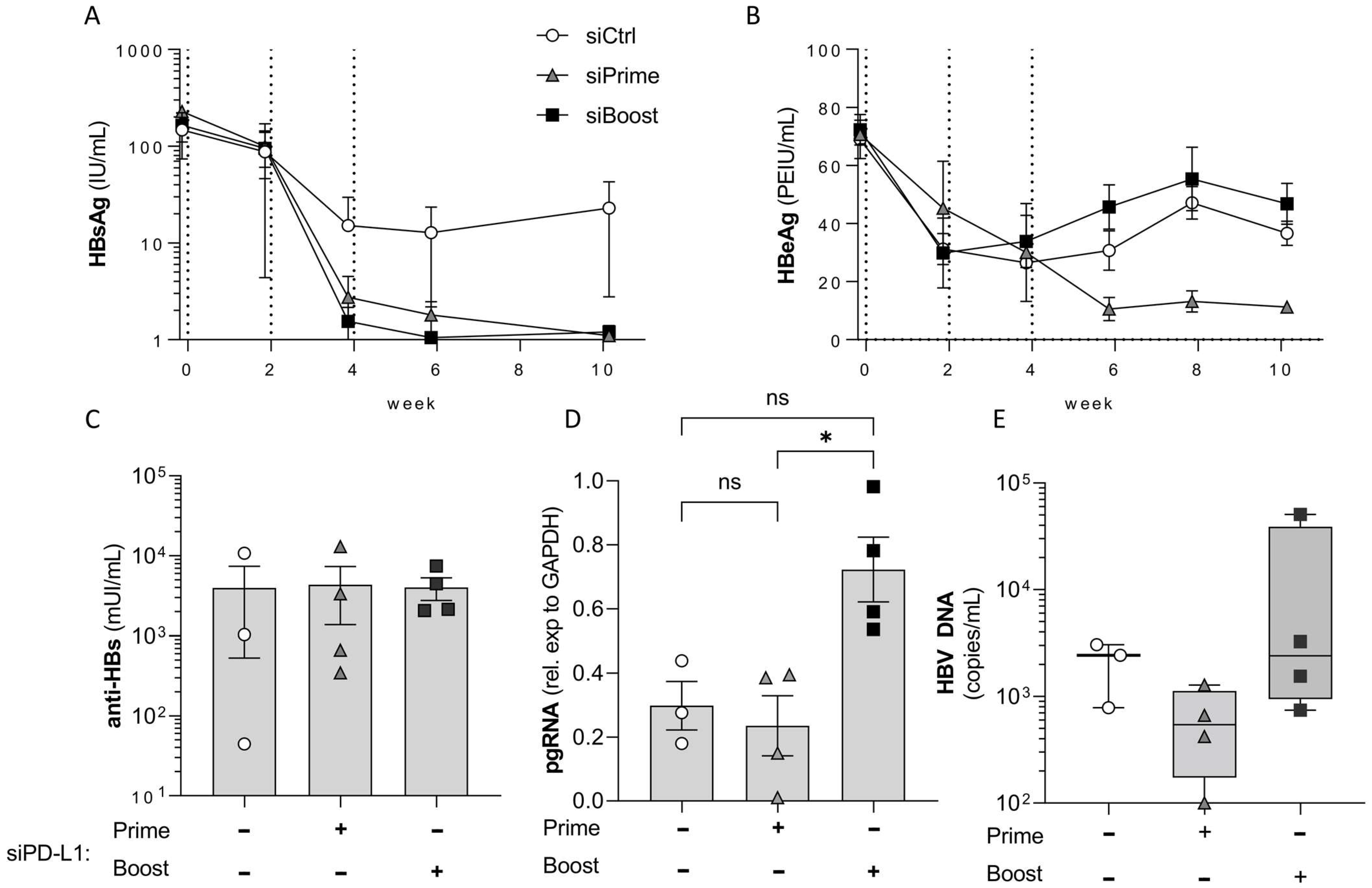
3.3. PD-L1 Knock-Down in the Liver Elicits Its Main Effect during Prime and Not during Boost Vaccination
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Global Progress Report on HIV, Viral Hepatitis and Sexually Transmitted Infections. 2021. Available online: http://apps.who.int/iris/bitstream/hadle/10665/342808/9789240030985-eng.pdf (accessed on 18 January 2022).
- Chen, J.; Yang, H.; Iloeje, U.H.; You, S.-L.; Lu, S.; Wang, L.; Su, J.; Sun, C.; Liaw, Y.; Chen, C. Carriers of Inactive Hepatitis B Virus Are Still at Risk for Hepatocellular Carcinoma and Liver-Related Death. Gastroenterology 2010, 138, 1747–1754. [Google Scholar] [CrossRef] [PubMed]
- Bertoletti, A.; Ferrari, C. Adaptive immunity in HBV infection. J. Hepatol. 2016, 64, S71–S83. [Google Scholar] [CrossRef] [PubMed]
- Protzer, U.; Maini, M.K.; Knolle, P.A. Living in the liver: Hepatic infections. Nat. Rev. Immunol. 2012, 12, 201–213. [Google Scholar] [CrossRef]
- Gane, E.; Verdon, D.J.; Brooks, A.E.; Gaggar, A.; Nguyen, A.H.; Subramanian, G.M.; Schwabe, C.; Dunbar, R. Anti-PD-1 blockade with nivolumab with and without therapeutic vaccination for virally suppressed chronic hepatitis B: A pilot study. J. Hepatol. 2019, 71, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Vandepapelière, P.; Lau, G.K.; Leroux-Roels, G.; Horsmans, Y.; Gane, E.; Tawandee, T.; bin Merican, M.I.; Win, K.M.; Trepo, C.; Cooksley, G.; et al. Therapeutic vaccination of chronic hepatitis B patients with virus suppression by antiviral therapy: A randomized, controlled study of co-administration of HBsAg/AS02 candidate vaccine and lamivudine. Vaccine 2007, 25, 8585–8597. [Google Scholar] [CrossRef]
- Backes, S.; Jäger, C.; Dembek, C.J.; Kosinska, A.D.; Bauer, T.; Stephan, A.-S.; Dišlers, A.; Mutwiri, G.; Busch, D.H.; Babiuk, L.A.; et al. Protein-prime/modified vaccinia virus Ankara vector-boost vaccination overcomes tolerance in high-antigenemic HBV-transgenic mice. Vaccine 2016, 34, 923–932. [Google Scholar] [CrossRef]
- Michler, T.; Kosinska, A.D.; Festag, J.; Bunse, T.; Su, J.; Ringelhan, M.; Imhof, H.; Grimm, D.; Steiger, K.; Mogler, C.; et al. Knockdown of Virus Antigen Expression Increases Therapeutic Vaccine Efficacy in High-titer HBV Carrier Mice. Gastroenterology 2020, 158, 1762–1775. [Google Scholar] [CrossRef]
- Kosinska, A.D.; Liu, J.; Lu, M.; Roggendorf, M. Therapeutic vaccination and immunomodulation in the treatment of chronic hepatitis B: Preclinical studies in the woodchuck. Med. Microbiol. Immunol. 2014, 204, 103–114. [Google Scholar] [CrossRef] [Green Version]
- Gehring, A.J.; Protzer, U. Targeting Innate and Adaptive Immune Responses to Cure Chronic HBV Infection. Gastroenterology 2019, 156, 325–337. [Google Scholar] [CrossRef] [Green Version]
- Hoogeveen, R.C.; Boonstra, A. Checkpoint Inhibitors and Therapeutic Vaccines for the Treatment of Chronic HBV Infection. Front. Immunol. 2020, 11, 401. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Zhang, E.; Ma, Z.; Wu, W.; Kosinska, A.; Zhang, X.; Möller, I.; Seiz, P.; Glebe, D.; Wang, B.; et al. Enhancing Virus-Specific Immunity In Vivo by Combining Therapeutic Vaccination and PD-L1 Blockade in Chronic Hepadnaviral Infection. PLoS Pathog. 2014, 10, e1003856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gane, E.J. Future anti-HBV strategies. Liver Int. 2017, 37 (Suppl. 1), 40–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schonrich, G.; Raftery, M.J. The PD-1/PD-L1 Axis and Virus Infections: A Delicate Balance. Front. Cell. Infect. Microbiol. 2019, 9, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cova, L. Present and future DNA vaccines for chronic hepatitis B treatment. Expert Opin. Biol. Ther. 2016, 17, 185–195. [Google Scholar] [CrossRef]
- Spain, L.; Diem, S.; Larkin, J. Management of toxicities of immune checkpoint inhibitors. Cancer Treat. Rev. 2016, 44, 51–60. [Google Scholar] [CrossRef]
- Dolina, J.S.; Sung, S.S.; Novobrantseva, T.I.; Nguyen, T.M.; Hahn, Y.S. Lipidoid Nanoparticles Containing PD-L1 siRNA Delivered In VivoEnter Kupffer Cells and Enhance NK and CD8+ T Cell-mediated Hepatic Antiviral Immunity. Mol. Ther. Nucleic Acids 2013, 2, e72. [Google Scholar] [CrossRef]
- Springer, A.D.; Dowdy, S.F. GalNAc-siRNA Conjugates: Leading the Way for Delivery of RNAi Therapeutics. Nucleic Acid Ther. 2018, 28, 109–118. [Google Scholar] [CrossRef]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012, 3, e00049. [Google Scholar] [CrossRef]
- Dion, S.; Bourgine, M.; Godon, O.; Levillayer, F.; Michel, M.L. Adeno-associated virus-mediated gene transfer leads to persistent hepatitis B virus replication in mice expressing HLA-A2 and HLA-DR1 molecules. J. Virol. 2013, 87, 5554–5563. [Google Scholar] [CrossRef] [Green Version]
- Upton, C.; Mossman, K.; McFadden, G. Encoding of a homolog of the IFN-gamma receptor by myxoma virus. Science 1992, 258, 1369–1372. [Google Scholar] [CrossRef]
- Maier, H.; Isogawa, M.; Freeman, G.J.; Chisari, F.V. PD-1:PD-L1 Interactions Contribute to the Functional Suppression of Virus-Specific CD8+T Lymphocytes in the Liver. J. Immunol. 2007, 178, 2714–2720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Bates, T.M.; Palma, M.L.; Shen, C.; Gambotto, A.; Macatangay, B.J.; Ferris, R.L.; Rinaldo, C.R.; Mailliard, R.B. Contrasting Roles of the PD-1 Signaling Pathway in Dendritic Cell-Mediated Induction and Regulation of HIV-1-Specific Effector T Cell Functions. J. Virol. 2019, 93, e02035-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Target | Sequence (5′ to 3′) |
|---|---|---|
| HBV3.5 kbRNA_fw a | HBV 3.5 kb RNA | GAGTGTGGATTCGCACTCC |
| HBV3.5 kbRNA_rev a mGAPDH_fw b mGAPDH_rev b | HBV 3.5 kb RNA Murine gapdh gene Murine gapdh gene | GAGGCGAGGGAGTTCTTCT ACCAACTGCTTAGCCC CCACGACGGACACATT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bunse, T.; Kosinska, A.D.; Michler, T.; Protzer, U. PD-L1 Silencing in Liver Using siRNAs Enhances Efficacy of Therapeutic Vaccination for Chronic Hepatitis B. Biomolecules 2022, 12, 470. https://doi.org/10.3390/biom12030470
Bunse T, Kosinska AD, Michler T, Protzer U. PD-L1 Silencing in Liver Using siRNAs Enhances Efficacy of Therapeutic Vaccination for Chronic Hepatitis B. Biomolecules. 2022; 12(3):470. https://doi.org/10.3390/biom12030470
Chicago/Turabian StyleBunse, Till, Anna D. Kosinska, Thomas Michler, and Ulrike Protzer. 2022. "PD-L1 Silencing in Liver Using siRNAs Enhances Efficacy of Therapeutic Vaccination for Chronic Hepatitis B" Biomolecules 12, no. 3: 470. https://doi.org/10.3390/biom12030470
APA StyleBunse, T., Kosinska, A. D., Michler, T., & Protzer, U. (2022). PD-L1 Silencing in Liver Using siRNAs Enhances Efficacy of Therapeutic Vaccination for Chronic Hepatitis B. Biomolecules, 12(3), 470. https://doi.org/10.3390/biom12030470