
Homocysteine, Vitamins B6 and Folic Acid in Experimental Models of Myocardial Infarction and Heart Failure—How Strong Is That Link?

 ,
,  , ,
, ,  , and
, and 
{kind=link}
{kind=link}
Abstract
:1. Introduction
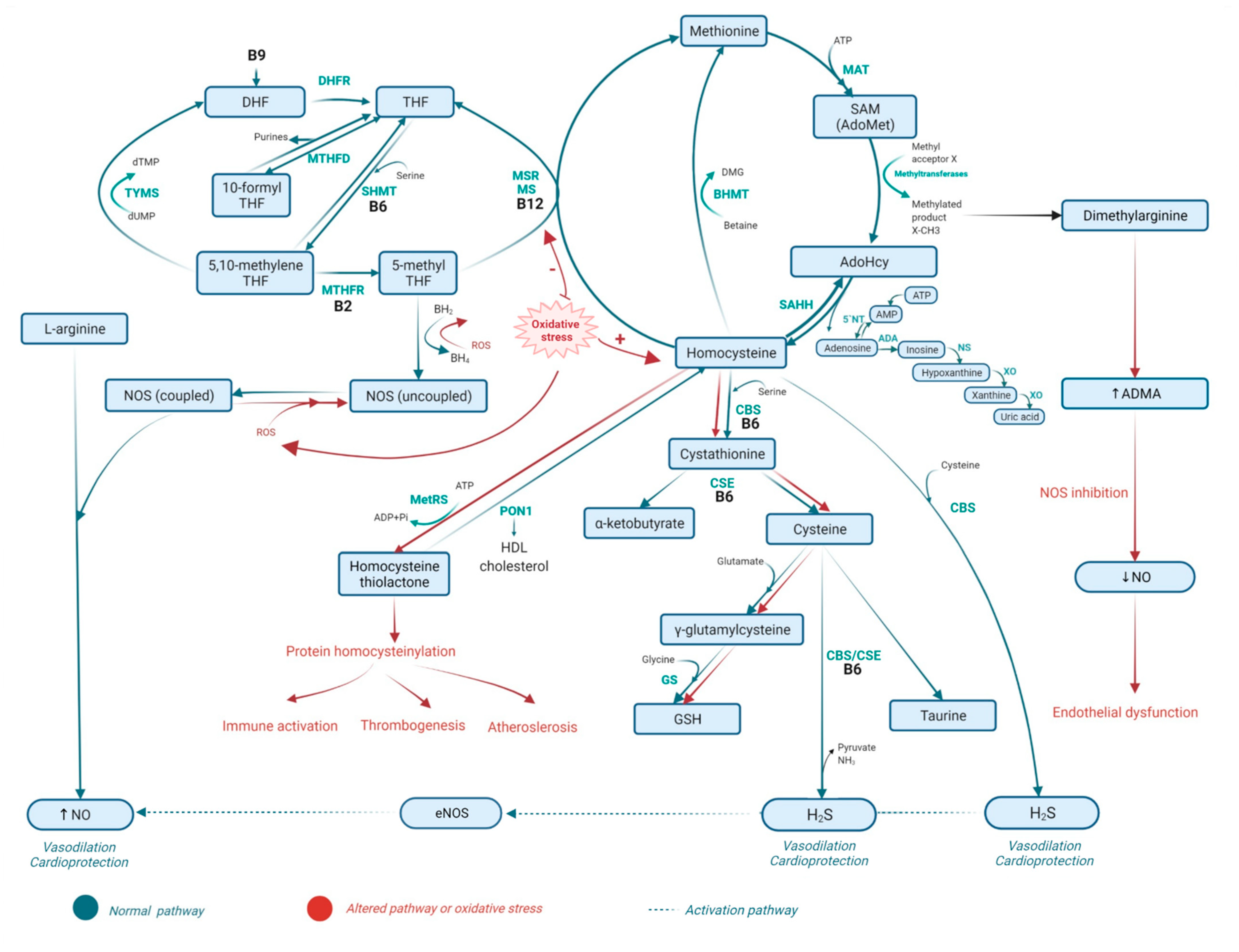
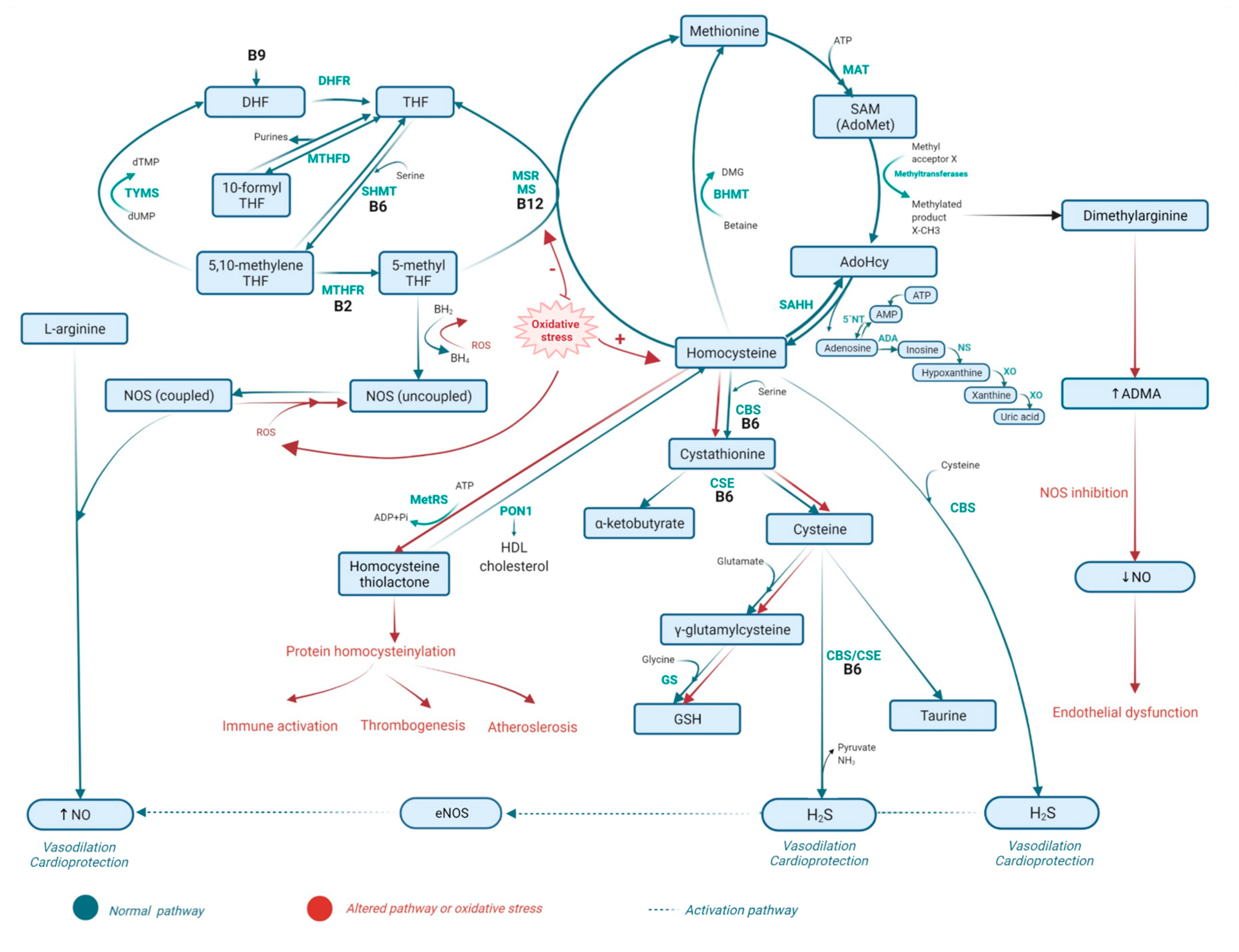
2. Metabolism of Homocysteine
2.1. Homocysteine Methabolism Pathaway
2.2. Folate Metabolism
2.3. The Role of Vitamins B6 and B12 in Homocysteine Metabolism and the Folate Cycle
2.4. Metabolic Disorders Related to Altered Homocysteine Metabolism
2.5. Consequences of Homocysteine, Folate, and Vitamin B6 Metabolism-Related Disorders
2.5.1. Homocysteine and CVD
2.5.2. Homocysteine and Diabetes
2.5.3. Homocysteine and Drugs
3. Oxidative Stress in Animal Models of MI and HF—The Importance of Homocysteine, Vitamin B6, and Folic Acid
4. Inflammation in Animal Models of Myocardial Infarction and Heart Failure—Significance of Homocysteine, Vitamin B6 and Folic Acid
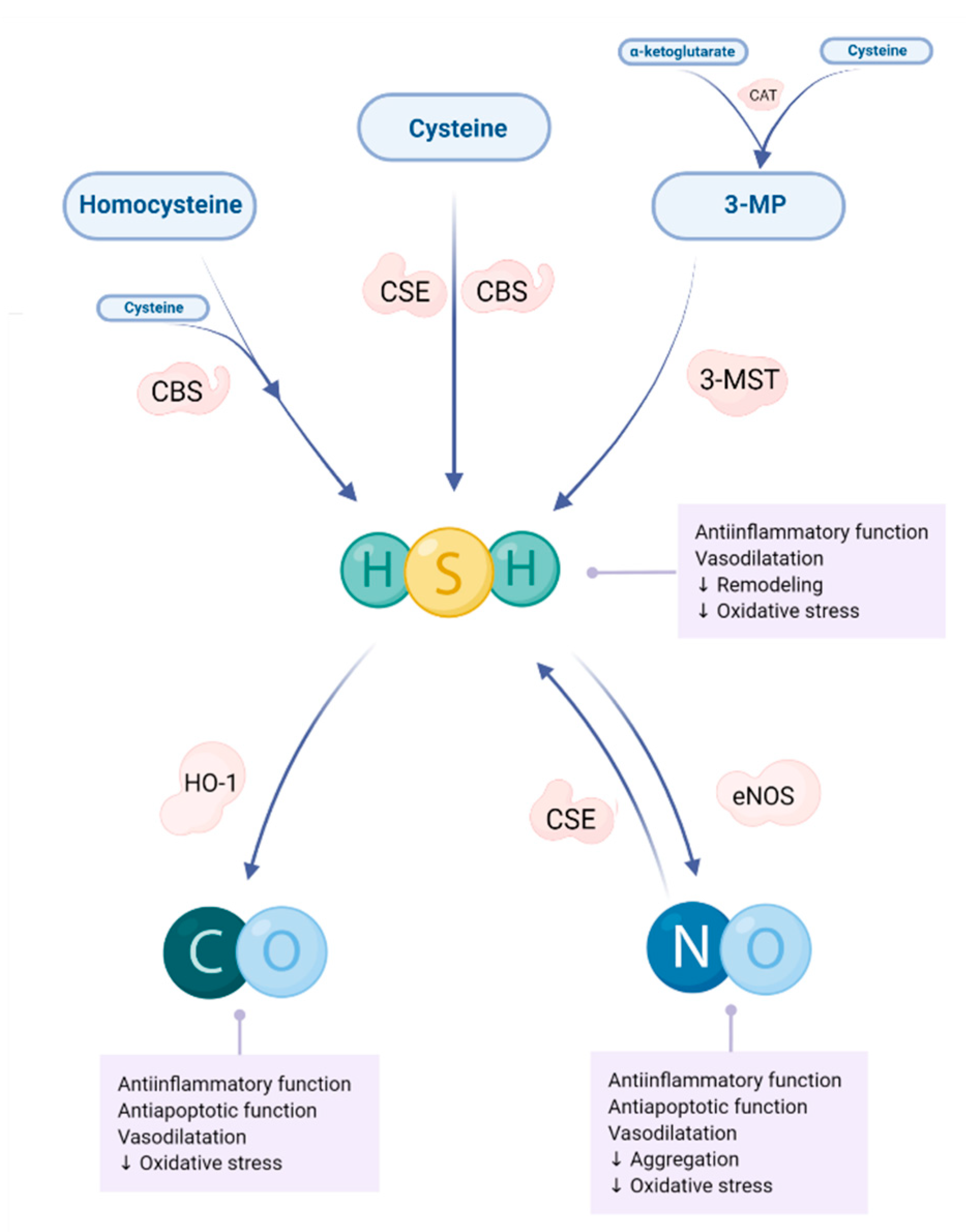
5. Gasotransmitters in Animal Models of Myocardial Infarction and Heart Failure—Significance of Homocysteine, Vitamin B6 and Folic Acid
5.1. Nitric Oxide
5.2. Hydrogen Sylfide
5.3. Carbon Monoxide
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 3MP | 3-mercaptopyruvate |
| 3-MST | 3-mercaptopyruvate suphurtrasferase |
| 5-MTHF | 5-methyltetrahydrofolate |
| 5′NT | Nucleotidase |
| ADA | Adenosine deaminase |
| ADMA | Asymetric dimethylarginine |
| AdoMet | S-adenosyl methionine |
| ALP | Alkaline phosphatase |
| AMI | Acute myocardial infarction |
| AST | Aspartate aminotransferase |
| BH4 | Tetrahydrobiopterin |
| BHMT | Betaine homocysteine methyltransferase |
| BNP | B-type natriuretic peptide |
| CAD | Coronary artery disease |
| Cbl | Cobalamin |
| Cat | Cationic amino acids |
| CAT | Cysteine aminotransferase |
| CBS | Cystathionine β-syntase |
| CK | Creatine kinase |
| CO | Cabon monoxide |
| CORM | CO-releasing molecules |
| CRP | C-reactive protein |
| CSE | Cystathionine γ-lyase |
| DALYs | Disability-adjusted life years |
| DATS | Diallyl trisulfide |
| DDAH | Dimethylarginine dimethylaminohydrolase |
| DHF | Dihydrofolate |
| DHFR | Dihydrofolate reductase |
| DMG | Dimethylglycine |
| dTMP | Deoxythymidine monophosphate |
| dUMP | Deoxyuridine mnonophosphate |
| DVT | Deep venous thrombosis |
| eNOS | Endothelial NOS |
| Gal-3 | Galectin-3 |
| GC | Guanylate cyclase |
| GDF-15 | Growth differentiation factor-15 |
| GGT | Gamma-glutamyl transferase |
| GPx | Glutathione peroxidase |
| GR | Glutathione reductase |
| GSH | Reduced glutathione |
| GSSG | Oxidized glutathione |
| GST | Glutathion S-transferase |
| H2S | Hydrogen sulphide |
| Hcy | Homocysteine |
| HDL | High density lipoprotein |
| HF | Heart failure |
| H-FABP | Heart fatty acid-binding protein |
| HHcy | Hyperhomocysteinemia |
| HO | Haem oxygenase |
| hs-cTN | High sensitive cardiac troponin |
| hs-Tn | High sensitive troponin |
| HTL | Homocystein thiolactone |
| IL-1 | Inerleukin-1 |
| IL-1β | Interleukin-1β |
| IL-18 | Interleukin-18 |
| IL-6 | Interleukin-6 |
| iNOS | Inducibile NOS |
| IP3 | Inositol triphosphate |
| ISO | Isoprenaline |
| LAD | Left anterior descending |
| LCA | Left coronary artery |
| LDH | Lactate dehydrogenase |
| LDL | Low density lipoprotein |
| LDLR | Low density lipoprotein receptor |
| Lp-PLA A2 | Phospholipase A2 associated lipoprotein |
| LVEDV | Left venticle end-diastolic volume |
| LVEF | Left ventricular ejection fraction |
| MAPK | Mitogen-activated protein kinases |
| MAT | Methionine adenosyltransferase |
| MDA | Malondialdehyde |
| MetRS | Methionyl-tRNA synthetase |
| MI | Myocardial infarction |
| MIP-1β | Macrophage inflammatory protein-1β |
| MMP-2 | Matrix metalloproteinase-2 |
| MMP-9 | Matrix metalloproteinase-9 |
| MMPs | Matrix metalloproteinases |
| MS | Methionine syntase |
| MSR | Methionine syntase reductase) |
| MYO | Myoglobin |
| NAD+ | Nicotinamide adenine dinucleotide |
| NF-κB | Nuclear factor kappa B |
| NMDA | N-methyl-D-aspartate |
| Nmnat | Nicotinamide mononucleotide adenylyl transferase |
| nNOS | Neuronal NOS |
| NO | Nitric oxide |
| NOS | Nitric oxide synthase |
| NS | Nucleosidase |
| NT-proBNP | N-terminal prohormone B-type natriuretic peptide |
| PKA | Protein kinase type A |
| PKC | Protein kinase type C |
| PLP | Pyridoxal phosphate |
| PON 1 | Paraoxonase 1 |
| PTX-3 | Petraxin |
| RAAS | Renin-angiotensin-aldosterone system |
| ROS | Reactive oxygen species |
| RSNOs | S-nitrosothiols |
| SAHH | S-adenosyl homocysteine hydrolase |
| SAM | S-adenosyl methionine |
| sCD40L | Soluble ligand CD40 |
| SERCA2a | Sarcoplasmatic reticulum atpase 2a |
| SHMT | Serine hydroxymethyltransferase |
| SNOCys | S-nitrocysteine |
| SNOHyc | S-nitrosohomocysteine |
| SNP | Sodium nitroprusside |
| SNS | Sympathetic nervous system |
| SOD | Superoxide dismutase |
| THF | Tetrahydrofolate |
| TNFR 1 | TNF-α receptor-1 |
| TNFR 2 | TNF-α receptor-2 |
| TNF-α | Tumor necrosis factor-α |
| TYMS | Thymidylate syntase |
| VCAM-1 | Vascular cell adhesion molecule/protein 1 |
| VLDL | Very low-density lipoprotein |
| vWF | Von Willebrand factor |
| ECG | Electrocardiography |
| XO | Xanthine oxidase |
References
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.; Benjamin, E.J.; Benziger, C.P.; et al. Global burden of cardiovascular diseases and risk factors, 1990–2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef]
- Paganelli, F.; Mottola, G.; Fromonot, J.; Marlinge, M.; Deharo, P.; Guieu, R.; Ruf, J. Hyperhomocysteinemia and cardiovascular disease: Is the adenosinergic system the missing link? Int. J. Mol. Sci. 2021, 22, 1690. [Google Scholar] [CrossRef] [PubMed]
- Mundi, S.; Massaro, M.; Scoditti, E.; Carluccio, M.A.; Van Hinsbergh, V.W.M.; Iruela-Arispe, M.L.; De Caterina, R. Endothelial permeability, LDL deposition, and cardiovascular risk factors—A review. Cardiovasc. Res. 2018, 114, 35–52. [Google Scholar] [CrossRef]
- Van Guldener, C.; Nanayakkara, P.W.B.; Stehouwer, C.D.A. Homocysteine and blood pressure. Curr. Hypertens. Rep. 2003, 5, 26–31. [Google Scholar] [CrossRef]
- Sreckovic, B.; Sreckovic, V.D.; Soldatovic, I.; Colak, E.; Sumarac-Dumanovic, M.; Janeski, H.; Janeski, N.; Gacic, J.; Mrdovic, I. Homocysteine is a marker for metabolic syndrome and atherosclerosis. Diabetes Metab. Syndr. Clin. Res. Rev. 2017, 11, 179–182. [Google Scholar] [CrossRef]
- Kim, J.; Kim, H.; Roh, H.; Kwon, Y. Causes of hyperhomocysteinemia and its pathological significance. Arch. Pharm. Res. 2018, 41, 372–383. [Google Scholar] [CrossRef]
- Djurovic, Z.; Jovanovic, V.; Obrenovic, R.; Djurovic, B.; Soldatovic, I.; Vranic, A.; Jakovljevic, V.; Djuric, D.; Zivkovic, V. The importance of the blood levels of homocysteine, folate and vitamin B12 in patients with primary malignant brain tumors. J. BUON 2021, 25, 2600–2607. [Google Scholar]
- Aleksic, D.; Djokic, D.; Golubicic, I.; Jakovljevic, V.; Djuric, D. The importance of the blood levels of homocysteine, folio acic and vitamin B12 in children with malignant diseases. J. BUON 2013, 18, 1019–1025. [Google Scholar]
- Blom, H.J.; Smulders, Y. Overview of homocysteine and folate metabolism. With special references to cardiovascular disease and neural tube defects. J. Inherit. Metab. Dis. 2011, 34, 75–81. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.N.; Gulati, S.; Baker, P.J.; Brody, L.C.; Banerjee, R.; Kruger, W.D. Cloning, mapping and RNA analysis of the human methionine synthase gene. Hum. Mol. Genet. 1996, 5, 1851–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreadou, I.; Iliodromitis, E.K.; Rassaf, T.; Schulz, R.; Papapetropoulos, A.; Ferdinandy, P. The role of gasotransmitters NO, H2S and CO in myocardial ischaemia/reperfusion injury and cardioprotection by preconditioning, postconditioning and remote conditioning. Br. J. Pharmacol. 2015, 172, 1587–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djuric, D.; Jakovljevic, V.; Zivkovic, V.; Srejovic, I. Homocysteine and homocysteine-related compounds: An overview of the roles in the pathology of the cardiovascular and nervous systems. Can. J. Physiol. Pharmacol. 2018, 96, 991–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolluru, G.K.; Shen, X.; Kevil, C.G. A tale of two gases: NO and H2S, foes or friends for life? Redox Biol. 2013, 1, 313–318. [Google Scholar] [CrossRef] [Green Version]
- Azzini, E.; Ruggeri, S.; Polito, A. Homocysteine: Its possible emerging role in at-risk population groups. Int. J. Mol. Sci. 2020, 21, 1421. [Google Scholar] [CrossRef] [Green Version]
- Vitvitsky, V.; Mosharov, E.; Tritt, M.; Ataullakhanov, F.; Banerjee, R. Redox regulation of homocysteine-dependent glutathione synthesis. Redox Rep. 2003, 8, 57–63. [Google Scholar] [CrossRef]
- Chubarov, A.S. Homocysteine thiolactone: Biology and chemistry. Encyclopedia 2021, 1, 445–459. [Google Scholar] [CrossRef]
- Jakubowski, H. Homocysteine modification in protein structure / function and human disease. Physiol. Rev. 2019, 99, 555–604. [Google Scholar] [CrossRef]
- Perła-Kaján, J.; Włoczkowska, O.; Zioła-Frankowska, A.; Frankowski, M.; Smith, A.D.; De Jager, C.A.; Refsum, H.; Jakubowski, H. Paraoxonase 1, B vitamins supplementation, and mild cognitive impairment. J. Alzheimer’s Dis. 2021, 81, 1211–1229. [Google Scholar] [CrossRef]
- Borowczyk, K.; Shih, D.M.; Jakubowski, H. Metabolism and neurotoxicity of homocysteine thiolactone in mice: Evidence for a protective role of paraoxonase 1. J. Alzheimer’s Dis. 2012, 30, 225–231. [Google Scholar] [CrossRef]
- Shih, D.M.; Gu, L.; Xia, Y.R.; Navab, M.; Li, W.F.; Hama, S.; Castellani, L.W.; Furlong, C.E.; Costa, L.G.; Fogelman, A.M.; et al. Mice lacking serum paraoxonase are susceptible to organophosphate toxicity and atherosclerosis. Nature 1998, 394, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, T.; Nicholls, S.J.; Topol, E.J.; Schmitt, D.; Shao, M.; Brennan, D.M.; Ellis, S.G. Relationship of paraoxonase 1 (PON1) gene polymorphisms and functional activity with systemic oxidative stress. JAMA 2008, 299, 1265–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanhewicz, A.E.; Kenney, W.L. Role of folic acid in nitric oxide bioavailability and vascular endothelial function. Nutr. Rev. 2017, 75, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Stanhewicz, A.E.; Alexander, L.M.; Kenney, W.L. Folic acid supplementation improves microvascular function in older adults through nitric oxide-dependent mechanisms. Clin. Sci. 2015, 129, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Pietrzik, K.; Bailey, L.; Shane, B. Folic acid and l-5-methyltetrahydrofolate: Comparison of clinical pharmacokinetics and pharmacodynamics. Clin. Pharmacokinet. 2010, 49, 535–548. [Google Scholar] [CrossRef]
- Jayedi, A.; Zargar, M.S. Intake of vitamin B6, folate, and vitamin B12 and risk of coronary heart disease: A systematic review and dose-response meta-analysis of prospective cohort studies. Crit. Rev. Food Sci. Nutr. 2019, 59, 2697–2707. [Google Scholar] [CrossRef]
- Ntaios, G.; Savopoulos, C.; Grekas, D.; Hatzitolios, A. The controversial role of B-vitamins in cardiovascular risk: An update. Arch. Cardiovasc. Dis. 2009, 102, 847–854. [Google Scholar] [CrossRef] [Green Version]
- Pusceddu, I.; Herrmann, W.; Kleber, M.E.; Scharnagl, H.; Hoffmann, M.M.; Winklhofer-Roob, B.M.; März, W.; Herrmann, M. Subclinical inflammation, telomere shortening, homocysteine, vitamin B6, and mortality: The Ludwigshafen Risk and Cardiovascular Health Study. Eur. J. Nutr. 2020, 59, 1399–1411. [Google Scholar] [CrossRef] [Green Version]
- Desouza, C.; Keebler, M.; McNamara, D.B.; Fonseca, V. Drugs affecting homocysteine metabolism. Drugs 2002, 62, 605–616. [Google Scholar] [CrossRef]
- Miller, J.W.; Nadeau, M.R.; Smith, D.; Selhub, J. Vitamin B-6 deficiency vs folate deficiency: Comparison of responses to methionine loading in rats. Am. J. Clin. Nutr. 1994, 59, 1033–1039. [Google Scholar] [CrossRef]
- Miller, A. The Methionine-Homocysteine Cycle and Its Effects on Cognitive Diseases. Altern. Med. Rev. 2003, 8, 7–19. [Google Scholar] [PubMed]
- Yakub, M.; Iqbal, M.P.; Kakepoto, G.N.; Rafique, G.; Memon, Y.; Azam, I.; Mehboobali, N.; Parveen, S.; Haider, G. High prevalence of mild hyperhomocysteinemia and folate, B12 and B6 deficiencies in an urban population in Karachi, Pakistan. Pak. J. Med. Sci. 2010, 26, 923–929. [Google Scholar]
- Chwatko, G.; Boers, G.H.J.; Strauss, K.A.; Shih, D.M.; Jakubowski, H. Mutations in methylenetetrahydrofolate reductase or cystathionine β-syntase gene, or a high-methionine diet, increase homocysteine thiolactone levels in humans and mice. FASEB J. 2007, 21, 1707–1713. [Google Scholar] [CrossRef]
- Van Guldener, C.; Robinson, K. Homocysteine and renal disease. Semin. Thromb. Hemost. 2000, 26, 313–324. [Google Scholar] [CrossRef]
- Van Guldener, C. Why is homocysteine elevated in renal failure and what can be expected from homocysteine-lowering? Nephrol. Dial. Transplant. 2006, 21, 1161–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, M.S.; Bostom, A.G.; Jacques, P.F.; Selhub, J.; Rosenberg, I.H. Hyperhomocysteinemia and hypercholesterolemia associated with hypothyroidism in the third US National Health and Nutrition Examination Survey. Atherosclerosis 2001, 155, 195–200. [Google Scholar] [CrossRef]
- Bamashmoos, S.A.; Al-nuzaily, M.A.K.; Al-meeri, A.M.; Ali, F.H.H. Relationship between total homocysteine, total cholesterol and creatinine levels in overt hypothyroid patients. Springerplus 2013, 2, 423. [Google Scholar] [CrossRef] [Green Version]
- Cicone, F.; Santaguida, M.; My, G.; Mancuso, G.; Papa, A.; Persechino, R.; Virili, C.; Brusca, N.; Tofani, A.; Scopinaro, F.; et al. Hyperhomocysteinemia in acute iatrogenic hypothyroidism: The relevance of thyroid autoimmunity. J. Endocrinol. Investig. 2017, 41, 831–837. [Google Scholar] [CrossRef]
- Almadori, G.; Bussu, F.; Galli, J.; Cadoni, G.; Zappacosta, B.; Persichilli, S.; Minucci, A.; Giardina, B. Serum folate and homocysteine levels in head and neck squamous cell carcinoma. Cancer 2002, 94, 1006–1011. [Google Scholar] [CrossRef] [Green Version]
- Kato, I.; Dnistrian, A.M.; Schwartz, M.; Toniolo, P.; Koenig, K.; Shore, R.E.; Akhmedkhanov, A.; Zeleniuch-Jacquotte, A.; Riboli, E. Serum folate, homocysteine and colorectal cancer risk in women: A nested case-control study. Br. J. Cancer 1999, 79, 1917–1922. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.F.; Haven, T.R.; Wu, T.L.; Tsao, K.C.; Wu, J.T. Serum total homocysteine increases with the rapid proliferation rate of tumor cells and decline upon cell death: A potential new tumor marker. Clin. Chim. Acta 2002, 321, 55–62. [Google Scholar] [CrossRef]
- Govoruskina, N.; Jakovljevic, V.; Zivkovic, V.; Milosavljevic, I.; Jeremic, J.; Bradic, J.; Bolevich, S.; Omarov, I.A.; Djuric, D.; Radonjic, K.; et al. The role of cardiac N-methyl-D-aspartate receptors in heart conditioning—effects on heart function and oxidative stress. Biomolecules 2020, 10, 1065. [Google Scholar] [CrossRef] [PubMed]
- Srejovic, I.; Jakovljevic, V.; Zivkovic, V.; Barudzic, N.; Radovanovic, A.; Stanojlovic, O.; Djuric, D. The effects of the modulation of NMDA receptors by homocysteine thiolactone and dizocilpine on cardiodynamics and oxidative stress in isolated rat heart. Mol. Cell. Biochem. 2015, 401, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Milovanovic, P.; Hrncic, D.; Radotic, K.; Stankovic, M.; Mutavdzic, D.; Djonic, D.; Rasic-Markovic, A.; Djuric, D.; Stanojlovic, O.; Djuric, M. Moderate hyperhomocysteinemia induced by short-term dietary methionine overload alters bone microarchitecture and collagen features during growth. Life Sci. 2017, 191, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Djurić, M.; Mutavdzin, S.; Loncar-Stojiljkovic, D.; Kostić, S.; Čolović, M.; Krstic, D.; Zivković, V.; Jakovljevic, V.; Djurić, D. The effects of certain gasotransmitters inhibition on homocysteine acutely induced changes on rat cardiac acetylcholinesterase activity. Scr. Med. 2019, 50, 112–116. [Google Scholar] [CrossRef]
- Furchgott, R.; Vanhoutte, P. Endothelium-derived relaxing and contractiong factors. FASEB J. 1989, 3, 2007–2018. [Google Scholar] [CrossRef]
- Jacobsen, D.W. Homocysteine and vitamins in cardiovascular disease. Clin. Chem. 1998, 44, 1833–1843. [Google Scholar] [CrossRef]
- Karolczak, K.; Olas, B. Mechanism of action of homocysteine and its thiolactone in hemostasis system. Physiol. Res. 2009, 58, 623–633. [Google Scholar] [CrossRef]
- Lentz, S.R.; Piegors, D.J.; Fernández, J.A.; Erger, R.A.; Arning, E.; Malinow, M.R.; Griffin, J.H.; Bottiglieri, T.; Haynes, W.G.; Heistad, D.D. Effect of hyperhomocysteinemia on protein C activation and activity. Blood 2002, 100, 2108–2112. [Google Scholar] [CrossRef] [Green Version]
- Raposo, B.; Rodríguez, C.; Martínez-González, J.; Badimon, L. High levels of homocysteine inhibit lysyl oxidase (LOX) and downregulate LOX expression in vascular endothelial cells. Atherosclerosis 2004, 177, 1–8. [Google Scholar] [CrossRef]
- Zivkovic, V.; Jakovljevic, V.; Djordjevic, D.; Vuletic, M.; Barudzic, N.; Djuric, D. The effects of homocysteine-related compounds on cardiac contractility, coronary flow, and oxidative stress markers in isolated rat heart. Mol. Cell. Biochem. 2012, 370, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Zivkovic, V.; Jakovljevic, V.; Pechanova, O.; Srejovic, I.; Joksimovic, J.; Selakovic, D.; Barudzic, N.; Djuric, D.M. Effects of DL-homocysteine thiolactone on cardiac contractility, coronary flow, and oxidative stress markers in the isolated rat heart: The role of different gasotransmitters. BioMed Res. Int. 2013, 2013, 318471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uzelac, J.J.; Stanić, M.; Krstić, D.; Čolović, M.; Djurić, D. Effects of homocysteine and its related compounds on oxygen consumption of the rat heart tissue homogenate: The role of different gasotransmitters. Mol. Cell. Biochem. 2018, 444, 143–148. [Google Scholar] [CrossRef]
- Radenković, M.; Djurić, D.; Janković, R.; Prostran, M. The analysis of transduction mechanisms associated with an acute action of homocysteine on isolated rat femoral artery. Acta Physiol. Hung. 2014, 101, 448–460. [Google Scholar] [CrossRef] [PubMed]
- Toroser, D.; Sohal, R.S. Age-associated perturbations in glutathione synthesis in mouse liver. Biochem. J. 2007, 405, 583–589. [Google Scholar] [CrossRef]
- Prasad, A.; Andrews, N.P.; Padder, F.A.; Husain, M.; Quyyumi, A.A. Glutathione reverses endothelial dysfunction and improves nitric oxide bioavailability. J. Am. Coll. Cardiol. 1999, 34, 507–514. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Chen, Y.; Xie, X.; Liu, J.; Wang, Q.; Kong, W.; Zhu, Y. Homocysteine activates vascular smooth muscle cells by DNA demethylation of platelet-derived growth factor in endothelial cells. J. Mol. Cell. Cardiol. 2012, 53, 487–496. [Google Scholar] [CrossRef]
- Majors, A.; Ehrhart, A.; Pezacka, E. Hyperhomocysteine as a risk factor for vascular disease Enhanced Collagen Production and Accumulation by Smooth Muscle Cells. Atheroscler. Thromb. Vasc. Biol. 1997, 17, 2074–2081. [Google Scholar] [CrossRef]
- Todorovic, D.; Stojanovic, M.; Medic, A.; Gopcevic, K.; Mutavdzin, S.; Stankovic, S.; Djuric, D. Four weeks of aerobic training affects cardiac tissue matrix metalloproteinase, lactate dehydrogenase and malate dehydrogenase enzymes activities, and hepatorenal biomarkers in experimental hyperhomocysteinemia in rats. Int. J. Mol. Sci. 2021, 22, 6792. [Google Scholar] [CrossRef]
- Kostić, S.; Mićovic, Ž.; Andrejević, L.; Cvetković, S.; Stamenković, A.; Stanković, S.; Obrenović, R.; Labudović-Borović, M.; Hrnčić, D.; Jakovljević, V.; et al. The effects of l-cysteine and N-acetyl-l-cysteine on homocysteine metabolism and haemostatic markers, and on cardiac and aortic histology in subchronically methionine-treated Wistar male rats. Mol. Cell. Biochem. 2019, 451, 43–54. [Google Scholar] [CrossRef]
- Jeremic, J.; Nikolic Turnic, T.; Zivkovic, V.; Jeremic, N.; Milosavljevic, I.; Srejovic, I.; Obrenovic, R.; Jancic, S.; Rakocevic, M.; Matic, S.; et al. Vitamin B complex mitigates cardiac dysfunction in high-methionine diet-induced hyperhomocysteinemia. Clin. Exp. Pharmacol. Physiol. 2018, 45, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Nikolic Turnic, T.; Jakovljevic, V.; Djuric, D.; Jeremic, N.; Jeremic, J.; Milosavljevic, I.; Srejovic, I.; Selakovic, D.; Zivkovic, V. Efficency of atorvastatin and simvastatin in improving of cardiac function during the different degree of hyperhomocysteinemia. Can. J. Physiol. Pharmacol. 2018, 96, 1040–1049. [Google Scholar] [CrossRef] [PubMed]
- Turnic, T.N.; Arsic, A.; Vucic, V.; Petrovic, S.; Ristic-Medic, D.; Zivkovic, V.; Srejovic, I.; Jeremic, J.; Radonjic, T.; Milosavljevic, I.; et al. Hydroxymethylglutaryl coenzyme a reductase inhibitors differentially modulate plasma fatty acids in rats with diet-induced-hyperhomocysteinemia: Is ω-3 fatty acids supplementation necessary? Front. Physiol. 2019, 10, 892. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Chen, H.; Liu, N.; Chen, J.; Gu, Y.; Chen, J.; Yang, K. Role of hyperhomocysteinemia and hyperuricemia in pathogenesis of atherosclerosis. J. Stroke Cerebrovasc. Dis. 2017, 26, 2695–2699. [Google Scholar] [CrossRef]
- Wen, J.; Wen, Y.; Zhiliang, L.; Lingling, C.; Longxing, C.; Ming, W.; Qiang, F. A decrease in the percentage of circulating mDC precursors in patients with coronary heart disease: A relation to the severity and extent of coronary artery lesions? Heart Vessel. 2013, 28, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Peng, D.; Liu, C.; Huang, C.; Luo, J. Serum high concentrations of homocysteine and low levels of folic acid and vitamin B12 are significantly correlated with the categories of coronary artery diseases. BMC Cardiovasc. Disord. 2017, 17, 37. [Google Scholar] [CrossRef] [Green Version]
- Ebbing, M.; Bleie, Ø.; Ueland, P.; Nordrehaug, J.; Vollset, S.; Refsum, H.; Pederson, E.; Nygard, O. Mortality and cardiovascular events in patients treated with homocysteine-lowering B vitamins after coronary angiography. JAMA 2008, 300, 795–804. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.M.; Zhou, B.; Nie, Z.L.; Gao, W.; Wang, Y.S.; Zhao, H.; Zhu, J.; Yan, J.J.; Yang, Z.J.; Wang, L.S. Folate and risk of coronary heart disease: A meta-analysis of prospective studies. Nutr. Metab. Cardiovasc. Dis. 2012, 22, 890–899. [Google Scholar] [CrossRef]
- Cui, R.; Iso, H.; Date, C.; Kikuchi, S.; Tamakoshi, A. Dietary folate and vitamin B6 and B12 intake in relation to mortality from cardiovascular diseases: Japan collaborative cohort study. Stroke 2010, 41, 1285–1289. [Google Scholar] [CrossRef] [Green Version]
- Cheng, D.; Kong, H.; Pang, W.; Yang, H.; Lu, H.; Huang, C.; Jiang, Y. Supplementation b vitamins improves cognitive function in the middle-aged and elderly with hyperhomocysteinemia. Nutr. Neurosci. 2016, 19, 461–466. [Google Scholar] [CrossRef]
- Guo, H.; Lee, J.D.; Ueda, T.; Cheng, J.; Shan, J.; Wang, J. Hyperhomocysteinaemia & folic acid supplementation in patients with high risk of coronary artery disease. Indian J. Med. Res. 2004, 119, 33–37. [Google Scholar] [PubMed]
- Djurić, P.; Mladenović, Z.; Spasić, M.; Jović, Z.; Marić-Kocijančić, J.; Prokić, D.; Subota, V.; Radojičić, Z.; Djurić, D. Hyperhomocysteinemia and inflammatory biomarkers are associated with higher clinical SYNTAX score in patients with stable coronary artery disease. Vojnosanit. Pregl. 2021, 78, 736–744. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, M.P.; Ishaq, M.; Kazmi, K.A.; Yousuf, F.A.; Mehboobali, N.; Ali, S.A.; Khan, A.H.; Waqar, M.A. Role of vitamins B6, B12 and folic acid on hyperhomocysteinemia in a Pakistani population of patients with acute myocardial infarction. Nutr. Metab. Cardiovasc. Dis. 2005, 15, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, T.; Zheng, Y.; Muka, T.; Troup, J.; Hu, F.B. Folic acid supplementation and the risk of cardiovascular diseases: A meta-analysis of randomized controlled trials. J. Am. Heart Assoc. 2016, 5, e003768. [Google Scholar] [CrossRef] [Green Version]
- Huo, Y.; Li, J.; Qin, X.; Huang, Y.; Wang, X.; Gottesman, R.F.; Tang, G.; Wang, B.; Chen, D.; He, M.; et al. Efficacy of folic acid therapy in primary prevention of stroke among adults with hypertension in China: The CSPPT randomized clinical trial. JAMA J. Am. Med. Assoc. 2015, 313, 1325–1335. [Google Scholar] [CrossRef]
- Park, J.H.; Saposnik, G.; Ovbiagele, B.; Markovic, D.; Towfighi, A. Effect of B-vitamins on stroke risk among individuals with vascular disease who are not on antiplatelets: A meta-analysis. Int. J. Stroke 2016, 11, 206–211. [Google Scholar] [CrossRef]
- Toole, J.F.; Malinow, M.R.; Chambless, L.E.; Spence, J.D.; Pettigrew, L.C.; Howard, V.J.; Sides, E.G.; Wang, C.-H.; Stampfer, M. Lowering homocysteine in patients with ischemic stroke to prevent recurrent stroke, myocardial infarction, and death. JAMA 2004, 291, 565–575. [Google Scholar] [CrossRef] [Green Version]
- The VITATOPS Trial Study B vitamins in patients with recent transient ischaemic attack or stroke in the VITAmins to prevent stroke (VITATOPS) trial: A randomised, double-blind, parallel, placebo-controlled trial. Lancet Neurol. 2010, 9, 855–865. [CrossRef] [Green Version]
- Saposnik, G.; Ray, J.G.; Sheridan, P.; McQueen, M.; Lonn, E. Homocysteine-lowering therapy and stroke risk, severity, and disability: Additional findings from the HOPE 2 trial. Stroke 2009, 40, 1365–1372. [Google Scholar] [CrossRef]
- Chrysant, S.G.; Chrysant, G.S. The current status of homocysteine as a risk factor for cardiovascular disease: A mini review. Expert Rev. Cardiovasc. Ther. 2018, 16, 559–565. [Google Scholar] [CrossRef]
- Hsu, C.-Y.; Chiu, S.-W.; Hong, K.-S.; Saver, J.L.; Wu, Y.-L.; Lee, J.-D.; Lee, M.; Ovbiagele, B. Folic acid in stroke prevention in countries without mandatory folic acid food fortification: A meta-analysis of randomized controlled trials. J. Stroke 2018, 20, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Zhou, K.; Zhao, R.; Geng, Z.; Jiang, L.; Cao, Y.; Xu, D.; Liu, Y.; Huang, L.; Zhou, J. Association between B-group vitamins and venous thrombosis: Systematic review and meta-analysis of epidemiological studies. J. Thromb. Thrombolysis 2012, 34, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Kasthuri, R.S.; Key, N.S. Medical management of venous thromboembolism: What the interventional radiologist needs to know. Semin. Interv. Radiol. 2012, 29, 3–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekim, M.; Ekim, H.; Yilmaz, Y.K.; Kulah, B.; Polat, M.F.; Gocmen, A.Y. Study on relationships among deep vein thrombosis, homocysteine & related B group vitamins. Pak. J. Med. Sci. 2015, 31, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Petersen, J.F.; Larsen, B.S.; Sabbah, M.; Nielsen, O.W.; Kumarathurai, P.; Sajadieh, A. Long-term prognostic significance of homocysteine in middle-aged and elderly. Biomarkers 2016, 21, 490–496. [Google Scholar] [CrossRef]
- Vizzardi, E.; Bonadei, I.; Zanini, G.; Frattini, S.; Fiorina, C.; Raddino, R.; Dei Cas, L. Homocysteine and heart failure: An overview. Recent Pat. Cardiovasc. Drug Discov. 2009, 4, 15–21. [Google Scholar] [CrossRef]
- Chen, P.; Poddar, R.; Tipa, E.V.; Dibello, P.M.; Moravec, C.D.; Robinson, K.; Green, R.; Kruger, W.D.; Garrow, T.A.; Jacobsen, D.W. Homocysteine metabolism in cardiovascular cells and tissues: Implications for hyperhomocysteinemia and cardiovascular disease. Adv. Enzym. Regul. 1999, 39, 93–109. [Google Scholar] [CrossRef]
- Fournier, P.; Fourcade, J.; Roncalli, J.; Salvayre, R.; Galinier, M.; Causse, E. Homocysteine in chronic heart failure. Clin. Lab. 2015, 61, 1137–1145. [Google Scholar] [CrossRef]
- Qujeq, D.; Omran, T.S.; Hosini, L. Correlation between total homocysteine, low-density lipoprotein cholesterol and high-density lipoprotein cholesterol in the serum of patients with myocardial infarction. Clin. Biochem. 2001, 34, 97–101. [Google Scholar] [CrossRef]
- Iqbal, N.S.; Wu, Y.; Hazen, S.; Tang, W.H.W. Elevated plasma homocysteine identifies patients with chronic heart failure at increased cardiovascular risk. J. Card. Fail. 2012, 18, S87. [Google Scholar] [CrossRef]
- Zheng, M.; Zou, C.; Li, M.; Huang, G.; Gao, Y.; Liu, H. Folic acid reduces tau phosphorylation by regulating PP2A methylation in streptozotocin-induced diabetic mice. Int. J. Mol. Sci. 2017, 18, 861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, K.T.; Garrow, T.A.; Schalinske, K.L. Type I diabetes leads to tissue-specific DNA hypomethylation in male rats. J. Nutr. 2008, 138, 2064–2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudchada, P.; Saokaew, S.; Sridetch, S.; Incampa, S.; Jaiyen, S.; Khaithong, W. Effect of folic acid supplementation on plasma total homocysteine levels and glycemic control in patients with type 2 diabetes: A systematic review and meta-analysis. Diabetes Res. Clin. Pract. 2012, 98, 151–158. [Google Scholar] [CrossRef]
- Lind, V.; Lauritzen, L.; Kristensen, M.; Ross, A.; Eriksen, J. Effect of folate supplementation on insulin sensitivity and type 2 diabetes: A meta-analysis of randomized controlled trials. Am. J. Clin. Nutr. 2019, 109, 1233. [Google Scholar] [CrossRef]
- Mutavdzin, S.; Gopcevic, K.; Stankovic, S.; Jakovljevic Uzelac, J.; Labudovic Borovic, M.; Djuric, D.M. The effect of folic acid administration on cardiac tissue matrix metalloproteinase activity and hepatorenal biomarkers in diabetic rats. Can. J. Physiol. Pharmacol. 2019, 97, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Mutavdzin, S.; Gopcevic, K.; Stankovic, S.; Jakovljevic, J.U.; Borovic, M.L.; Djuric, D.D. The effects of folic acid administration on cardiac oxidative stress and cardiovascular biomarkers in diabetic rats. Oxid. Med. Cell. Longev. 2019, 2019, 1342549. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.G.; Li, W.H.; Lin, Z.Q.; Wang, L.X. Effects of folic acid on cardiac myocyte apoptosis in rats with streptozotocin-induced diabetes mellitus. Cardiovasc. Drugs Ther. 2008, 22, 299–304. [Google Scholar] [CrossRef]
- Nix, W.A.; Zirwes, R.; Bangert, V.; Kaiser, R.P.; Schilling, M.; Hostalek, U.; Obeid, R. Vitamin B status in patients with type 2 diabetes mellitus with and without incipient nephropathy. Diabetes Res. Clin. Pract. 2015, 107, 157–165. [Google Scholar] [CrossRef] [Green Version]
- Abdullah, K.M.; Abul Qais, F.; Hasan, H.; Naseem, I. Anti-diabetic study of vitamin B6 on hyperglycaemia induced protein carbonylation, DNA damage and ROS production in alloxan induced diabetic rats. Toxicol. Res. 2019, 8, 568–579. [Google Scholar] [CrossRef]
- Mascolo, E.; Vernì, F. Vitamin B6 and diabetes: Relationship and molecular mechanisms. Int. J. Mol. Sci. 2020, 21, 3669. [Google Scholar] [CrossRef]
- van der Pol, A.; van Gilst, W.H.; Voors, A.A.; van der Meer, P. Treating oxidative stress in heart failure: Past, present and future. Eur. J. Heart Fail. 2019, 21, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Takimoto, E.; Kass, D.A. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension 2007, 49, 241–248. [Google Scholar] [CrossRef]
- Hill, M.F.; Singal, P.K. Antioxidant and oxidative stress changes during heart failure subsequent to myocardial infarction in rats. Am. J. Pathol. 1996, 148, 291–300. [Google Scholar] [PubMed]
- Khaper, N.; Singal, P.K. Effects of afterload-reducing drugs on pathogenesis of antioxidant changes and congestive heart failure in rats. J. Am. Coll. Cardiol. 1997, 29, 856–861. [Google Scholar] [CrossRef] [Green Version]
- Khaper, N.; Kaur, K.; Li, T.; Farahmand, F.; Singal, P.K. Antioxidant enzyme gene expression in congestive heart failure following myocardial infarction. Mol. Cell. Biochem. 2003, 251, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Van Deel, E.D.; Lu, Z.; Xu, X.; Zhu, G.; Hu, X.; Oury, T.D.; Bache, R.J.; Duncker, D.J.; Chen, Y. Extracellular SOD protects the heart against oxidative stress and hypertrophy after myocardial infarction. Free Radic. Biol. Med. 2008, 44, 1305–1313. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Li, Y.L.; Zimmerman, M.C.; Davisson, R.L.; Schultz, H.D. Role of CuZn superoxide dismutase on carotid body function in heart failure rabbits. Cardiovasc. Res. 2009, 81, 678–685. [Google Scholar] [CrossRef]
- Shiomi, T.; Tsutsui, H.; Matsusaka, H.; Murakami, K.; Hayashidani, S.; Ikeuchi, M.; Wen, J.; Kubota, T.; Utsumi, H.; Takeshita, A. Overexpression of glutathione peroxidase prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation 2004, 109, 544–549. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Watanabe, M.; Engelman, D.T.; Engelman, R.M.; Schley, J.A.; Maulik, N.; Ho, Y.; Oberley, T.D.; Das, D.K. Glutathione peroxidase are resistant to myocardial ischemia reperfusion injury. J. Mol. Cell. Cardiol. 1996, 28, 1759–1767. [Google Scholar] [CrossRef]
- Forgione, M.A.; Cap, A.; Liao, R.; Moldovan, N.I.; Eberhardt, R.T.; Lim, C.C.; Jones, J.; Goldschmidt-Clermont, P.J.; Loscalzo, J. Heterozygous cellular glutathione peroxidase deficiency in the mouse: Abnormalities in vascular and cardiac function and structure. Circulation 2002, 106, 1154–1158. [Google Scholar] [CrossRef] [Green Version]
- Diguet, N.; Trammell, S.; Tannous, C.; Deloux, R.; Piquereau, J.; Maougenot, N.; Gouge, A.; Grassete, M.; Manoury, B.; Blanc, J.; et al. Nicotinamide riboside preserves cardiac function in a mouse model of dilated cardiomyopathy. Circulation 2018, 137, 2256–2273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamy, C.; Mulder, P.; Khouzami, L.; Andrieu-abadie, N.; Defer, N.; Candiani, G.; Pavoine, C.; Caramelle, P.; Souktani, R.; Le Corvoisier, P.; et al. Neutral sphingomyelinase inhibition participates to the benefits of N-acetylcysteine treatment in post-myocardial infarction failing heart rats. J. Mol. Cell. Cardiol. 2007, 43, 344–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourraindeloup, M.; Adamy, C.; Candiani, G.; Cailleret, M.; Bourin, M.C.; Badoual, T.; Su, J.B.; Adubeiro, S.; Roudot-Thoraval, F.; Dubois-Rande, J.L.; et al. N-Acetylcysteine Treatment normalizes serum tumor necrosis factor-α level and hinders the progression of cardiac injury in hypertensive rats. Circulation 2004, 110, 2003–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danielyan, K.E.; Simonyan, A.A. Protective abilities of pyridoxine in experimental oxidative stress settings in vivo and in vitro. Biomed. Pharmacother. 2017, 86, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Hille, R.; Nishino, T. Xanthine oxidase and xanthine dehydrogenase. FASEB J. 1995, 9, 995–1003. [Google Scholar] [CrossRef]
- Dhalla, N.S.; Takeda, S.; Elimban, V. Mechanisms of the beneficial effects of vitamin B6 and pyridoxal 5-phosphate on cardiac performance in ischemic heart disease. Clin. Chem. Lab. Med. 2013, 51, 535–543. [Google Scholar] [CrossRef]
- Endo, N.; Nishiyama, K.; Otsuka, A.; Kanouchi, H.; Taga, M.; Oka, T. Antioxidant activity of vitamin B 6 delays homocysteine-induced atherosclerosis in rats. Br. J. Nutr. 2006, 95, 1088–1093. [Google Scholar] [CrossRef] [Green Version]
- Deth, R.; Muratore, C.; Benzeery, J.; Pawer-Charnitsky, V.-A.; Waly, M. How environmental and genetic factors combine to cause autism: A redox/methylation hypothesis. Neurotoxicology 2010, 29, 190–201. [Google Scholar] [CrossRef]
- Mendes, R.; Candido, G.; Mostarda, C.; Sirvente, R.; D’Almeida, V.; Ribeiro, M.; Araujo, A.; Salemi, V.; Rigatto, K.; Irigoyen, M.; et al. Effects od isolated vitamin B6 supplementation on oxidative stress and heart function parameters in experimental hyperhomocysteinemia. Clin. Biomed. Res. 2017, 37, 73–80. [Google Scholar] [CrossRef]
- Stroes, E.S.G.; Van Faassen, E.E.; Yo, M.; Martasek, P.; Boer, P.; Govers, R.; Rabelink, T.J. Folic acid reverts dysfunction of endothelial nitric oxide synthase. Circ. Res. 2000, 86, 1129–1134. [Google Scholar] [CrossRef]
- Shirodaria, C.; Antoniades, C.; Lee, J.; Jackson, C.E.; Robson, M.D.; Francis, J.M.; Moat, S.J.; Ratnatunga, C.; Pillai, R.; Refsum, H.; et al. Global improvement of vascular function and redox state with low-dose folic acid: Implications for folate therapy in patients with coronary artery disease. Circulation 2007, 115, 2262–2270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Tang, R.; Ouyang, S.; Ma, F.; Liu, Z.; Wu, J. Folic acid prevents cardiac dysfunction and reduces myocardial fibrosis in a mouse model of high-fat diet-induced obesity. Nutr. Metab. 2017, 14, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, A.; Mujumdar, V.; Palmer, L.; Bower, J.D.; Tyagi, S.C. Reversal of endocardial endothelial dysfunction by folic acid in homocysteinemic hypertensive rats. Am. J. Hypertens. 2002, 15, 157–163. [Google Scholar] [CrossRef]
- Guo, X.; Cui, H.; Zhang, H.; Guan, X.; Zhang, Z.; Jia, C.; Wu, J.; Yang, H.; Qiu, W.; Zhang, C.; et al. Protective Effect of Folic Acid on Oxidative DNA Damage. Medicine 2015, 94, e1872. [Google Scholar] [CrossRef]
- Uzelac, J.J.; Djukic, T.; Mutavdzin, S.; Stankovic, S.; Borovic, M.L.; Rakocevic, J.; Milic, N.; Radojevic, A.S.; Vasic, M.; Zigon, N.J.; et al. The influence of subchronic co-application of vitamins B6 and folic acid on cardiac oxidative stress and biochemical markers in monocrotaline-induced heart failure in male wistar albino rats. Can. J. Physiol. Pharmacol. 2020, 98, 93–102. [Google Scholar] [CrossRef]
- Uzelac, J.J.; Djukic, T.; Radic, T.; Mutavdzin, S.; Stankovic, S.; Rakocevic, J.K.; Borovic, M.L.; Milic, N.; Simic, T.; Savic-Radojevic, A.; et al. Folic acid affects cardiometabolic, oxidative stress, and immunohistochemical parameters in monocrotaline-induced rat heart failure. Can. J. Physiol. Pharmacol. 2020, 98, 708–716. [Google Scholar] [CrossRef]
- Octavia, Y.; Kararigas, G.; de Boer, M.; Chrifi, I.; Kietadisorn, R.; Swinnen, M.; Duimel, H.; Verheyen, F.K.; Brandt, M.M.; Fliegner, D.; et al. Folic acid reduces doxorubicin-induced cardiomyopathy by modulating endothelial nitric oxide synthase. J. Cell. Mol. Med. 2017, 21, 3277–3287. [Google Scholar] [CrossRef] [Green Version]
- Djurić, D.; Vušanović, A.; Jakovljević, V. The effects of folic acid and nitric oxide synthase inhibition on coronary flow and oxidative stress markers in isolated rat heart. Mol. Cell. Biochem. 2007, 300, 177–183. [Google Scholar] [CrossRef]
- Tawakol, A.; Migrino, R.; Aziz, K.; Waitkowska, J.; Holmvang, G.; Alpert, N.; Muller, J.; Fischman, A.; Gewirtz, H. High-dose folic acid acutely improves coronary vasodilator function in patients with coronary artery disease. J. Am. Coll. Cardiol. 2005, 45, 1580–1584. [Google Scholar] [CrossRef] [Green Version]
- Doshi, S.N.; McDowell, I.F.W.; Moat, S.J.; Payne, N.; Durrant, H.J.; Lewis, M.J.; Goodfellow, J. Folic acid improves endothelial function in coronary artery disease via mechanisms largely independent of homocysteine lowering. Circulation 2002, 105, 22–26. [Google Scholar] [CrossRef] [Green Version]
- Scicchitano, P.; Cortese, F.; Gesualdo, M.; De Palo, M.; Massari, F.; Giordano, P.; Ciccone, M.M. The role of endothelial dysfunction and oxidative stress in cerebrovascular diseases. Free Radic. Res. 2019, 53, 579–595. [Google Scholar] [CrossRef] [PubMed]
- Minatoguchi, S.; Takemura, G.; Chen, X.H.; Wang, N.; Uno, Y.; Koda, M.; Arai, M.; Misao, Y.; Lu, C.; Suzuki, K.; et al. Acceleration of the healing process and myocardial regeneration may be important as a mechanism of improvement of cardiac function and remodeling by postinfarction granulocyte colony-stimulating factor treatment. Circulation 2004, 109, 2572–2580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barandon, L.; Couffinhal, T.; Dufourcq, P.; Ezan, J.; Costet, P.; Daret, D.; Deville, C.; Duplàa, C. Frizzled A, a novel angiogenic factor: Promises for cardiac repair. Eur. J. Cardio-Thorac. Surg. 2004, 25, 76–83. [Google Scholar] [CrossRef] [Green Version]
- Bonvini, R.F.; Hendiri, T.; Camenzind, E. Inflammatory response post-myocardial infarction and reperfusion: A new therapeutic target? Eur. Heart J. Suppl. 2005, 7, 127–136. [Google Scholar] [CrossRef] [Green Version]
- Ilic, A.; Todorovic, D.; Mutavdzin, S.; Boricic, N.; Nedeljkovic, B.B.; Stankovic, S.; Simic, T.; Stevanovic, P.; Celic, V.; Djuric, D. Translocator protein modulation by 4′-chlorodiazepam and no synthase inhibition affect cardiac oxidative stress, cardiometabolic and inflammatory markers in isoprenaline-induced rat myocardial infarction. Int. J. Mol. Sci. 2021, 22, 2867. [Google Scholar] [CrossRef]
- Griselli, M.; Herbert, J.; Hutchinson, W.L.; Taylor, K.M.; Sohail, M.; Krausz, T.; Pepys, M.B. C-reactive protein and complement are important mediators of tissue damage in acute myocardial infarction. J. Exp. Med. 1999, 190, 1733–1739. [Google Scholar] [CrossRef] [Green Version]
- Sekido, N.; Mukaida, N.; Harada, A.; Nakanishi, I.; Watanabe, Y.; Matsushima, K. Prevention of lung reperfusion injury in rabbits by a monoclonal antibody against interleukin-8. Nature 1993, 365, 654–657. [Google Scholar] [CrossRef]
- Kereiakes, D.J. Inflammation as a therapeutic target: A unique role for abciximab. Am. Heart J. 2003, 146, S1–S4. [Google Scholar] [CrossRef]
- Serini, G.; Gabbiani, G. Mechanisms of myofibroblast activity and phenotypic modulation. Exp. Cell Res. 1999, 250, 273–283. [Google Scholar] [CrossRef]
- Paoletti, R.; Gotto, A.M.; Hajjar, D.P. Inflammation in atherosclerosis and implications for therapy. Circulation 2004, 109, 20–26. [Google Scholar] [CrossRef] [Green Version]
- Liuzzo, G.; Buffon, A.; Biasucci, L.M.; Gallimore, J.R.; Caligiuri, G.; Vitelli, A.; Altamura, S.; Ciliberto, G.; Rebuzzi, A.G.; Crea, F.; et al. Enhanced inflammatory response to coronary angioplasty in patients with severe unstable angina. Circulation 1998, 98, 2370–2376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cusack, M.R.; Marber, M.S.; Lambiase, P.D.; Bucknall, C.A.; Redwood, S.R. Systemic inflammation in unstable angina is the result of myocardial necrosis. J. Am. Coll. Cardiol. 2002, 39, 1917–1923. [Google Scholar] [CrossRef] [Green Version]
- Bujak, M.; Dobaczewski, M.; Chatila, K.; Mendoza, L.H.; Li, N.; Reddy, A.; Frangogiannis, N.G. Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am. J. Pathol. 2008, 173, 57–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbate, A.; Salloum, F.N.; van Tassell, B.W.; Vecile, E.; Toldo, S.; Seropian, I.; Mezzaroma, E.; Dobrina, A. Alterations in the interleukin-1/interleukin-1 receptor antagonist balance modulate cardiac remodeling following myocardial infarction in the mouse. PLoS ONE 2011, 6, e27923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauro, A.G.; Bonaventura, A.; Mezzaroma, E.; Quader, M.; Toldo, S. NLRP3 Inflammasome in acute myocardial infarction. J. Cardiovasc. Pharmacol. 2019, 74, 175–187. [Google Scholar] [CrossRef]
- Venkatachalam, K.; Prabhu, S.D.; Reddy, V.S.; Boylston, W.H.; Valente, A.J.; Chandrasekar, B. Neutralization of interleukin-18 ameliorates ischemia/reperfusion-induced myocardial injury. J. Biol. Chem. 2009, 284, 7853–7865. [Google Scholar] [CrossRef] [Green Version]
- Morris, M.S.; Sakakeeny, L.; Jacques, P.F.; Picciano, M.F.; Selhub, J. Vitamin B-6 intake is inversely related to, and the requirement is affected by, inflammation status. J. Nutr. 2010, 140, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Vermaak, W.J.H.; Barnard, H.C.; Van Dalen, E.M.S.P.; Potgieter, G.M.; Van Jaarsveld, H.; Myburgh, S.J.S. Compartmentalization of pyridoxal-5′-phosphate during the acute phase of myocardial infarction. Klin. Wochenschr. 1988, 66, 428–433. [Google Scholar] [CrossRef]
- Vermaak, J.H.; Barnard, H.C.; Potgieter, G.M.; Marx, J. Plasma pyridoxal-5’-phosphate levels in myocardial infarction. S. Afr. Med. J. 1986, 70, 195–196. [Google Scholar]
- Friso, S.; Jacques, P.F.; Wilson, P.W.F.; Rosenberg, I.H.; Selhub, J. Low circulating vitamin B6 is associated with elevation of the inflammation marker C-reactive protein independently of plasma homocysteine levels. Circulation 2001, 103, 2788–2791. [Google Scholar] [CrossRef] [Green Version]
- Chiang, E.P.I.; Selhub, J.; Bagley, P.J.; Dallal, G.; Roubenoff, R. Pyridoxine supplementation corrects vitamin B6 deficiency but does not improve inflammation in patients with rheumatoid arthritis. Arthritis Res. Ther. 2005, 7, R1404–R1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dusitanond, P.; Eikelboom, J.W.; Hankey, G.J.; Thom, J.; Gilmore, G.; Loh, K.; Yi, Q.; Klijn, C.J.M.; Langton, P.; Van Bockxmeer, F.M.; et al. Homocysteine-lowering treatment with folic acid, cobalamin, and pyridoxine does not reduce blood markers of inflammation, endothelial dysfunction, or hypercoagulability in patients with previous transient ischemic attack or stroke: A randomized substudy of the VITATOPS trial. Stroke 2005, 36, 144–146. [Google Scholar] [CrossRef]
- Sood, S.; Chelu, M.; Van Oort, R.; Skapura, D.; Santonastasi, M.; Dobrev, D.; Wehrens, X. Intracellular calcium leak due to FKBP12.6 deficiency in mice facilitates the inducibility of atrial fibrillation. Heart Rhythm 2008, 5, 1047–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Chen, J.; Li, Y.S.; Feng, Y.B.; Gu, X.; Shi, C.Z. Folic acid reduces adhesion molecules VCAM-1 expession in aortic of rats with hyperhomocysteinemia. Int. J. Cardiol. 2006, 106, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Holt, E.M.; Steffen, L.M.; Moran, A.; Basu, S.; Steinberger, J.; Ross, J.A.; Hong, C.-P.; Sinaiko, A.R. Fruit and vegetable consumption and its relation to markers of inflammation and oxidative stress in adolescents. J. Am. Diet. Assoc. 2009, 109, 414–421. [Google Scholar] [CrossRef] [Green Version]
- Tousoulis, D.; Kourkouti, P.; Briasoulis, A.; Vogiatzi, G.; Valatsou, A.; Pantopoulou, A.; Oikonomou, E.; Perrea, D.; Stefanadis, C. Divergent effects of folic acid administration on inflammatory status and cholesterol levels in apoE deficient mice. Int. J. Cardiol. 2014, 173, 608–609. [Google Scholar] [CrossRef]
- Hofmann, M.A.; Lalla, E.; Lu, Y.; Gleason, M.R.; Wolf, B.M.; Tanji, N.; Ferran, L.J.; Kohl, B.; Rao, V.; Kisiel, W.; et al. Hyperhomocysteinemia enhances vascular inflammation and accelerates atherosclerosis in a murine model. J. Clin. Investig. 2001, 107, 675–683. [Google Scholar] [CrossRef] [Green Version]
- Marui, N.; Offermann, M.K.; Swerlick, R.; Kunsch, C.; Rosen, C.A.; Ahmad, M.; Wayne Alexander, R.; Medford, R.M. Vascular cell adhesion molecule-1 (VCAM-1) gene transcription and expression are regulated through an antioxidant-sensitive mechanism in human vascular endothelial cells. J. Clin. Investig. 1993, 92, 1866–1874. [Google Scholar] [CrossRef]
- Neish, A.S.; Williams, A.J.; Palmer, H.J.; Whitley, M.Z.; Collins, T. Functional analysis of the human vascular cell adhesion molecule 1 promoter. J. Exp. Med. 1992, 176, 1583–1593. [Google Scholar] [CrossRef]
- Schreck, R.; Albermann, K.; Baeuerle, P.A. Nuclear factor kb: An oxidative stress-responsive transcription factor of eukaryotic cells (a review). Free Radic. Res. 1992, 17, 221–237. [Google Scholar] [CrossRef]
- Kretz-Remy, C.; Mehlen, P.; Mirault, M.E.; Arrigo, A.P. Inhibition of IκB-α phosphorylation and degradation and subsequent NF-κB activation by glutathione peroxidase overexpression. J. Cell Biol. 1996, 133, 1083–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, C. Gaseotransmitters: New frontiers for translational science. Sci. Transl. Med. 2010, 2, 59ps54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coletta, C.; Papapetropoulos, A.; Erdelyi, K.; Olah, G.; Módis, K.; Panopoulos, P.; Asimakopoulou, A.; Gerö, D.; Sharina, I.; Martin, E.; et al. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc. Natl. Acad. Sci. USA 2012, 109, 9161–9166. [Google Scholar] [CrossRef] [Green Version]
- Muchova, L.; Wong, R.J.; Hsu, M.; Morioka, I.; Vitek, L.; Zelenka, J.; Schröder, H.; Stevenson, D.K. Statin treatment increases formation of carbon monoxide and bilirubin in mice: A novel mechanism of in vivo antioxidant protection. Can. J. Physiol. Pharmacol. 2007, 85, 800–810. [Google Scholar] [CrossRef] [PubMed]
- Djurić, M.; Kostic, S.; Loncar-Stojiljkovic, D.; Mutavdzin, S.; Colović, M.; Krstic, D.; Stevanovic, P.; Djuric, D. The effects of gasotransmitters inhibition on homocysteine acutely induced changes in oxidative stress markers in rat plasma. Scr. Med. 2019, 50, 6–12. [Google Scholar] [CrossRef]
- Jones, S.P.; Girod, W.G.; Palazzo, A.J.; Granger, D.N.; Grisham, M.B.; Jourd’heuil, D.; Huang, P.L.; Lefer, D.J. Myocardial ischemia-reperfusion injury is exacerbated in absence of endothelial cell nitric oxide synthase. Am. J. Physiol. Heart Circ. Physiol. 1999, 276, 1567–1573. [Google Scholar] [CrossRef] [Green Version]
- Calvert, J.W.; Lefer, D.J. Myocardial protection by nitrite. Cardiovasc. Res. 2009, 83, 195–203. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Caldwell, R.B.; Li-Masters, T.; Caldwell, R.W. Homocysteine induces endothelial dysfunction via inhibition of arginine transport. J. Physiol. Pharmacol. 2007, 58, 191–206. [Google Scholar]
- Topal, G.; Brunet, A.; Millanvoye, E.; Boucher, J.L.; Rendu, F.; Devynck, M.A.; David-Dufilho, M. Homocysteine induces oxidative stress by uncoupling of no synthase activity through reduction of tetrahydrobiopterin. Free Radic. Biol. Med. 2004, 36, 1532–1541. [Google Scholar] [CrossRef]
- Lai, W.K.C.; Kan, M.Y. Homocysteine-induced endothelial dysfunction. Ann. Nutr. Metab. 2015, 67, 1–12. [Google Scholar] [CrossRef]
- Bir, S.; Kolluru, G.; Fang, K.; Kevil, C. Redox balance dynamically regulates vascular growth and remodeling. Semin. Cell Dev. Biol. 2012, 23, 745–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leiper, J.M.; Vallance, P. The synthesis and metabolism of asymmetric dimethylarginine (ADMA). Eur. J. Clin. Pharmacol. 2006, 62, 33–38. [Google Scholar] [CrossRef]
- Böger, R.H.; Bode-Böger, S.M.; Tsao, P.S.; Lin, P.S.; Chan, J.R.; Cooke, J.P. An endogenous inhibitor of nitric oxide synthase regulates endothelial adhesiveness for monocytes. J. Am. Coll. Cardiol. 2000, 36, 2287–2295. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Palfrey, H.A.; Pathak, R.; Kadowitz, P.J.; Gettys, T.W.; Murthy, S.N. The metabolism and significance of homocysteine in nutrition and health. Nutr. Metab. 2017, 14, 78. [Google Scholar] [CrossRef] [Green Version]
- Johnson, G.; Tsao, P.; Leffer, A. Cardioprotective effects of authentic nitric oxide in myocardial ischemia with reperfusion. Crit. Care Med. 1991, 19, 244–252. [Google Scholar] [CrossRef]
- Loke, K.E.; Laycock, S.K.; Mital, S.; Wolin, M.S.; Bernstein, R.; Oz, M.; Addonizio, L.; Kaley, G.; Hintze, T.H. Nitric oxide modulates mitochondrial respiration in failing human heart. Circulation 1999, 100, 1291–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, D.D. The biological lifetime of nitric oxide: Implications for the perivascular dynamics of NO and O2. Proc. Natl. Acad. Sci. USA 2001, 98, 355–360. [Google Scholar] [CrossRef]
- Ma, X.I.; Weyrich, A.S.; Lefer, D.J.; Lefer, A.M. Diminished basal nitric oxide release after myocardial ischemia and reperfusion promotes neutrophil adherence to coronary endothelium. Circ. Res. 1993, 72, 403–412. [Google Scholar] [CrossRef] [Green Version]
- Palazzo, A.J.; Jones, S.P.; Girod, W.G.; Anderson, D.C.; Granger, D.N.; Lefer, D.J.; Anthony, J.; Jones, S.P.; Wesley, G.; Anderson, D.C.; et al. Myocardial ischemia-reperfusion injury in CD18- and ICAM-1-deficient mice. Am. J. Physiol. Heart Circ. Physiol. 1998, 275, H2300–H2307. [Google Scholar] [CrossRef]
- Walter, U.; Gambaryan, S. Roles of cGMP/cGMP-dependent protein kinase in platelet activation. Blood 2004, 104, 2609. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Bombeck, C.A.; Yang, S.; Kim, Y.M.; Billiar, T.R. Nitric oxide suppresses apoptosis via interrupting caspase activation and mitochondrial dysfunction in cultured hepatocytes. J. Biol. Chem. 1999, 274, 17325–17333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dezfulian, C.; Shiva, S.; Alekseyenko, A.; Pendyal, A.; Beiser, D.G.; Munasinghe, J.P.; Anderson, S.A.; Chesley, C.F.; Vanden Hoek, T.L.; Gladwin, M.T. Nitrite therapy after cardiac arrest reduces reactive oxygen species generation, improves cardiac and neurological function, and enhances survival via reversible inhibition of mitochondrial complex I. Circulation 2009, 120, 897–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendgen-Cotta, U.B.; Merx, M.W.; Shiva, S.; Schmitz, J.; Becher, S.; Klare, J.P.; Steinhoff, H.J.; Goedecke, A.; Schrader, J.; Gladwin, M.T.; et al. Nitrite reductase activity of myoglobin regulates respiration and cellular viability in myocardial ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA 2008, 105, 10256–10261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, F.; Shiva, S.; Vincent, P.; Ringwood, L.; Hsu, L.; Hon, Y.; Alteras, A.; Cannon, R.O.; Gladwin, M.; Arai, A. Nitrite anion (NO2−) provides potent cytoprotective and anti- apoptotic effects as adjunctive therapy to reperfusion for acute myocardial infarction. Circulation 2008, 117, 2886–2994. [Google Scholar] [CrossRef] [Green Version]
- Webb, A.; Bond, R.; McLean, P.; Uppal, R.; Benjamin, N.; Ahluwalia, A. Reduction of nitrite to nitric oxide during ischemia protects against myocardial ischemia-reperfusion damage. Proc. Natl. Acad. Sci. USA 2004, 101, 13683–13688. [Google Scholar] [CrossRef] [Green Version]
- Duranski, M.R.; Greer, J.J.M.; Dejam, A.; Jaganmohan, S.; Hogg, N.; Langston, W.; Patel, R.P.; Yet, S.F.; Wang, X.; Kevil, C.G.; et al. Cytoprotective effects of nitrite during in vivo ischemia-reperfusion of the heart and liver. J. Clin. Investig. 2005, 115, 1232–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, W.Y.; Dudman, N.P.B.; Perry, M.A.; Wang, X.L. Homocysteine attenuates hemodynamic responses to nitric oxide in vivo. Atherosclerosis 2002, 161, 169–176. [Google Scholar] [CrossRef]
- Upchurch, G.R.; Welche, G.N.; Fabian, A.J.; Freedman, J.E.; Johnson, J.L.; Keaney, J.F.; Loscalzo, J. Homocyst(e)ine decreases bioavailable nitric oxide by a mechanism involving glutathione peroxidase. J. Biol. Chem. 1997, 272, 17012–17017. [Google Scholar] [CrossRef] [Green Version]
- Sauls, D.L.; Lockhart, E.; Warren, M.E.; Lenkowski, A.; Wilhelm, S.E.; Hoffman, M. Modification of fibrinogen by homocysteine thiolactone increases resistance to fibrinolysis: A potential mechanism of the thrombotic tendency in hyperhomocysteinemia. Biochemistry 2006, 45, 2480–2487. [Google Scholar] [CrossRef]
- Tamura, Y.; Inoue, A.; Ijiri, Y.; Naemura, A.; Yamamoto, J. Short- and long-term treatment with folic acid suppresses thrombus formation in atherogenic mice in vivo. Pathophysiology 2014, 21, 169–175. [Google Scholar] [CrossRef]
- Vermeulen, E.G.J.; Stehouwer, C.D.A.; Twisk, J.W.R.; Van Den Berg, M.; De Jong, S.C.; Mackaay, A.J.C.; Van Campen, C.M.C.; Visser, F.C.; Jakobs, C.A.J.M.; Bulterijs, E.J.; et al. Effect of homocysteine-lowering treatment with folic acid plus vitamin B6 on progression of subclinical atherosclerosis: A randomised, placebo-controlled trial. Lancet 2000, 355, 517–522. [Google Scholar] [CrossRef]
- Welch, G.; Loscalzo, J. Homocysteine and atherothrombosis. N. Engl. J. Med. 1998, 338, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Lynn, E.G.; Austin, R.C. Hydrogen sulfide in the pathogenesis of atherosclerosis and its therapeutic potential. Expert Rev. Clin. Pharmacol. 2011, 4, 97–108. [Google Scholar] [CrossRef]
- Wang, R. Physiological implications of hydrogen sulfide: A whiff exploration that blossomed. Physiol. Rev. 2012, 92, 791–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Citi, V.; Martelli, A.; Gorica, E.; Brogi, S.; Testai, L.; Calderone, V. Role of hydrogen sulfide in endothelial dysfunction: Pathophysiology and therapeutic approaches. J. Adv. Res. 2021, 27, 99–113. [Google Scholar] [CrossRef]
- Yang, R.; Jia, Q.; Ma, S.F.; Wang, Y.; Mehmood, S.; Chen, Y. Exogenous H2S mitigates myocardial fibrosis in diabetic rats through suppression of the canonical Wnt pathway. Int. J. Mol. Med. 2019, 44, 549–558. [Google Scholar] [CrossRef] [Green Version]
- Gorini, F.; Bustaffa, E.; Chatzianagnostou, K.; Bianchi, F.; Vassalle, C. Hydrogen sulfide and cardiovascular disease: Doubts, clues, and interpretation difficulties from studies in geothermal areas. Sci. Total Environ. 2020, 743, 140818. [Google Scholar] [CrossRef]
- Polhemus, D.J.; Kondo, K.; Bhushan, S.; Bir, S.C.; Kevil, C.G.; Murohara, T.; Lefer, D.J.; Calvert, J.W. Hydrogen sulfide attenuates cardiac dysfunction after heart failure via induction of angiogenesis. Circ. Heart Fail. 2013, 6, 1077–1086. [Google Scholar] [CrossRef] [Green Version]
- Elrod, J.W.; Calvert, J.W.; Morrison, J.; Doeller, J.E.; Kraus, D.W.; Tao, L.; Jiao, X.; Scalia, R.; Kiss, L.; Szabo, C.; et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc. Natl. Acad. Sci. USA 2007, 104, 15560–15565. [Google Scholar] [CrossRef] [Green Version]
- Polhemus, D.J.; Calvert, J.W.; Butler, J.; Lefer, D.J. The cardioprotective actions of hydrogen sulfide in acute myocardial infarction and heart failure. Scientifica 2014, 2014, 768607. [Google Scholar] [CrossRef] [Green Version]
- Blackstone, E.; Morrison, M.; Roth, M.B. H2S induces a suspended animation-like state in mice. Science 2005, 308, 518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Predmore, B.L.; Kondo, K.; Bhushan, S.; Zlatopolsky, M.A.; King, A.L.; Aragon, J.P.; Bennett Grinsfelder, D.; Condit, M.E.; Lefer, D.J. The polysulfide diallyl trisulfide protects the ischemic myocardium by preservation of endogenous hydrogen sulfide and increasing nitric oxide bioavailability. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H2410–H2418. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; He, G.W. Imbalance of homocysteine and H2S: Significance, mechanisms, and therapeutic promise in vascular injury. Oxid. Med. Cell. Longev. 2019, 2019, 7629673. [Google Scholar] [CrossRef] [Green Version]
- Li, J.J.; Li, Q.; Du, H.P.; Wang, Y.L.; You, S.J.; Wang, F.; Xu, X.S.; Cheng, J.; Cao, Y.J.; Liu, C.F.; et al. Homocysteine triggers inflammatory responses in macrophages through inhibiting CSE-H2S signaling via DNA hypermethylation of CSE promoter. Int. J. Mol. Sci. 2015, 16, 12560–12577. [Google Scholar] [CrossRef]
- Li, M.H.; Tang, J.P.; Zhang, P.; Li, X.; Wang, C.Y.; Wei, H.J.; Yang, X.F.; Zou, W.; Tang, X.Q. Disturbance of endogenous hydrogen sulfide generation and endoplasmic reticulum stress in hippocampus are involved in homocysteine-induced defect in learning and memory of rats. Behav. Brain Res. 2014, 262, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Peers, C.; Steele, D.S. Carbon monoxide: A vital signalling molecule and potent toxin in the myocardium. J. Mol. Cell. Cardiol. 2012, 52, 359–365. [Google Scholar] [CrossRef]
- Johnson, R.A.; Teran, F.J.; Durante, W.; Peyton, K.J.; Johnson, F.K. Enhanced heme oxygenase-mediated coronary vasodilation in dahl salt-sensitive hypertension. Am. J. Hypertens. 2004, 17, 25–30. [Google Scholar] [CrossRef] [Green Version]
- Stein, A.B.; Bolli, R.; Dawn, B.; Sanganalmath, S.K.; Zhu, Y.; Wang, O.L.; Guo, Y.; Motterlini, R.; Xuan, Y.T. Carbon monoxide induces a late preconditioning-mimetic cardioprotective and antiapoptotic milieu in the myocardium. J. Mol. Cell. Cardiol. 2012, 52, 228–236. [Google Scholar] [CrossRef] [Green Version]
- Parviz, Y.; Waleed, M.; Vijayan, S.; Adlam, D.; Lavi, S.; Al Nooryani, A.; Iqbal, J.; Stone, G.W. Cellular and molecular approaches to enhance myocardial recovery after myocardial infarction. Cardiovasc. Revascularization Med. 2019, 20, 351–364. [Google Scholar] [CrossRef]
- Wang, G.; Hamid, T.; Keith, R.J.; Zhou, G.; Partridge, C.R.; Xiang, X.; Kingery, J.R.; Lewis, R.K.; Li, Q.; Rokosh, D.G.; et al. Cardioprotective and antiapoptotic effects of heme oxygenase-1 in the failing heart. Circulation 2010, 121, 1912–1925. [Google Scholar] [CrossRef] [Green Version]
- Stein, A.B.; Guo, Y.; Tan, W.; Wu, W.J.; Zhu, X.; Li, Q.; Luo, C.; Dawn, B.; Johnson, T.R.; Motterlini, R.; et al. Administration of a CO-releasing molecule induces late preconditioning against myocardial infarction. J. Mol. Cell. Cardiol. 2005, 38, 127–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segersvärd, H.; Lakkisto, P.; Hänninen, M.; Forsten, H.; Siren, J.; Immonen, K.; Kosonen, R.; Sarparanta, M.; Laine, M.; Tikkanen, I. Carbon monoxide releasing molecule improves structural and functional cardiac recovery after myocardial injury. Eur. J. Pharmacol. 2018, 818, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Mahan, V. Cardiac function dependence on carbon monoxide. Med. Gas Res. 2020, 10, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Głowacka, U.; Brzozowski, T.; Magierowski, M. Synergisms, discrepancies and interactions between hydrogen sulfide and carbon monoxide in the gastrointestinal and digestive system physiology, pathophysiology and pharmacology. Biomolecules 2020, 10, 445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramochkin, D.V.; Haertdinov, N.N.; Porokhnya, M.V.; Zefirov, A.L.; Sitdikova, G.F. Carbon monoxide affects electrical and contractile activity of rat myocardium. J. Biomed. Sci. 2011, 18, 40. [Google Scholar] [CrossRef] [Green Version]
- Dallas, M.L.; Yang, Z.; Boyle, J.P.; Boycott, H.E.; Scragg, J.L.; Milligan, C.J.; Elies, J.; Duke, A.; Thireau, J.; Reboul, C.; et al. Carbon monoxide induces cardiac arrhythmia via induction of the late Na+ current. Am. J. Respir. Crit. Care Med. 2012, 186, 648–656. [Google Scholar] [CrossRef] [Green Version]
- Farrugia, G.; Szurszewski, J. Carbon monoxide, hydrogen sulfide, and nitric oxide as signaling molecules in the gastrointestinal tract. Gastroenterology 2014, 147, 303–313. [Google Scholar] [CrossRef] [Green Version]
- Hishiki, T.; Yamamoto, T.; Morikawa, T.; Kubo, A.; Kajimura, M.; Suematsu, M. Carbon monoxide: Impact on remethylation/transsulfuration metabolism and its pathophysiologic implications. J. Mol. Med. 2012, 90, 245–254. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bajic, Z.; Sobot, T.; Skrbic, R.; Stojiljkovic, M.P.; Ponorac, N.; Matavulj, A.; Djuric, D.M. Homocysteine, Vitamins B6 and Folic Acid in Experimental Models of Myocardial Infarction and Heart Failure—How Strong Is That Link? Biomolecules 2022, 12, 536. https://doi.org/10.3390/biom12040536
Bajic Z, Sobot T, Skrbic R, Stojiljkovic MP, Ponorac N, Matavulj A, Djuric DM. Homocysteine, Vitamins B6 and Folic Acid in Experimental Models of Myocardial Infarction and Heart Failure—How Strong Is That Link? Biomolecules. 2022; 12(4):536. https://doi.org/10.3390/biom12040536
Chicago/Turabian StyleBajic, Zorislava, Tanja Sobot, Ranko Skrbic, Milos P. Stojiljkovic, Nenad Ponorac, Amela Matavulj, and Dragan M. Djuric. 2022. "Homocysteine, Vitamins B6 and Folic Acid in Experimental Models of Myocardial Infarction and Heart Failure—How Strong Is That Link?" Biomolecules 12, no. 4: 536. https://doi.org/10.3390/biom12040536
APA StyleBajic, Z., Sobot, T., Skrbic, R., Stojiljkovic, M. P., Ponorac, N., Matavulj, A., & Djuric, D. M. (2022). Homocysteine, Vitamins B6 and Folic Acid in Experimental Models of Myocardial Infarction and Heart Failure—How Strong Is That Link? Biomolecules, 12(4), 536. https://doi.org/10.3390/biom12040536