
Advanced Strategies for Therapeutic Targeting of Wild-Type and Mutant p53 in Cancer


 ,
,  ,
, 
Abstract
:1. Introduction
2. p53 Structure and Mutations Exploited for Drug Development
2.1. p53 Structure Exploited for Drugs Reactivating p53 Signaling
2.2. Conformational Changes of p53 Mutants Utilized for Drugs That Restore Wild-Type Function
2.3. p53 Transgenic Mouse Models (p53 Knockout, Mutant p53 Knock-Ins, and Inducible Models) Provide a Powerful Tool to Investigate p53 Function
3. Strategies for Boosting Wild-Type p53 Activity in Cancer: Gene Therapy, Cytotoxic Chemotherapy, MDM2/MDMX(MDM4) Inhibitors, p53-Binding Compounds, Targeting p53 PTMs
3.1. Small Molecules Can Directly Target Wild-Type p53 Protein
3.2. Small Molecules Can Activate p53 via Targeting MDM2/MDMX(MDM4)
3.3. Small Molecules Can Induce p53 Transcription via Post-Translational Modifications
4. p53-Dependent and p53-Independent Strategies for Targeting p53 Pathway Restoration in p53-Mutated Cancers
4.1. Restoration of Wild-Type Function in Tumors Expressing Mutant p53
4.2. Therapeutic Induction of Mutant p53 Degradation
4.3. Activation of p73 to Bypassing Mutant p53 in Restoration of Wild-Type p53 Function
4.4. Restoration of p53 Pathway Signaling Independent of p53 through Non-Canonical Regulatory Pathways
5. Targeting p53 Function in Immunotherapy: Bispecific Antibodies, Gene Therapy, Small Molecule Combinations
5.1. Activation of Wild-Type p53 for Immunotherapy Using Immune Checkpoint Inhibitors
5.2. Targeting Mutant p53 in Immunotherapy
6. Combination Therapies Targeting p53 Cell Cycle Checkpoints, MDM2-p53 Inhibition, or Mutant p53
6.1. Targeting p53 in Combination with Cell Stress Signaling
6.2. Targeting p53 in Combination with Inhibition of Raf/MEK/ERK Pathways
6.3. Targeting p53 (Wild-Type and Mutant) in Combination with Conventional Chemotherapy
6.4. Combination Targeting p53 Cell Cycle Checkpoints in p53 Mutated Cancer Cells
7. Summary and Prospects
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in Context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.S.; Ramos, H.; Inga, A.; Sousa, E.; Saraiva, L. Structural and Drug Targeting Insights on Mutant p53. Cancers 2021, 13, 3344. [Google Scholar] [CrossRef]
- Joerger, A.C.; Fersht, A.R. The tumor suppressor p53: From structures to drug discovery. Cold Spring Harb. Perspect. Biol. 2010, 2, a000919. [Google Scholar] [CrossRef]
- Blandino, G.; Di Agostino, S. New therapeutic strategies to treat human cancers expressing mutant p53 proteins. J. Exp. Clin. Cancer Res. 2018, 37, 30. [Google Scholar] [CrossRef] [Green Version]
- Lopes, E.A.; Gomes, S.; Saraiva, L.; Santos, M.M.M. Small Molecules Targeting Mutant P53: A Promising Approach for Cancer Treatment. Curr. Med. Chem. 2020, 26, 7323–7336. [Google Scholar] [CrossRef]
- Huang, J. Current developments of targeting the p53 signaling pathway for cancer treatment. Pharmacol. Ther. 2021, 220, 107720. [Google Scholar] [CrossRef]
- Valente, J.F.A.; Queiroz, J.A.; Sousa, F. p53 as the Focus of Gene Therapy: Past, Present and Future. Curr. Drug Targets 2018, 19, 1801–1817. [Google Scholar] [CrossRef]
- Soragni, A.; Janzen, D.M.; Johnson, L.M.; Lindgren, A.G.; Nguyen, T.Q.A.; Tiourin, E.; Soriaga, A.B.; Lu, J.; Jiang, L.; Faull, K.F.; et al. A Designed Inhibitor of p53 Aggregation Rescues p53 Tumor Suppression in Ovarian Carcinomas. Cancer Cell 2016, 29, 90–103. [Google Scholar] [CrossRef] [Green Version]
- Duffy, M.J.; Synnott, N.C.; O’Grady, S.; Crown, J. Targeting p53 for the treatment of cancer. Semin. Cancer Biol. 2020, 79, 58–67. [Google Scholar] [CrossRef]
- Sabapathy, K.; Lane, D. Therapeutic targeting of p53: All mutants are equal, but some mutants are more equal than others. Nat. Rev. Clin. Oncol. 2018, 15, 13–30. [Google Scholar] [CrossRef]
- Lane, D.P.; Cheok, C.F.; Lain, S. p53-based cancer therapy. Cold Spring Harb. Perspect. Biol. 2010, 2, a001222. [Google Scholar] [CrossRef] [Green Version]
- Menichini, P.; Monti, P.; Speciale, A.; Cutrona, G.; Matis, S.; Fais, F.; Taiana, E.; Neri, A.; Bomben, R.; Gentile, M.; et al. Antitumor Effects of PRIMA-1 and PRIMA-1(Met) (APR246) in Hematological Malignancies: Still a Mutant P53-Dependent Affair? Cells 2021, 10, 98. [Google Scholar] [CrossRef]
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.; McGraw, K.L.; et al. Eprenetapopt (APR-246) and Azacitidine in TP53-Mutant Myelodysplastic Syndromes. J. Clin. Oncol. 2021, 39, 1584–1594. [Google Scholar] [CrossRef]
- Lindemann, A.; Patel, A.A.; Tang, L.; Liu, Z.; Wang, L.; Silver, N.L.; Tanaka, N.; Rao, X.; Takahashi, H.; Maduka, N.K.; et al. COTI-2, a novel thiosemicarbazone derivative, exhibits antitumor activity in HNSCC through p53-dependent and -independent mechanisms. Clin. Cancer Res. 2019, 25, 5650–5662. [Google Scholar]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 Mutations in Human Cancers: Origins, Consequences, and Clinical Use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [Green Version]
- Barta, J.A.; McMahon, S.B. Lung-Enriched Mutations in the p53 Tumor Suppressor: A Paradigm for Tissue-Specific Gain of Oncogenic Function. Mol. Cancer Res. 2019, 17, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Robles, A.; Harris, C.C. Clinical Outcomes and Correlates of TP53 Mutations and Cancer. Cold Spring Harb. Perspect. Biol. 2009, 2, a001016. [Google Scholar] [CrossRef] [Green Version]
- Brosh, R.; Rotter, V. When mutants gain new powers: News from the mutant p53 field. Nat. Cancer 2009, 9, 701–713. [Google Scholar] [CrossRef]
- El-Deiry, W.S.; Kern, S.E.; Pietenpol, J.A.; Kinzler, K.W.; Vogelstein, B. Definition of a consensus binding site for p53. Nat. Genet. 1992, 1, 45–49. [Google Scholar] [CrossRef]
- Wang, W.; Kim, S.-H.; El-Deiry, W.S. Small-molecule modulators of p53 family signaling and antitumor effects in p53-deficient human colon tumor xenografts. Proc. Natl. Acad. Sci. USA 2006, 103, 11003–11008. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Zhou, L.; Hong, B.; van den Heuvel, A.P.J.; Prabhu, V.V.; Warfel, N.A.; Kline, C.L.B.; Dicker, D.T.; Kopelovich, L.; El-Deiry, W.S. Small-Molecule NSC59984 Restores p53 Pathway Signaling and Antitumor Effects against Colorectal Cancer via p73 Activation and Degradation of Mutant p53. Cancer Res. 2015, 75, 3842–3852. [Google Scholar] [CrossRef] [Green Version]
- Hong, B.; Prabhu, V.V.; Zhang, S.; van den Heuvel, A.P.J.; Dicker, D.T.; Kopelovich, L.; El-Deiry, W.S. Prodigiosin Rescues Deficient p53 Signaling and Antitumor Effects via Upregulating p73 and Disrupting Its Interaction with Mutant p53. Cancer Res. 2014, 74, 1153–1165. [Google Scholar] [CrossRef] [Green Version]
- Richardson, C.; Zhang, S.; Borrero, L.J.H.; El-Deiry, W.S. Small-molecule CB002 restores p53 pathway signaling and represses colorectal cancer cell growth. Cell Cycle 2017, 16, 1719–1725. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Borrero, L.J.; Zhang, S.; Lulla, A.; Dicker, D.T.; El-Deiry, W.S. CB002, a novel p53 tumor suppressor pathway-restoring small molecule induces tumor cell death through the pro-apoptotic protein NOXA. Cell Cycle 2018, 17, 557–567. [Google Scholar] [CrossRef] [Green Version]
- Schlichtholz, B.; Trédaniel, J.; Lubin, R.; Zalcman, G.; Hirsch, A.; Soussi, T. Analyses of p53 antibodies in sera of patients with lung carcinoma define immunodominant regions in the p53 protein. Br. J. Cancer 1994, 69, 809–816. [Google Scholar] [CrossRef] [Green Version]
- Schlichtholz, B.; Legros, Y.; Gillet, D.; Gaillard, C.; Marty, M.; Lane, D.; Calvo, F.; Soussi, T. The immune response to p53 in breast cancer patients is directed against immunodominant epitopes unrelated to the mutational hot spot. Cancer Res. 1992, 52, 6380–6384. [Google Scholar]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef]
- Lain, S.; Hollick, J.J.; Campbell, J.; Staples, O.D.; Higgins, M.; Aoubala, M.; McCarthy, A.; Appleyard, V.; Murray, K.E.; Baker, L.; et al. Discovery, In Vivo Activity, and Mechanism of Action of a Small-Molecule p53 Activator. Cancer Cell 2008, 13, 454–463. [Google Scholar] [CrossRef] [Green Version]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef]
- Zhang, S.; Lou, J.; Li, Y.; Zhou, F.; Yan, Z.; Lyu, X.; Zhao, Y. Recent Progress and Clinical Development of Inhibitors that Block MDM4/p53 Protein-Protein Interactions. J. Med. Chem. 2021, 64, 10621–10640. [Google Scholar] [CrossRef]
- Espadinha, M.; Barcherini, V.; Lopes, E.A.; Santos, M.M.M. An Update on MDMX and Dual MDM2/X Inhibitors. Curr. Top Med. Chem. 2018, 18, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Graves, B.; Thompson, T.; Xia, M.; Janson, C.; Lukacs, C.; Deo, D.; Di Lello, P.; Fry, D.; Garvie, C.; Huang, K.-S.; et al. Activation of the p53 pathway by small-molecule-induced MDM2 and MDMX dimerization. Proc. Natl. Acad. Sci. USA 2012, 109, 11788–11793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvajal, L.A.; Ben Neriah, D.; Senecal, A.; Benard, L.; Thiruthuvanathan, V.; Yatsenko, T.; Narayanagari, S.-R.; Wheat, J.C.; Todorova, T.I.; Mitchell, K.; et al. Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci. Transl. Med. 2018, 10, eaao3003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, L.M.M.; Durell, S.R.; Mazur, S.J.; Appella, E. p53 N-terminal phosphorylation: A defining layer of complex regulation. Carcinogenesis 2012, 33, 1441–1449. [Google Scholar] [CrossRef] [Green Version]
- Carr, M.I.; Jones, S.N. Regulation of the Mdm2-p53 signaling axis in the DNA damage response and tumorigenesis. Transl. Cancer Res. 2016, 5, 707–724. [Google Scholar] [CrossRef] [Green Version]
- Liebl, M.C.; Hofmann, T.G. Cell fate regulation upon DNA damage: p53 Serine 46 kinases pave the cell death road. Bioessays 2019, 41, 1900127. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Dyson, H.J.; Wright, P.E. A phosphorylation-dependent switch in the disordered p53 transactivation domain regulates DNA binding. Proc. Natl. Acad. Sci. USA 2021, 118, e2021456118. [Google Scholar] [CrossRef]
- Vieler, M.; Sanyal, S. p53 Isoforms and Their Implications in Cancer. Cancers 2018, 10, 288. [Google Scholar] [CrossRef] [Green Version]
- Khoury, M.P.; Bourdon, J.-C. The Isoforms of the p53 Protein. Cold Spring Harb. Perspect. Biol. 2009, 2, a000927. [Google Scholar] [CrossRef] [Green Version]
- Bourdon, J.-C.; Surget, S.; Khoury, M.P. Uncovering the role of p53 splice variants in human malignancy: A clinical perspective. Onco. Targets Ther. 2013, 7, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Bourdon, J.-C.; Fernandes, K.; Murray-Zmijewski, F.; Liu, G.; Diot, A.; Xirodimas, D.P.; Saville, M.K.; Lane, D.P. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005, 19, 2122–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivares-Illana, V.; Fåhraeus, R. p53 isoforms gain functions. Oncogene 2010, 29, 5113–5119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joruiz, S.M.; Bourdon, J.-C. p53 Isoforms: Key Regulators of the Cell Fate Decision. Cold Spring Harb. Perspect. Med. 2016, 6, a026039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeckler, F.; Joerger, A.; Jaggi, G.; Rutherford, T.J.; Veprintsev, D.; Fersht, A.R. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc. Natl. Acad. Sci. USA 2008, 105, 10360–10365. [Google Scholar] [CrossRef] [Green Version]
- Bauer, M.; Jones, R.N.; Tareque, R.K.; Springett, B.; Dingler, F.; Verduci, L.; Patel, K.J.; Fersht, A.R.; Joerger, A.C.; Spencer, J. A structure-guided molecular chaperone approach for restoring the transcriptional activity of the p53 cancer mutant Y220C. Future Med. Chem. 2019, 11, 2491–2504. [Google Scholar] [CrossRef] [Green Version]
- Baud, M.G.; Bauer, M.; Verduci, L.; Dingler, F.A.; Patel, K.J.; Roy, D.H.; Joerger, A.; Fersht, A.R. Aminobenzothiazole derivatives stabilize the thermolabile p53 cancer mutant Y220C and show anticancer activity in p53-Y220C cell lines. Eur. J. Med. Chem. 2018, 152, 101–114. [Google Scholar] [CrossRef]
- Butler, J.S.; Loh, S.N. Structure, Function, and Aggregation of the Zinc-Free Form of the p53 DNA Binding Domain. Biochemistry 2003, 42, 2396–2403. [Google Scholar] [CrossRef]
- Yu, X.; Vazquez, A.; Levine, A.J.; Carpizo, D.R. Allele-Specific p53 Mutant Reactivation. Cancer Cell 2012, 21, 614–625. [Google Scholar] [CrossRef] [Green Version]
- Galea, C.; Bowman, P.; Kriwacki, R.W. Disruption of an intermonomer salt bridge in the p53 tetramerization domain results in an increased propensity to form amyloid fibrils. Protein Sci. 2005, 14, 2993–3003. [Google Scholar] [CrossRef] [Green Version]
- Zawacka-Pankau, J.; Selivanova, G. Pharmacological reactivation of p53 as a strategy to treat cancer. J. Intern. Med. 2014, 277, 248–259. [Google Scholar] [CrossRef] [Green Version]
- Pairawan, S.; Zhao, M.; Yuca, E.; Annis, A.; Evans, K.; Sutton, D.; Carvajal, L.; Ren, J.; Santiago, S.; Guerlavais, V. First in class dual MDM2/MDMX inhibitor ALRN-6924 enhances antitumor efficacy of chemotherapy in TP53 wild-type hormone receptor-positive breast cancer models. Breast Cancer Res. 2021, 23, 29. [Google Scholar] [CrossRef] [PubMed]
- Perdrix, A.; Najem, A.; Saussez, S.; Awada, A.; Journe, F.; Ghanem, G.; Krayem, M. PRIMA-1 and PRIMA-1Met (APR-246): From Mutant/Wild Type p53 Reactivation to Unexpected Mechanisms Underlying Their Potent Anti-Tumor Effect in Combinatorial Therapies. Cancers 2017, 9, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vareki, S.M.; Salim, K.Y.; Danter, W.R.; Koropatnick, J. Novel anti-cancer drug COTI-2 synergizes with therapeutic agents and does not induce resistance or exhibit cross-resistance in human cancer cell lines. PLoS ONE 2018, 13, e0191766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lampreht Tratar, U.; Horvat, S.; Cemazar, M. Transgenic Mouse Models in Cancer Research. Front. Oncol. 2018, 8, 268. [Google Scholar] [CrossRef] [Green Version]
- Kersten, K.; De Visser, K.E.; Van Miltenburg, M.H.; Jonkers, J. Genetically engineered mouse models in oncology research and cancer medicine. EMBO Mol. Med. 2016, 9, 137–153. [Google Scholar] [CrossRef]
- Brož, D.K.; Attardi, L.D. In vivo analysis of p53 tumor suppressor function using genetically engineered mouse models. Carcinogenesis 2010, 31, 1311–1318. [Google Scholar] [CrossRef] [Green Version]
- Harvey, M.; McArthur, M.J.; Montgomery, C.A., Jr.; Butel, J.S.; Bradley, A.; Donehower, L.A. Spontaneous and carcinogen-induced tumorigenesis in p53-deficient mice. Nat. Genet. 1993, 5, 225–229. [Google Scholar] [CrossRef]
- Schwitalla, S.; Ziegler, P.K.; Horst, D.; Becker, V.; Kerle, I.; Begus-Nahrmann, Y.; Lechel, A.; Rudolph, K.L.; Langer, R.; Slotta-Huspenina, J.; et al. Loss of p53 in Enterocytes Generates an Inflammatory Microenvironment Enabling Invasion and Lymph Node Metastasis of Carcinogen-Induced Colorectal Tumors. Cancer Cell 2013, 23, 93–106. [Google Scholar] [CrossRef] [Green Version]
- French, J.E.; Lacks, D.; Trempus, C.; Dunnick, J.K.; Foley, J.; Mahler, J.; Tice, R.R.; Tennant, R.W. Loss of heterozygosity frequency at the Trp53 locus in p53-deficient (+/−) mouse tumors is carcinogen-and tissue-dependent. Carcinogenesis 2001, 22, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Oda, H.; Zhang, S.; Tsurutani, N.; Shimizu, S.; Nakatsuru, Y.; Aizawa, S.; Ishikawa, T. Loss of p53 is an early event in induction of brain tumors in mice by transplacental carcinogen exposure. Cancer Res. 1997, 57, 646–650. [Google Scholar]
- Sakai, H.; Tsukamoto, T.; Yamamoto, M.; Shirai, N.; Iidaka, T.; Hirata, A.; Yanai, T.; Masegi, T.; Donehower, L.A.; Tatematsu, M. High susceptibility of nullizygous p53 knockout mice to colorectal tumor induction by 1,2-dimethylhydrazine. J. Cancer Res. Clin. Oncol. 2003, 129, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, K.; Sukata, T.; Yamamoto, S.; Uwagawa, S.; Seki, T.; Kawasaki, H.; Yoshitake, A.; Wanibuchi, H.; Koide, A.; Mori, Y. High susceptibility of p53(+/−) knockout mice in N-butyl-N-(4-hydroxybutyl)nitrosamine urinary bladder carcinogenesis and lack of frequent mutation in residual allele. Cancer Res. 1998, 58, 3806–3811. [Google Scholar] [PubMed]
- Kado, S.; Uchida, K.; Funabashi, H.; Iwata, S.; Nagata, Y.; Ando, M.; Onoue, M.; Matsuoka, Y.; Ohwaki, M.; Morotomi, M. Intestinal microflora are necessary for development of spontaneous adenocarcinoma of the large intestine in T-cell receptor beta chain and p53 double-knockout mice. Cancer Res. 2001, 61, 2395–2398. [Google Scholar] [PubMed]
- Yamamoto, M.; Tsukamoto, T.; Sakai, H.; Shirai, N.; Ohgaki, H.; Furihata, C.; Donehower, L.A.; Yoshida, K.; Tatematsu, M. p53 knockout mice (−/−) are more susceptible than (+/−) or (+/+) mice to N-methyl-N-nitrosourea stomach carcinogenesis. Carcinogenesis 2000, 21, 1891–1897. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Singh, M.; Santos, G.S.; Guerlavais, V.; Carvajal, L.A.; Aivado, M.; Zhan, Y.; Oliveira, M.M.S.; Westerberg, L.S.; Annis, D.A. Pharmacological activation of p53 triggers viral mimicry response thereby abolishing tumor immune evasion and promoting anti-tumor immunity. Cancer Discov. 2021. [Google Scholar] [CrossRef]
- Blagih, J.; Zani, F.; Chakravarty, P.; Hennequart, M.; Pilley, S.; Hobor, S.; Hock, A.K.; Walton, J.B.; Morton, J.P.; Gronroos, E.; et al. Cancer-Specific Loss of p53 Leads to a Modulation of Myeloid and T Cell Responses. Cell Rep. 2020, 30, 481–496.e6. [Google Scholar] [CrossRef]
- A Christophorou, M.; Martin-Zanca, D.; Soucek, L.; Lawlor, E.R.; Brown-Swigart, L.; Verschuren, E.; I Evan, G. Temporal dissection of p53 function in vitro and in vivo. Nat. Genet. 2005, 37, 718–726. [Google Scholar] [CrossRef]
- Martins, C.P.; Brown-Swigart, L.; Evan, G.I. Modeling the Therapeutic Efficacy of p53 Restoration in Tumors. Cell 2006, 127, 1323–1334. [Google Scholar] [CrossRef] [Green Version]
- Hanel, W.; Marchenko, N.D.; Xu, S.; Yu, S.X.; Weng, W.; Moll, U.M. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ. 2013, 20, 898–909. [Google Scholar] [CrossRef] [Green Version]
- Lang, G.A.; Iwakuma, T.; Suh, Y.-A.; Liu, G.; Rao, V.; Parant, J.M.; Valentin-Vega, Y.A.; Terzian, T.; Caldwell, L.C.; Strong, L.C.; et al. Gain of Function of a p53 Hot Spot Mutation in a Mouse Model of Li-Fraumeni Syndrome. Cell 2004, 119, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Olive, K.; Tuveson, D.A.; Ruhe, Z.C.; Yin, B.; Willis, N.A.; Bronson, R.T.; Crowley, D.; Jacks, T. Mutant p53 Gain of Function in Two Mouse Models of Li-Fraumeni Syndrome. Cell 2004, 119, 847–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrova, E.M.; Yallowitz, A.R.; Li, D.; Xu, S.; Schulz, R.; A Proia, D.; Lozano, G.; Dobbelstein, M.; Moll, U.M. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature 2015, 523, 352–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tchelebi, L.; Ashamalla, H.; Graves, P.R. Mutant p53 and the Response to Chemotherapy and Radiation. Subcell. Biochem. 2014, 85, 133–159. [Google Scholar] [CrossRef] [PubMed]
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef] [PubMed]
- Vega-Stromberg, T. Chemotherapy-induced Secondary Malignancies. J. Infus. Nurs. 2003, 26, 353–361. [Google Scholar] [CrossRef]
- Li, H.; Zhang, J.; Tong, J.H.M.; Chan, A.W.H.; Yu, J.; Kang, W.; To, K.F. Targeting the Oncogenic p53 Mutants in Colorectal Cancer and Other Solid Tumors. Int. J. Mol. Sci. 2019, 20, 5999. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Pan, C.; Bei, J.-X.; Li, B.; Liang, C.; Xu, Y.; Fu, X. Mutant p53 in Cancer Progression and Targeted Therapies. Front. Oncol. 2020, 10, 595187. [Google Scholar] [CrossRef]
- Issaeva, N.; Bozko, P.; Enge, M.; Protopopova, M.; Verhoef, L.G.G.C.; Masucci, M.; Pramanik, A.; Selivanova, G. Small molecule RITA binds to p53, blocks p53–HDM-2 interaction and activates p53 function in tumors. Nat. Med. 2004, 10, 1321–1328. [Google Scholar] [CrossRef]
- Sanz, G.; Singh, M.; Peuget, S.; Selivanova, G. Inhibition of p53 inhibitors: Progress, challenges and perspectives. J. Mol. Cell Biol. 2019, 11, 586–599. [Google Scholar] [CrossRef] [Green Version]
- Wurz, R.P.; Cee, V.J. Targeted Degradation of MDM2 as a New Approach to Improve the Efficacy of MDM2-p53 Inhibitors. J. Med. Chem. 2019, 62, 445–447. [Google Scholar] [CrossRef] [Green Version]
- Levine, A.J. Targeting Therapies for the p53 Protein in Cancer Treatments. Annu. Rev. Cancer Biol. 2019, 3, 21–34. [Google Scholar] [CrossRef]
- Heise, C.; Sampson-Johannes, A.; Williams, A.; Mccormick, F.; Von Hoff, D.D.; Kirn, D.H. ONYX-015, an E1B gene-attenuated adenovirus, causes tumor-specific cytolysis and antitumoral efficacy that can be augmented by standard chemotherapeutic agents. Nat. Med. 1997, 3, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.-W.; Li, L.; Li, D.; Liu, J.; Li, X.; Li, W.; Xu, X.; Zhang, M.; Chandler, L.A.; Lin, H.; et al. The First Approved Gene Therapy Product for Cancer Ad-p53(Gendicine): 12 Years in the Clinic. Hum. Gene Ther. 2018, 29, 160–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, S.; Jia, H.; Kandachi, K. China approves first gene therapy. Nat. Biotechnol. 2004, 22, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Foster, B.A. Pharmacological Rescue of Mutant p53 Conformation and Function. Science 1999, 286, 2507–2510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bykov, V.N.; Issaeva, N.; Shilov, A.; Hultcrantz, M.; Pugacheva, E.; Chumakov, P.; Bergman, J.; Wiman, K.; Selivanova, G. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002, 8, 282–288. [Google Scholar] [CrossRef]
- Bykov, V.N.; Zache, N.; Stridh, H.; Westman, J.; Bergman, J.; Selivanova, G.; Wiman, K.G. PRIMA-1MET synergizes with cisplatin to induce tumor cell apoptosis. Oncogene 2005, 24, 3484–3491. [Google Scholar] [CrossRef] [Green Version]
- Bykov, V.N.; Issaeva, N.; Zache, N.; Shilov, A.; Hultcrantz, M.; Bergman, J.; Selivanova, G.; Wiman, K. Reactivation of Mutant p53 and Induction of Apoptosis in Human Tumor Cells by Maleimide Analogs. J. Biol. Chem. 2005, 280, 30384–30391. [Google Scholar] [CrossRef] [Green Version]
- Zache, N.; Lambert, J.M.; Rökaeus, N.; Shen, J.; Hainaut, P.; Bergman, J.; Wiman, K.G.; Bykov, V.J. Mutant p53 targeting by the low molecular weight compound STIMA-1. Mol. Oncol. 2008, 2, 70–80. [Google Scholar] [CrossRef] [Green Version]
- Kaar, J.; Basse, N.; Joerger, A.; Stephens, E.; Rutherford, T.J.; Fersht, A.R. Stabilization of mutant p53 via alkylation of cysteines and effects on DNA binding. Protein Sci. 2010, 19, 2267–2278. [Google Scholar] [CrossRef] [Green Version]
- Punganuru, S.R.; Madala, H.R.; Venugopal, S.N.; Samala, R.; Mikelis, C.; Srivenugopal, K.S. Design and synthesis of a C7-aryl piperlongumine derivative with potent antimicrotubule and mutant p53-reactivating properties. Eur. J. Med. Chem. 2016, 107, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.; Joerger, A.; Fersht, A.R. 2-Sulfonylpyrimidines: Mild alkylating agents with anticancer activity toward p53-compromised cells. Proc. Natl. Acad. Sci. USA 2016, 113, E5271–E5280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Synnott, N.C.; O’Connell, D.; Crown, J.; Duffy, M.J. COTI-2 reactivates mutant p53 and inhibits growth of triple-negative breast cancer cells. Breast Cancer Res. Treat. 2019, 179, 47–56. [Google Scholar] [CrossRef]
- Gomes, A.S.; Ramos, H.; Gomes, S.; Loureiro, J.; Soares, J.; Barcherini, V.; Monti, P.; Fronza, G.; Oliveira, C.; Domingues, L.; et al. SLMP53-1 interacts with wild-type and mutant p53 DNA-binding domain and reactivates multiple hotspot mutations. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129440. [Google Scholar] [CrossRef] [PubMed]
- Soares, J.; Raimundo, L.; Pereira, N.A.L.; Monteiro, Â.; Gomes, S.; Bessa, C.; Pereira, C.; Queiroz, G.; Bisio, A.; Fernandes, J.; et al. Reactivation of wild-type and mutant p53 by tryptophanolderived oxazoloisoindolinone SLMP53-1, a novel anticancer small-molecule. Oncotarget 2016, 7, 4326–4343. [Google Scholar] [CrossRef] [Green Version]
- Gomes, S.; Bosco, B.; Loureiro, J.B.; Ramos, H.; Raimundo, L.; Soares, J.; Nazareth, N.; Barcherini, V.; Domingues, L.; Oliveira, C.; et al. SLMP53-2 Restores Wild-Type-Like Function to Mutant p53 through Hsp70: Promising Activity in Hepatocellular Carcinoma. Cancers 2019, 11, 1151. [Google Scholar] [CrossRef] [Green Version]
- Tal, P.; Eizenberger, S.; Cohen, E.; Goldfinger, N.; Pietrokovski, S.; Oren, M.; Rotter, V. Cancer therapeutic approach based on conformational stabilization of mutant p53 protein by small peptides. Oncotarget 2016, 7, 11817–11837. [Google Scholar] [CrossRef] [Green Version]
- Proia, D.A.; Bates, R.C. Ganetespib and HSP90: Translating Preclinical Hypotheses into Clinical Promise. Cancer Res. 2014, 74, 1294–1300. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Marchenko, N.; Moll, U.M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011, 18, 1904–1913. [Google Scholar] [CrossRef] [Green Version]
- Foggetti, G.; Ottaggio, L.; Russo, D.; Mazzitelli, C.; Monti, P.; Degan, P.; Miele, M.; Fronza, G.; Menichini, P. Autophagy induced by SAHA affects mutant P53 degradation and cancer cell survival. Biosci. Rep. 2019, 39, BSR20181345. [Google Scholar] [CrossRef] [Green Version]
- Parrales, A.; Ranjan, A.; Iyer, S.; Padhye, S.; Weir, S.J.; Roy, A.; Iwakuma, T. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell Biol. 2016, 18, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu-Norberg, H.; Kim, M.; Xia, H.G.; Iwanicki, M.P.; Ofengeim, D.; Coloff, J.L.; Pan, L.; Ince, T.A.; Kroemer, G.; Brugge, J.S.; et al. Chaperone-mediated autophagy degrades mutant p53. Genes Dev. 2013, 27, 1718–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kravchenko, J.E.; Ilyinskaya, G.V.; Komarov, P.G.; Agapova, L.S.; Kochetkov, D.V.; Strom, E.; Frolova, E.I.; Kovriga, I.; Gudkov, A.; Feinstein, E.; et al. Small-molecule RETRA suppresses mutant p53-bearing cancer cells through a p73-dependent salvage pathway. Proc. Natl. Acad. Sci. USA 2008, 105, 6302–6307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsiue, E.H.-C.; Wright, K.M.; Douglass, J.; Hwang, M.S.; Mog, B.J.; Pearlman, A.H.; Paul, S.; DiNapoli, S.R.; Konig, M.F.; Wang, Q.; et al. Targeting a neoantigen derived from a common TP53 mutation. Science 2021, 371, eabc8697. [Google Scholar] [CrossRef]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef]
- Duffy, M.J.; Synnott, N.C.; Crown, J. Mutant p53 as a target for cancer treatment. Eur. J. Cancer 2017, 83, 258–265. [Google Scholar] [CrossRef]
- Grinkevich, V.V.; Nikulenkov, F.; Shi, Y.; Enge, M.; Bao, W.; Maljukova, A.; Gluch, A.; Kel, A.; Sangfelt, O.; Selivanova, G. Ablation of key oncogenic pathways by RITA-reactivated p53 is required for efficient apoptosis. Cancer Cell 2009, 15, 441–453. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, A.; Yang, J.; Maya-Mendoza, A.; Jackson, D.A.; Ashcroft, M. Pharmacological activation of a novel p53-dependent S-phase checkpoint involving CHK-1. Cell Death Dis. 2011, 2, e160. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Ahmed, A.; Poon, E.; Perusinghe, N.; Brandon, A.D.H.; Box, G.; Valenti, M.; Eccles, S.; Rouschop, K.; Wouters, B.; et al. Small-Molecule Activation of p53 Blocks Hypoxia-Inducible Factor 1α and Vascular Endothelial Growth Factor Expression In Vivo and Leads to Tumor Cell Apoptosis in Normoxia and Hypoxia. Mol. Cell. Biol. 2009, 29, 2243–2253. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Nikulenkov, F.; Zawacka-Pankau, J.; Li, H.; Gabdoulline, R.; Xu, J.; Eriksson, S.; Hedström, E.; Issaeva, N.; Kel, A.; et al. ROS-dependent activation of JNK converts p53 into an efficient inhibitor of oncogenes leading to robust apoptosis. Cell Death Differ. 2014, 21, 612–623. [Google Scholar] [CrossRef] [Green Version]
- Wiegering, A.; Matthes, N.; Mühling, B.; Koospal, M.; Quenzer, A.; Peter, S.; Germer, C.-T.; Linnebacher, M.; Otto, C. Reactivating p53 and Inducing Tumor Apoptosis (RITA) Enhances the Response of RITA-Sensitive Colorectal Cancer Cells to Chemotherapeutic Agents 5-Fluorouracil and Oxaliplatin. Neoplasia 2017, 19, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Weilbacher, A.; Gutekunst, M.; Oren, M.; E Aulitzky, W.; Van Der Kuip, H. RITA can induce cell death in p53-defective cells independently of p53 function via activation of JNK/SAPK and p38. Cell Death Dis. 2014, 5, e1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.; Li, X.; Wang, W.; Zhang, L.; Tao, L.; Dong, X.; Sheng, R.; Yang, B.; Hu, Y. Design, synthesis, and biological evaluation of imidazoline derivatives as p53–MDM2 binding inhibitors. Bioorganic Med. Chem. 2011, 19, 5454–5461. [Google Scholar] [CrossRef] [PubMed]
- Ray-Coquard, I.; Blay, J.-Y.; Italiano, A.; Le Cesne, A.; Penel, N.; Zhi, J.; Heil, F.; Rueger, R.; Graves, B.; Ding, M.; et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: An exploratory proof-of-mechanism study. Lancet Oncol. 2012, 13, 1133–1140. [Google Scholar] [CrossRef]
- Ding, Q.; Zhang, Z.; Liu, J.-J.; Jiang, N.; Zhang, J.; Ross, T.M.; Chu, X.-J.; Bartkovitz, D.; Podlaski, F.; Janson, C.; et al. Discovery of RG7388, a Potent and Selective p53–MDM2 Inhibitor in Clinical Development. J. Med. Chem. 2013, 56, 5979–5983. [Google Scholar] [CrossRef] [PubMed]
- Verreault, M.; Schmitt, C.; Goldwirt, L.; Pelton, K.; Haidar, S.; Levasseur, C.; Guehennec, J.; Knoff, D.; Labussière, M.; Marie, Y.; et al. Preclinical Efficacy of the MDM2 Inhibitor RG7112 in MDM2-Amplified and TP53 Wild-type Glioblastomas. Clin. Cancer Res. 2016, 22, 1185–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreeff, M.; Kelly, K.R.; Yee, K.; Assouline, S.; Strair, R.; Popplewell, L.; Bowen, D.; Martinelli, G.; Drummond, M.W.; Vyas, P.; et al. Results of the Phase I Trial of RG7112, a Small-Molecule MDM2 Antagonist in Leukemia. Clin. Cancer Res. 2016, 22, 868–876. [Google Scholar] [CrossRef] [Green Version]
- Patnaik, A.; Tolcher, A.; Beeram, M.; Nemunaitis, J.; Weiss, G.J.; Bhalla, K.; Agrawal, M.; Nichols, G.; Middleton, S.; Beryozkina, A.; et al. Clinical pharmacology characterization of RG7112, an MDM2 antagonist, in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2015, 76, 587–595. [Google Scholar] [CrossRef]
- Her, N.-G.; Oh, J.-W.; Oh, Y.J.; Han, S.; Cho, H.J.; Lee, Y.; Ryu, G.H.; Nam, D.-H. Potent effect of the MDM2 inhibitor AMG232 on suppression of glioblastoma stem cells. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Shadfan, M.; Lopez-Pajares, V.; Yuan, Z.-M. MDM2 and MDMX: Alone and together in regulation of p53. Transl. Cancer Res. 2012, 1, 88–89. [Google Scholar]
- Reed, D.; Shen, Y.; Shelat, A.A.; Arnold, L.A.; Ferreira, A.M.; Zhu, F.; Mills, N.; Smithson, D.C.; Regni, C.A.; Bashford, D.; et al. Identification and Characterization of the First Small Molecule Inhibitor of MDMX. J. Biol. Chem. 2010, 285, 10786–10796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleh, M.N.; Patel, M.R.; Bauer, T.M.; Goel, S.; Falchook, G.S.; Shapiro, G.I.; Chung, K.Y.; Infante, J.R.; Conry, R.M.; Rabinowits, G.; et al. Phase 1 Trial of ALRN-6924, a Dual Inhibitor of MDMX and MDM2, in Patients with Solid Tumors and Lymphomas Bearing Wild-type TP53. Clin. Cancer Res. 2021, 27, 5236–5247. [Google Scholar] [CrossRef] [PubMed]
- Gasparian, A.V.; Burkhart, C.A.; Purmal, A.A.; Brodsky, L.; Pal, M.; Saranadasa, M.; Bosykh, D.A.; Commane, M.; Guryanova, O.A.; Pal, S.; et al. Curaxins: Anticancer compounds that simultaneously suppress NF-kappaB and activate p53 by targeting FACT. Sci. Transl. Med. 2011, 3, 95ra74. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.K.; Gwak, J.; Song, I.-S.; Park, H.-S.; Oh, S. Induction of apoptosis in colon cancer cells by a novel topoisomerase I inhibitor TopIn. Biochem. Biophys. Res. Commun. 2011, 409, 75–81. [Google Scholar] [CrossRef]
- Lambert, J.M.; Gorzov, P.; Veprintsev, D.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.N. PRIMA-1 Reactivates Mutant p53 by Covalent Binding to the Core Domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Bykov, V.J.N.; Wiman, K.G.; Zawacka-Pankau, J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 2018, 9, 439. [Google Scholar] [CrossRef] [Green Version]
- Demma, M.; Maxwell, E.; Ramos, R.; Liang, L.; Li, C.; Hesk, D.; Rossman, R.; Mallams, A.; Doll, R.; Liu, M.; et al. SCH529074, a Small Molecule Activator of Mutant p53, Which Binds p53 DNA Binding Domain (DBD), Restores Growth-suppressive Function to Mutant p53 and Interrupts HDM2-mediated Ubiquitination of Wild Type p53. J. Biol. Chem. 2010, 285, 10198–10212. [Google Scholar] [CrossRef] [Green Version]
- Salim, K.Y.; Vareki, A.M.; Danter, W.R.; Koropatnick, J. COTI-2, a novel small molecule that is active against multiple human cancer cell lines in vitro and in vivo. Oncotarget 2016, 7, 41363–41379. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.; Rockliffe, N.; Johnson, G.G.; Sherrington, P.D.; Pettitt, A.R. Hsp90 inhibition has opposing effects on wild-type and mutant p53 and induces p21 expression and cytotoxicity irrespective of p53/ATM status in chronic lymphocytic leukaemia cells. Oncogene 2008, 27, 2445–2455. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Marchenko, N.D.; Schulz, R.; Fischer, V.; Velasco-Hernandez, T.; Talos, F.; Moll, U.M. Functional Inactivation of Endogenous MDM2 and CHIP by HSP90 Causes Aberrant Stabilization of Mutant p53 in Human Cancer Cells. Mol. Cancer Res. 2011, 9, 577–588. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Chen, L.; Li, C.; Lu, W.; Chen, J. Inhibition of MDM2 by hsp90 Contributes to Mutant p53 Stabilization. J. Biol. Chem. 2001, 276, 40583–40590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Z.; Zhang, Y.; He, Y.; Cao, Q.; Zhang, T.; Lou, L.; Cai, Q. Full-Length Transcriptome Assembly of Italian Ryegrass Root Integrated with RNA-Seq to Identify Genes in Response to Plant Cadmium Stress. Int. J. Mol. Sci. 2020, 21, 1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Z.; Yang, G.; Deng, T.; Wang, J.; Zhou, H.; Popov, S.A.; Shults, E.E.; Wang, C. Design and linkage optimization of ursane-thalidomide-based PROTACs and identification of their targeted-degradation properties to MDM2 protein. Bioorganic Chem. 2021, 111, 104901. [Google Scholar] [CrossRef]
- Zhang, S.; Zhou, L.; El-Deiry, W.S. Small-Molecule NSC59984 Induces Mutant p53 Degradation through a ROS-ERK2-MDM2 Axis in Cancer Cells. Mol. Cancer Res. 2022. [Google Scholar] [CrossRef] [PubMed]
- Galbraith, M.D.; Andrysik, Z.; Sullivan, K.D.; Espinosa, J.M. Global Analyses to Identify Direct Transcriptional Targets of p53. Methods Pharmacol. Toxicol. 2021, 2267, 19–56. [Google Scholar] [CrossRef]
- Allen, M.A.; Andrysik, Z.; Dengler, V.L.; Mellert, H.S.; Guarnieri, A.; A Freeman, J.; Sullivan, K.D.; Galbraith, M.D.; Luo, X.; Kraus, W.L.; et al. Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms. eLife 2014, 3, e02200. [Google Scholar] [CrossRef] [PubMed]
- Broz, D.K.; Mello, S.S.; Bieging, K.T.; Jiang, D.; Dusek, R.L.; Brady, C.A.; Sidow, A.; Attardi, L.D. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 2013, 27, 1016–1031. [Google Scholar] [CrossRef] [Green Version]
- Menendez, D.; Nguyen, T.-A.; Freudenberg, J.M.; Mathew, V.J.; Anderson, C.W.; Jothi, R.; Resnick, M.A. Diverse stresses dramatically alter genome-wide p53 binding and transactivation landscape in human cancer cells. Nucleic Acids Res. 2013, 41, 7286–7301. [Google Scholar] [CrossRef] [Green Version]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef] [Green Version]
- Li, M. The role of P53 up-regulated modulator of apoptosis (PUMA) in ovarian development, cardiovascular and neurodegenerative diseases. Apoptosis 2021, 26, 235–247. [Google Scholar] [CrossRef]
- Sharma, K.; Vu, T.; Cook, W.; Naseri, M.; Zhan, K.; Nakajima, W.; Harada, H. p53-independent Noxa induction by cisplatin is regulated by ATF3/ATF4 in head and neck squamous cell carcinoma cells. Mol. Oncol. 2018, 12, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.L.; Veal, G.J.; Redfern, C.P.F.; Lovat, P. Role of Noxa in p53-independent fenretinide-induced apoptosis of neuroectodermal tumours. Apoptosis 2007, 12, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Galán, P.; Roué, G.; Villamor, N.; Montserrat, E.; Campo, E.; Colomer, D. The proteasome inhibitor bortezomib induces apoptosis in mantle-cell lymphoma through generation of ROS and Noxa activation independent of p53 status. Blood 2006, 107, 257–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warfel, N.A.; El-Deiry, W.S. p21WAF1 and tumourigenesis: 20 years after. Curr. Opin. Oncol. 2013, 25, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Ahsan, N.; Lulla, A.; Lev, A.; Abbosh, P.; Dicker, D.T.; Zhang, S.; El-Deiry, W.S. P53-independent partial restoration of the p53 pathway in tumors with mutated p53 through ATF4 transcriptional modulation by ERK1/2 and CDK9. Neoplasia 2021, 23, 304–325. [Google Scholar] [CrossRef]
- Borrero, L.H.; Dicker, D.T.; Santiago, J.; Sanders, J.; Tian, X.; Ahsan, N.; Lev, A.; Zhou, L.; El-Deiry, W.S. A subset of CB002 xanthine analogs bypass p53-signaling to restore a p53 transcriptome and target an S-phase cell cycle checkpoint in tumors with mutated-p53. eLife 2021, 10, 10. [Google Scholar] [CrossRef]
- DeLeo, A.B.; Appella, E. The p53 Saga: Early Steps in the Development of Tumor Immunotherapy. J. Immunol. 2020, 204, 2321–2328. [Google Scholar] [CrossRef]
- Kunimura, N.; Kitagawa, K.; Sako, R.; Narikiyo, K.; Tominaga, S.; Bautista, D.S.; Wei Xu, M.F.; Shirakawa, T. Combination of rAd-p53 in situ gene therapy and anti-PD-1 antibody immunotherapy induced anti-tumor activity in mouse syngeneic urogenital cancer models. Sci. Rep. 2020, 10, 17464. [Google Scholar] [CrossRef]
- Hardwick, N.R.; Carroll, M.; Kaltcheva, T.; Qian, D.; Lim, D.; Leong, L.; Chu, P.; Kim, J.; Chao, J.; Fakih, M.; et al. p53MVA Therapy in Patients with Refractory Gastrointestinal Malignancies Elevates p53-Specific CD8+ T-cell Responses. Clin. Cancer Res. 2014, 20, 4459–4470. [Google Scholar] [CrossRef] [Green Version]
- Chung, V.; Kos, F.J.; Hardwick, N.; Yuan, Y.; Chao, J.; Li, D.; Waisman, J.; Li, M.; Zurcher, K.; Frankel, P.; et al. Evaluation of safety and efficacy of p53MVA vaccine combined with pembrolizumab in patients with advanced solid cancers. Clin. Transl. Oncol. 2018, 21, 363–372. [Google Scholar] [CrossRef]
- Hao, Q.; Lu, H.; Zhou, X. A potential synthetic lethal strategy with PARP inhibitors: Perspective on ‘Inactivation of the tumor suppressor p53 by long noncoding RNA RMRP’. J. Mol. Cell Biol. 2021, 13, 690–692. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Omlin, A.; De Bono, J.S. Development of Therapeutic Combinations Targeting Major Cancer Signaling Pathways. J. Clin. Oncol. 2013, 31, 1592–1605. [Google Scholar] [CrossRef] [PubMed]
- Grellety, T.; Laroche-Clary, A.; Chaire, V.; Lagarde, P.; Chibon, F.; Neuville, A.; Italiano, A. PRIMA-1(MET) induces death in soft-tissue sarcomas cell independent of p53. BMC Cancer 2015, 15, 684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujihara, K.M.; Benitez, M.C.; Cabalag, C.S.; Zhang, B.Z.; Ko, H.S.; Liu, D.S.; Simpson, K.J.; Haupt, Y.; Lipton, L.; Haupt, S.; et al. SLC7A11 Is a Superior Determinant of APR-246 (Eprenetapopt) Response than TP53 Mutation Status. Mol. Cancer Ther. 2021, 20, 1858–1867. [Google Scholar] [CrossRef]
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.B.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the system xC(-)/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 2017, 8, 14844. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Mao, Y.; Brandt-Rauf, P.W.; Williams, A.C.; Fine, R.L. Selective induction of apoptosis in mutant p53 premalignant and malignant cancer cells by PRIMA-1 through the c-Jun-NH2-kinase pathway. Mol. Cancer Ther. 2005, 4, 901–909. [Google Scholar] [CrossRef] [Green Version]
- Saha, M.N.; Jiang, H.; Yang, Y.; Zhu, X.; Wang, X.; Schimmer, A.D.; Qiu, L.; Chang, H. Targeting p53 via JNK pathway: A novel role of RITA for apoptotic signaling in multiple myeloma. PLoS ONE 2012, 7, e30215. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yamamoto, Y.; Imanishi, M.; Zhang, X.; Sato, M.; Sugaya, A.; Hirose, M.; Endo, S.; Natori, Y.; Moriwaki, T.; et al. Enhanced G1 arrest and apoptosis via MDM4/MDM2 double knockdown and MEK inhibition in wild-type TP53 colon and gastric cancer cells with aberrant KRAS signaling. Oncol. Lett. 2021, 22, 558. [Google Scholar] [CrossRef]
- Roy, S.; Laroche-Clary, A.; Verbeke, S.; Derieppe, M.A.; Italiano, A. MDM2 Antagonists Induce a Paradoxical Activation of Erk1/2 through a P53-Dependent Mechanism in Dedifferentiated Liposarcomas: Implications for Combinatorial Strategies. Cancers 2020, 12, 2253. [Google Scholar] [CrossRef]
- Ji, Z.; Kumar, R.; Taylor, M.J.; Rajadurai, A.; Marzuka-Alcalá, A.; Chen, Y.E.; Njauw, C.-N.J.; Flaherty, K.T.; Jönsson, G.B.; Tsao, H. Vemurafenib Synergizes with Nutlin-3 to Deplete Survivin and Suppresses Melanoma Viability and Tumor Growth. Clin. Cancer Res. 2013, 19, 4383–4391. [Google Scholar] [CrossRef] [Green Version]
- Vatsyayan, R.; Singhal, J.; Nagaprashantha, L.P.D.; Awasthi, S.; Singhal, S.S. Nutlin-3 enhances sorafenib efficacy in renal cell carcinoma. Mol. Carcinog. 2013, 52, 39–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bragado, P.; Armesilla, A.; Silva, A.; Porras, A. Apoptosis by cisplatin requires p53 mediated p38α MAPK activation through ROS generation. Apoptosis 2007, 12, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Grandela, C.; Pera, M.F.; Grimmond, S.M.; Kolle, G.; Wolvetang, E.J. p53 is required for etoposide-induced apoptosis of human embryonic stem cells. Stem. Cell Res. 2007, 1, 116–128. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Xia, P.; Zhang, H.; Liu, B.; Shi, Y. P53 is required for Doxorubicin-induced apoptosis via the TGF-beta signaling pathway in osteosarcoma-derived cells. Am. J. Cancer Res. 2015, 6, 114–125. [Google Scholar]
- Saha, M.N.; Chen, Y.; Chen, M.-H.; Chen, G.; Chang, H. Small molecule MIRA-1 induces in vitro and in vivo anti-myeloma activity and synergizes with current anti-myeloma agents. Br. J. Cancer 2014, 110, 2224–2231. [Google Scholar] [CrossRef] [Green Version]
- Xie, Q.; Wu, M.Y.; Zhang, D.X.; Yang, Y.M.; Wang, B.S.; Zhang, J.; Xu, J.; Zhong, W.; Hu, J. Synergistic anticancer effect of exogenous wild-type p53 gene combined with 5-FU in human colon cancer resistant to 5-FU in vivo. World J. Gastroenterol. 2016, 22, 7342–7352. [Google Scholar] [CrossRef]
- Magrini, R.; Russo, D.; Ottaggio, L.; Fronza, G.; Inga, A.; Menichini, P. PRIMA-1 synergizes with adriamycin to induce cell death in non-small cell lung cancer cells. J. Cell. Biochem. 2008, 104, 2363–2373. [Google Scholar] [CrossRef]
- Messina, R.L.; Sanfilippo, M.; Vella, V.; Pandini, G.; Vigneri, P.; Nicolosi, M.L.; Gianì, F.; Vigneri, R.; Frasca, F. Reactivation of p53 mutants by prima-1 [corrected] in thyroid cancer cells. Int. J. Cancer 2012, 130, 2259–2270. [Google Scholar] [CrossRef]
- Mohell, N.; Alfredsson, J.; Fransson, A.; Uustalu, M.; Bystrom, S.; Gullbo, J.; Hallberg, A.; Bykov, V.J.N.; Bjorklund, U.; Wiman, K. APR-246 overcomes resistance to cisplatin and doxorubicin in ovarian cancer cells. Cell Death Dis. 2015, 6, e1794. [Google Scholar] [CrossRef] [Green Version]
- Lin, A.B.; McNeely, S.C.; Beckmann, R.P. Achieving Precision Death with Cell-Cycle Inhibitors that Target DNA Replication and Repair. Clin. Cancer Res. 2017, 23, 3232–3240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Hunter, T. Roles of Chk1 in cell biology and cancer therapy. Int. J. Cancer 2014, 134, 1013–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadhikar, M.A.; Sciuto, M.R.; Alves, M.V.O.; Pickering, C.R.; Osman, A.A.; Neskey, D.M.; Mei Zhao, A.L.F.; Myers, J.N.; Frederick, M.J. Chk1/2 inhibition overcomes the cisplatin resistance of head and neck cancer cells secondary to the loss of functional p53. Mol. Cancer Ther. 2013, 12, 1860–1873. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.X.; Cai, S.; Li, S.; Ryan, C.E.; Guo, Z.; Schaiff, W.T.; Lin, L.; Hoog, J.; Goiffon, R.J.; Prat, A.; et al. Targeting Chk1 in p53-deficient triple-negative breast cancer is therapeutically beneficial in human-in-mouse tumor models. J. Clin. Investig. 2012, 122, 1541–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vance, S.; Liu, E.; Zhao, L.; Parsels, J.D.; Parsels, L.A.; Brown, J.L.; Maybaum, J.; Lawrence, T.S.; Morgan, M.A. Selective radiosensitization of p53 mutant pancreatic cancer cells by combined inhibition of Chk1 and PARP1. Cell Cycle 2011, 10, 4321–4329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geenen, J.J.; Schellens, J.H. Molecular Pathways: Targeting the Protein Kinase Wee1 in Cancer. Clin. Cancer Res. 2017, 23, 4540–4544. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Shen, Q.; Tao, R.; Chang, W.; Li, R.; Xie, G.; Liu, W.; Zhang, P.; Tao, K. Wee1 inhibition can suppress tumor proliferation and sensitize p53 mutant colonic cancer cells to the anticancer effect of irinotecan. Mol. Med. Rep. 2017, 17, 3344–3349. [Google Scholar] [CrossRef]
- RajeshKumar, N.; De Oliveira, E.; Ottenhof, N.; Watters, J.; Brooks, D.; DeMuth, T.; Shumway, S.D.; Mizuarai, S.; Hirai, H.; Maitra, A.; et al. MK-1775, a Potent Wee1 Inhibitor, Synergizes with Gemcitabine to Achieve Tumor Regressions, Selectively in p53-Deficient Pancreatic Cancer Xenografts. Clin. Cancer Res. 2011, 17, 2799–2806. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Bi, J.; Li, Y.; Yang, S.; Zhang, Y.; Li, M.; Liu, H.; Li, Y.; McDonald, M.E.; Thiel, K.W.; et al. AZD1775 Increases Sensitivity to Olaparib and Gemcitabine in Cancer Cells with p53 Mutations. Cancers 2018, 10, 149. [Google Scholar] [CrossRef] [Green Version]
- Oza, A.M.; Estevez-Diz, M.D.P.; Grischke, E.-M.; Hall, M.; Marmé, F.; Provencher, D.M.; Uyar, D.S.; Weberpals, J.I.; Wenham, R.M.; Laing, N.; et al. A Biomarker-enriched, Randomized Phase II Trial of Adavosertib (AZD1775) Plus Paclitaxel and Carboplatin for Women with Platinum-sensitive TP53-mutant Ovarian Cancer. Clin. Cancer Res. 2020, 26, 4767–4776. [Google Scholar] [CrossRef]
- Leijen, S.; Van Geel, R.M.J.M.; Sonke, G.; De Jong, D.; Rosenberg, E.; Marchetti, S.; Pluim, D.; van Werkhoven, E.; Rose, S.; Lee, M.A.; et al. Phase II Study of WEE1 Inhibitor AZD1775 Plus Carboplatin in Patients With TP53-Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy Within 3 Months. J. Clin. Oncol. 2016, 34, 4354–4361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorecki, L.; Andrs, M.; Korabecny, J. Clinical Candidates Targeting the ATR–CHK1–WEE1 Axis in Cancer. Cancers 2021, 13, 795. [Google Scholar] [CrossRef] [PubMed]
- Kong, A.; Mehanna, H. WEE1 Inhibitor: Clinical Development. Curr. Oncol. Rep. 2021, 23, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Haronikova, L.; Bonczek, O.; Zatloukalova, P.; Kokas-Zavadil, F.; Kucerikova, M.; Coates, P.J.; Fahraeus, R.; Vojtesek, B. Resistance mechanisms to inhibitors of p53-MDM2 interactions in cancer therapy: Can we overcome them? Cell Mol. Biol. Lett. 2021, 26, 53. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhao, Y.; Aguilar, A.; Bernard, D.; Yang, C.-Y. Targeting the MDM2–p53 Protein–Protein Interaction for New Cancer Therapy: Progress and Challenges. Cold Spring Harb. Perspect. Med. 2017, 7, a026245. [Google Scholar] [CrossRef] [Green Version]
- Chapeau, E.A.; Gembarska, A.; Durand, E.Y.; Mandon, E.; Estadieu, C.; Romanet, V.; Wiesmann, M.; Tiedt, R.; Lehar, J.; de Weck, A.; et al. Resistance mechanisms to TP53-MDM2 inhibition identified by in vivo piggyBac transposon mutagenesis screen in an Arf−/− mouse model. Proc. Natl. Acad. Sci. USA 2017, 114, 3151–3156. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Zawacka-Pankau, J. The p53/MDM2/MDMX-targeted therapies-a clinical synopsis. Cell Death Dis. 2020, 11, 237. [Google Scholar] [CrossRef]
- Sobol, R.E.; Menander, K.B.; Chada, S.; Wiederhold, D.; Sellman, B.; Talbott, M.; Nemunaitis, J.J. Analysis of Adenoviral p53 Gene Therapy Clinical Trials in Recurrent Head and Neck Squamous Cell Carcinoma. Front. Oncol. 2021, 11, 645745. [Google Scholar] [CrossRef]
- Moore, E.C.; Sun, L.; Clavijo, P.E.; Friedman, J.; Harford, J.B.; Saleh, A.D.; Van Waes, C.; Chang, E.H.; Allen, C.T. Nanocomplex-based TP53 gene therapy promotes anti-tumor immunity through TP53- and STING-dependent mechanisms. OncoImmunology 2018, 7, e1404216. [Google Scholar] [CrossRef] [Green Version]
- Batır, M.B.; Şahin, E.; Çam, F.S. Evaluation of the CRISPR/Cas9 directed mutant TP53 gene repairing effect in human prostate cancer cell line PC-3. Mol. Biol. Rep. 2019, 46, 6471–6484. [Google Scholar] [CrossRef]
- Chira, S.; Gulei, D.; Hajitou, A.; Berindan-Neagoe, I. Restoring the p53 ‘Guardian’ Phenotype in p53-Deficient Tumor Cells with CRISPR/Cas9. Trends Biotechnol. 2018, 36, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Mirgayazova, R.; Khadiullina, R.; Chasov, V.; Mingaleeva, R.; Miftakhova, R.; Rizvanov, A.; Bulatov, E. Therapeutic Editing of the TP53 Gene: Is CRISPR/Cas9 an Option? Genes 2020, 11, 704. [Google Scholar] [CrossRef] [PubMed]
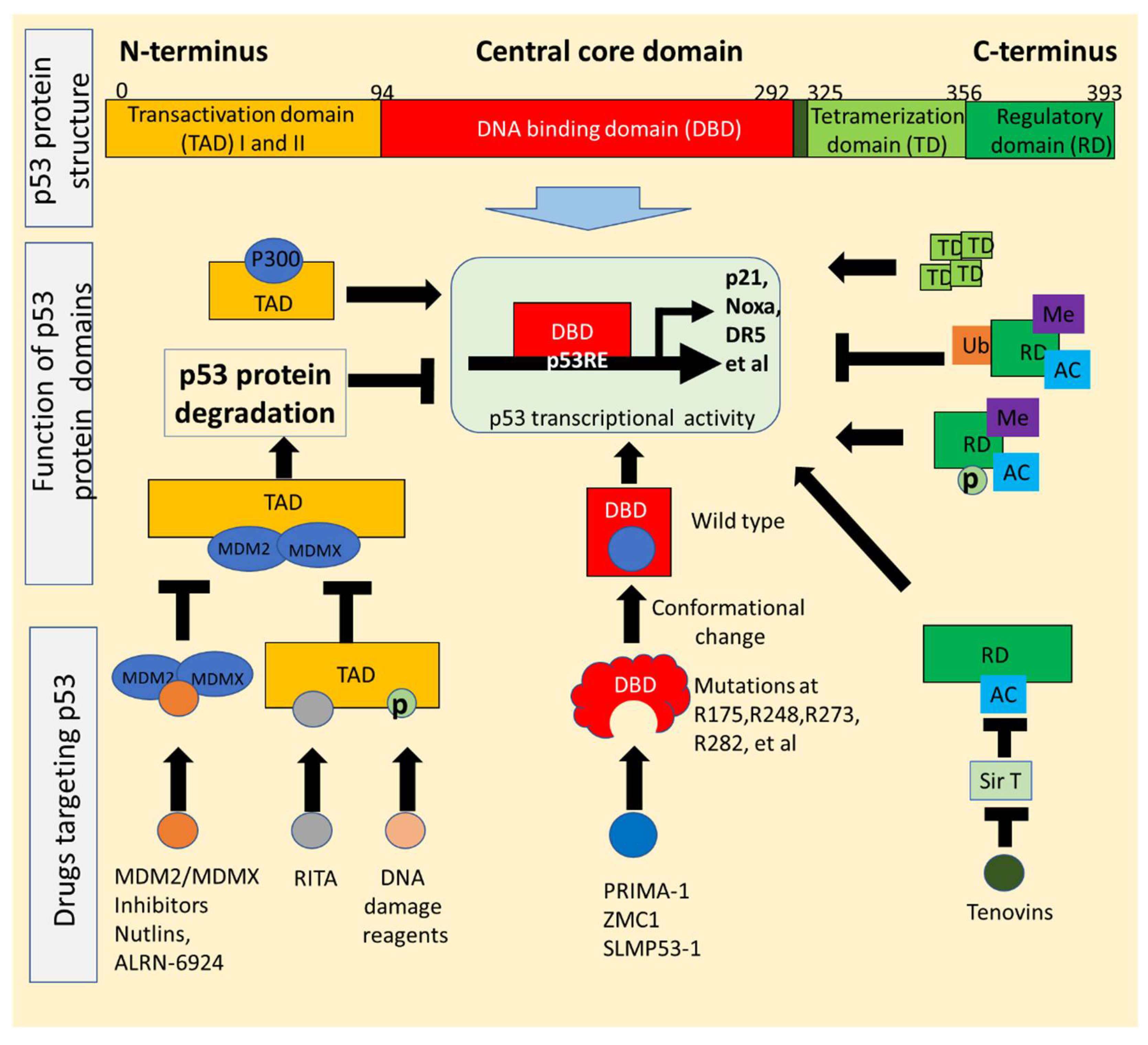

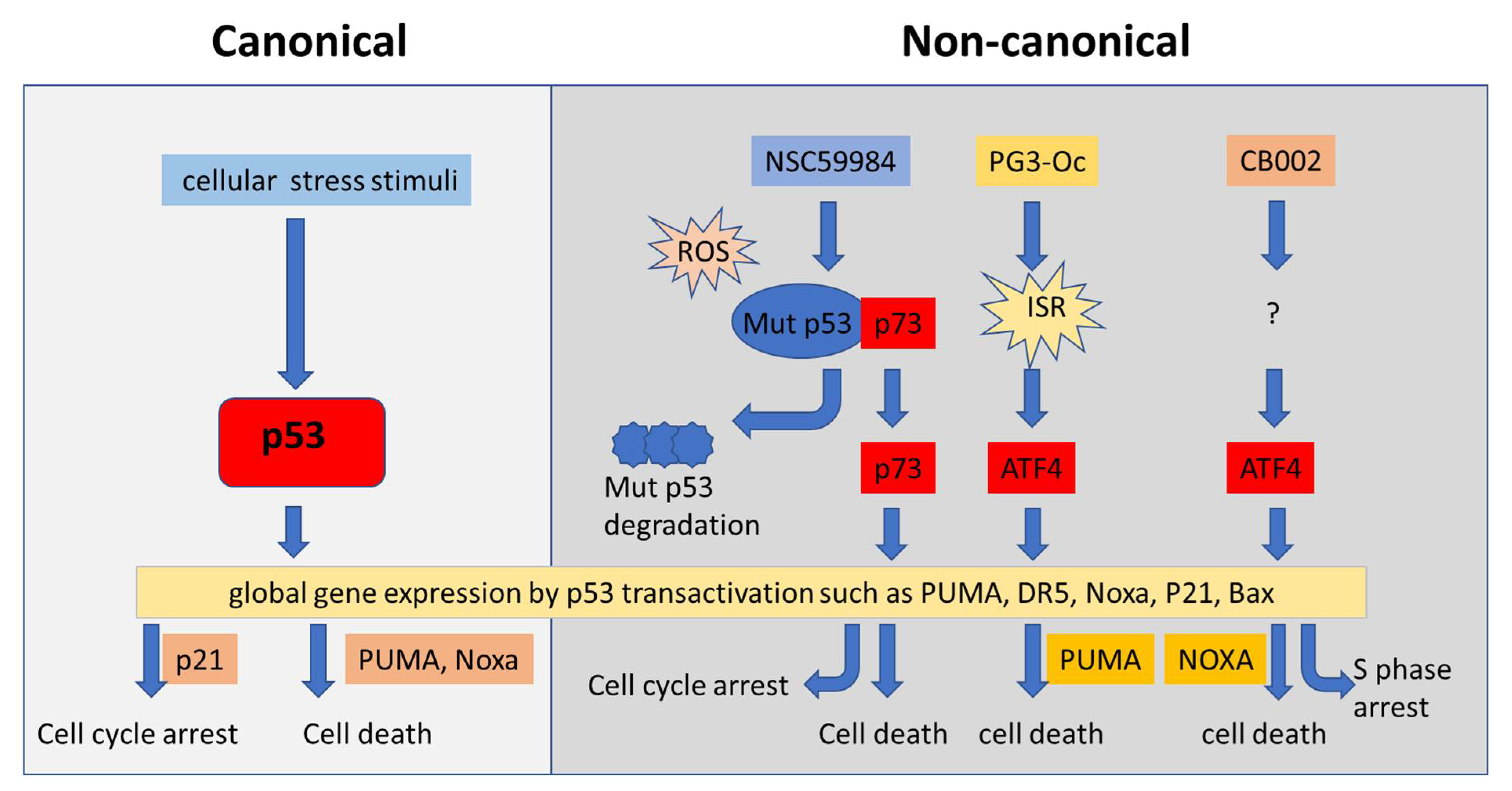

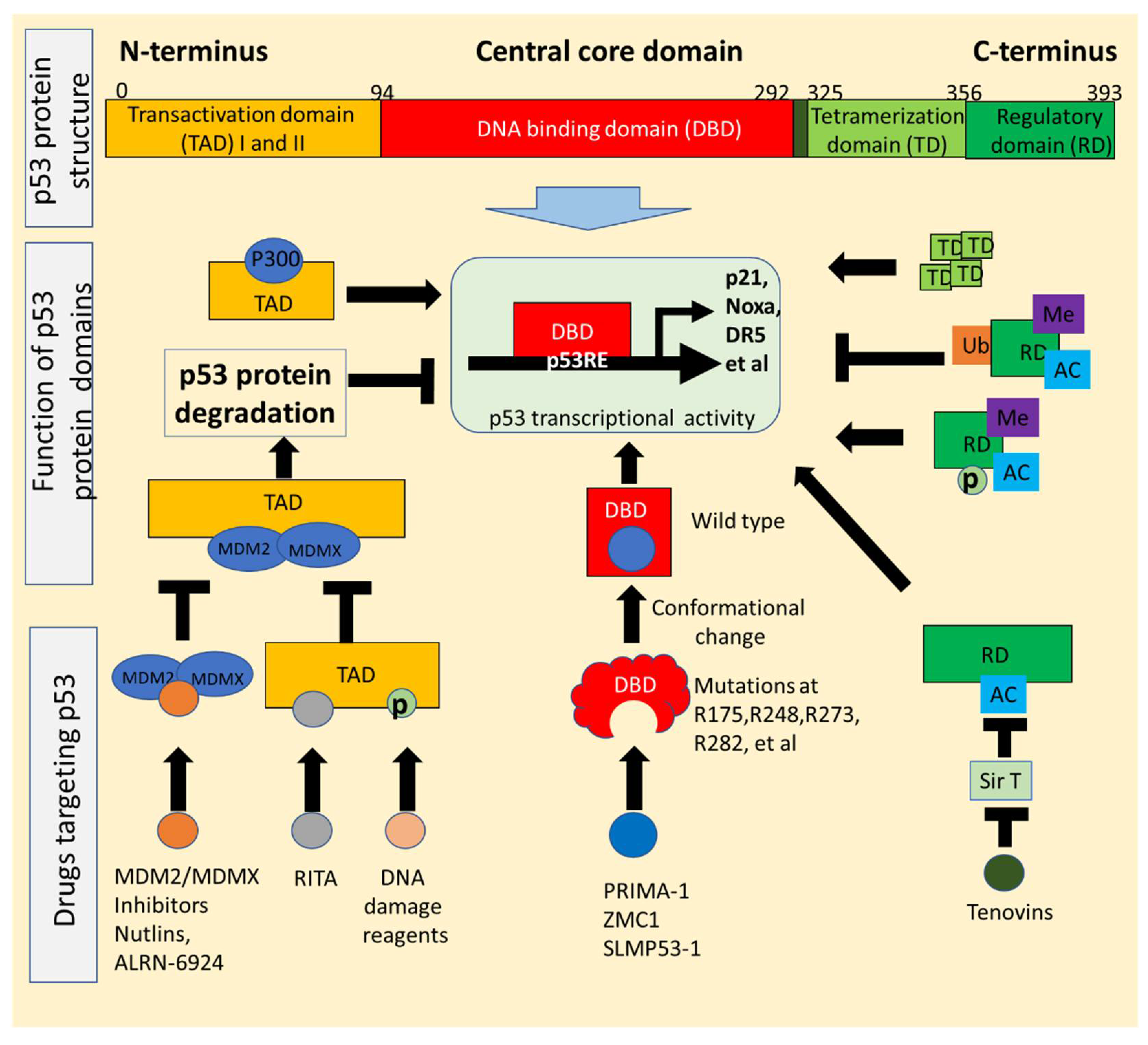{kind=link}
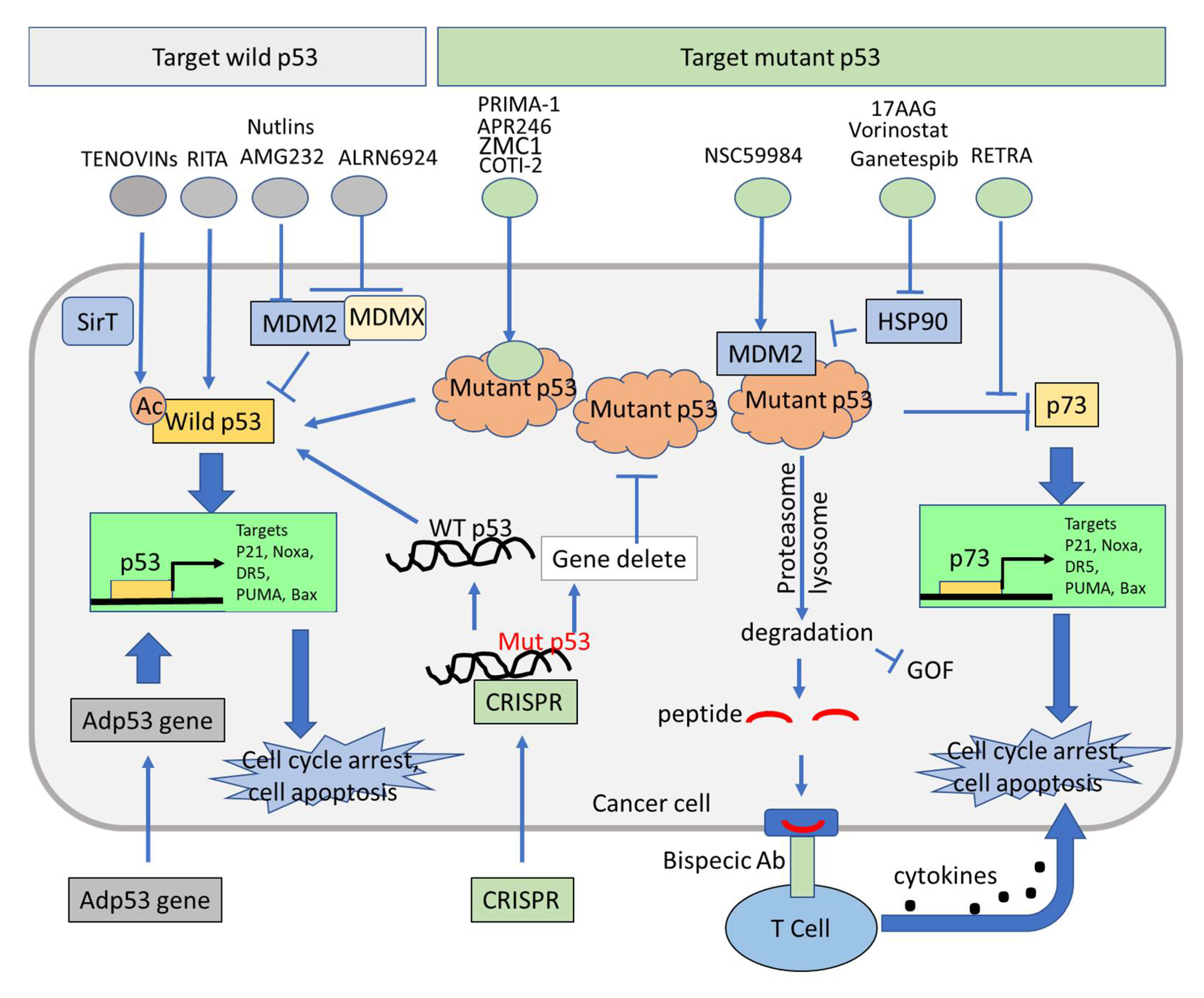{kind=link}
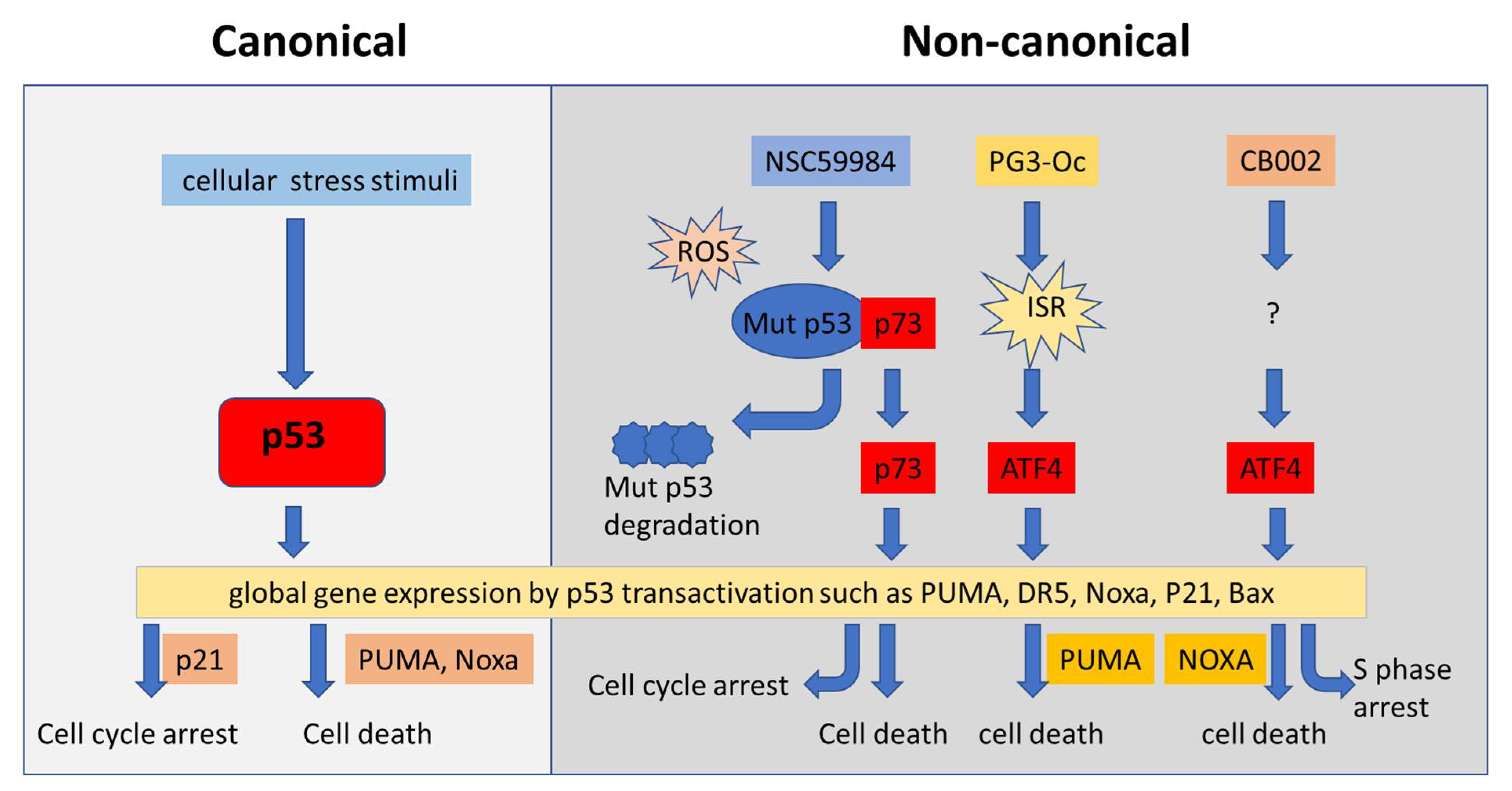{kind=link}
| Compound/Peptide/Antibody | Chemical Name and/or Class | Target/Mechanism | Clinical Development | References | ||
|---|---|---|---|---|---|---|
| Targeting wild p53 activation | p53 activator | RITA | Binds to p53 and prevents WT p53 degradation by blocking interaction with MDM2 | Experimental and/or preclinical | [78] | |
| MDM2 inhbitors | Nutlin-3a | Cis-imidazoline | Blocks the interactive binding sites of p53 and MDM2, dramatically increasing the half-life of p53 and activating p53-mediated transcription. | The listed inhibitors, except nutlin-3a, have undergone or are currently undergoing clinical trials | [9,79] | |
| RG7112 | Cis-imidazoline | |||||
| RG7388 | Cis-imidazoline | |||||
| RG7775 | Pegylated prodrug idasanutlin | |||||
| MI-77301 | Spirooxindole | |||||
| AMG232 | Piperidinone | |||||
| SAR405838 | Piperidinone | |||||
| MK-8242 | 2(1H)-Pyrimidinone | |||||
| CGM097 | Dihydroisoquinolinone | |||||
| DS-3032b | Unknown | |||||
| HDM201 | Imidazopyrrolidinone | |||||
| MDM2/MDMX(MDM4) dual inhibitors | ALRN-6924 | Stapled peptide | Blocks the interactive binding sites of p53 and MDM2/MDMX(MDM4), dramatically increasing the half-life of p53 and activating p53-mediated transcription. | currently undergoing clinical trials | ||
| RO-5963 RO-2443 | indolyl hydantoin | Binds to MDMX(MDM4)/MDM2 and blocks p53-MDM2/MDMX interaction | [32] | |||
| MDM2 degradators | PROTAC 8, A1874 | IMiD-based MDM2 | Targeted degradation of MDM2 using proteolysis targeting chimeras (PROTACs) | Experimental and/or preclinical | [9,80] | |
| Gene therapy- based on oncolytic Viruses | ONYX-015 | Recombinant adenovirus with wild-type p53 (Ad-p53) | A mutant adenovirus with a deleted E1B-55Kd gene commonly fails to replicate efficiently in cells with a wild-type p53 but replicates in many (but not all) cells with a mutant p53 gene. | In clinical trials | [9,81,82] | |
| Gendicine (Ad-53) | Recombinant adenovirus engineered to express wildtype-p53 (rAd-p53) | Gene replacement (gene therapy) | Approved in 2003 by the China Food and Drug Administration (CFDA) to treat head and neck cancer | [9,83,84] | ||
| Targeting mutant p53 | Restoration of wild-type function to mutant p53 | CP-31398 | Styrylquinazoline | Cysteine-binding compounds, Michael acceptor binding to mutant p53 | Experimental and/or preclinical | [85] |
| PRIMA-1 | Quinuclidinone | Cysteine-binding compound is converted to MQ, which binds mutant p53 by Michael addition | Experimental and/or preclinical | [86] | ||
| APR-246 | Quinuclidinone | Cysteine-binding compound is converted to MQ, which binds mutant p53 by Michael addition | Phase Ib/II for ovarian cancer, MDS, and oesophageal cancer | [87] | ||
| MIRA-1 | Maleimide | Michael acceptor binding to mutant p53 | Experimental and/or preclinical | [88] | ||
| STIMA-1 | Styrylquinazoline | Michael acceptor binding to mutant p53 | Experimental and/or preclinical | [89] | ||
| 3-Benzoylacrylic acid | Benzoylacrylate | Binds to mutant p53 by Michael addition | Experimental and/or preclinical | [90] | ||
| KSS-9 | Piperlongumine | Microtubule poison; redox; Michael acceptor binding to mutant p53 | Experimental and/or preclinical | [91] | ||
| PK11007 | Sulfonylpyrimidine | Binds to mutant p53 by nucleophilic aromatic substitution | Experimental and/or preclinical | [92] | ||
| ZMC1 ZMC2 ZMC3 ZN-1 | Thiosemicarbazone | Zn2+ chelator | Experimental and/or preclinical | [48] | ||
| COTI-2 | Thiosemicarbazone | Zn2+ chelator | Phase I for gynecological tumors and head and neck cancer | [93] | ||
| SLM P53-1 | Tryptophanol-derived oxazoloisoindolinone | restores wt-like DNA binding ability to mut p53R280K Bridges extra interaction between p53 and DNA that rescues DNA binding and transcription activity | Experimental and/or preclinical | [94,95] | ||
| SLM p53-2 | Tryptopha-nol-derived oxa-zoloisoindolinone | Restores wild-type-like conformation and DNA-binding ability, possibly by enhancing interaction with Hsp70. | Experimental and/or preclinical | [96] | ||
| MB725 MB710 | Aminobenzothiazole | Binds to Y220C of p53 DBD | Experimental and/or preclinical | [46] | ||
| PK083 Pk9318 | Carbazole | Binds to Y220C of p53 DBD | [44,45] | |||
| pCAPs | Peptides | Binds to mutant p53 and promotes refolding | Experimental and/or preclinical | [97] | ||
| Mutant p53 degradation | Ganetespib Onalespib Luminespib TAS-116 | Depletion of mutant p53 using HSP90 inhibitors or statins | In clinical trials | [9,98] | ||
| Vorinostat | Suberanilohydroxamic acid (SAHA) | Histone deacetylase (HDAC) inhibitor, destabilizes mut p53 through inhibition of the HDAC6-HSP90 chaperone axis, and at the same time, inhibit the transcription of mutant p53 through HDAC8 | In clinical trials | [72,77,99,100] | ||
| Atorvastatin Lovastatin | Statin drugs, Inhibition of mevalonate pathway | In clinical trials | [9,101] | |||
| NSC59984 | Activation of MDM2 | [21] | ||||
| Spaurtin | Chaperone-mediated autophagy (CMA) pathway | [102] | ||||
| Reacp53 | Peptide | Disrupts mutant-p53 aggregates | Experimental and/or preclinical | [8] | ||
| Interruption of mutant GOF | RETRA | 2-(4,5-Dihydro-1,3-thiazol-2-ylthio)-1-(3,4-dihydroxyphenyl) ethanone | Binds to mutant p53 and disrupts mutant-p53–p73 complexes | Experimental and/or preclinical | [103] | |
| Prodigiosin | Disrupts mutant-p53–p73 complexes | Experimental and/or preclinical | [22] | |||
| Immunotherapy | H2-scDb H2-Fab | Bispecific antibody | Bispecific antibody links T cells to cancer cells with one arm binding to T cell receptor and the other arm binding to HLA-mutant p53 R175H peptide on cancer cell surface. | Experimental and/or preclinical | [104] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Carlsen, L.; Hernandez Borrero, L.; Seyhan, A.A.; Tian, X.; El-Deiry, W.S. Advanced Strategies for Therapeutic Targeting of Wild-Type and Mutant p53 in Cancer. Biomolecules 2022, 12, 548. https://doi.org/10.3390/biom12040548
Zhang S, Carlsen L, Hernandez Borrero L, Seyhan AA, Tian X, El-Deiry WS. Advanced Strategies for Therapeutic Targeting of Wild-Type and Mutant p53 in Cancer. Biomolecules. 2022; 12(4):548. https://doi.org/10.3390/biom12040548
Chicago/Turabian StyleZhang, Shengliang, Lindsey Carlsen, Liz Hernandez Borrero, Attila A. Seyhan, Xiaobing Tian, and Wafik S. El-Deiry. 2022. "Advanced Strategies for Therapeutic Targeting of Wild-Type and Mutant p53 in Cancer" Biomolecules 12, no. 4: 548. https://doi.org/10.3390/biom12040548