
Roles of mTOR in the Regulation of Pancreatic β-Cell Mass and Insulin Secretion

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Basic Knowledge of mTOR
3. mTOR and Insulin Secretion
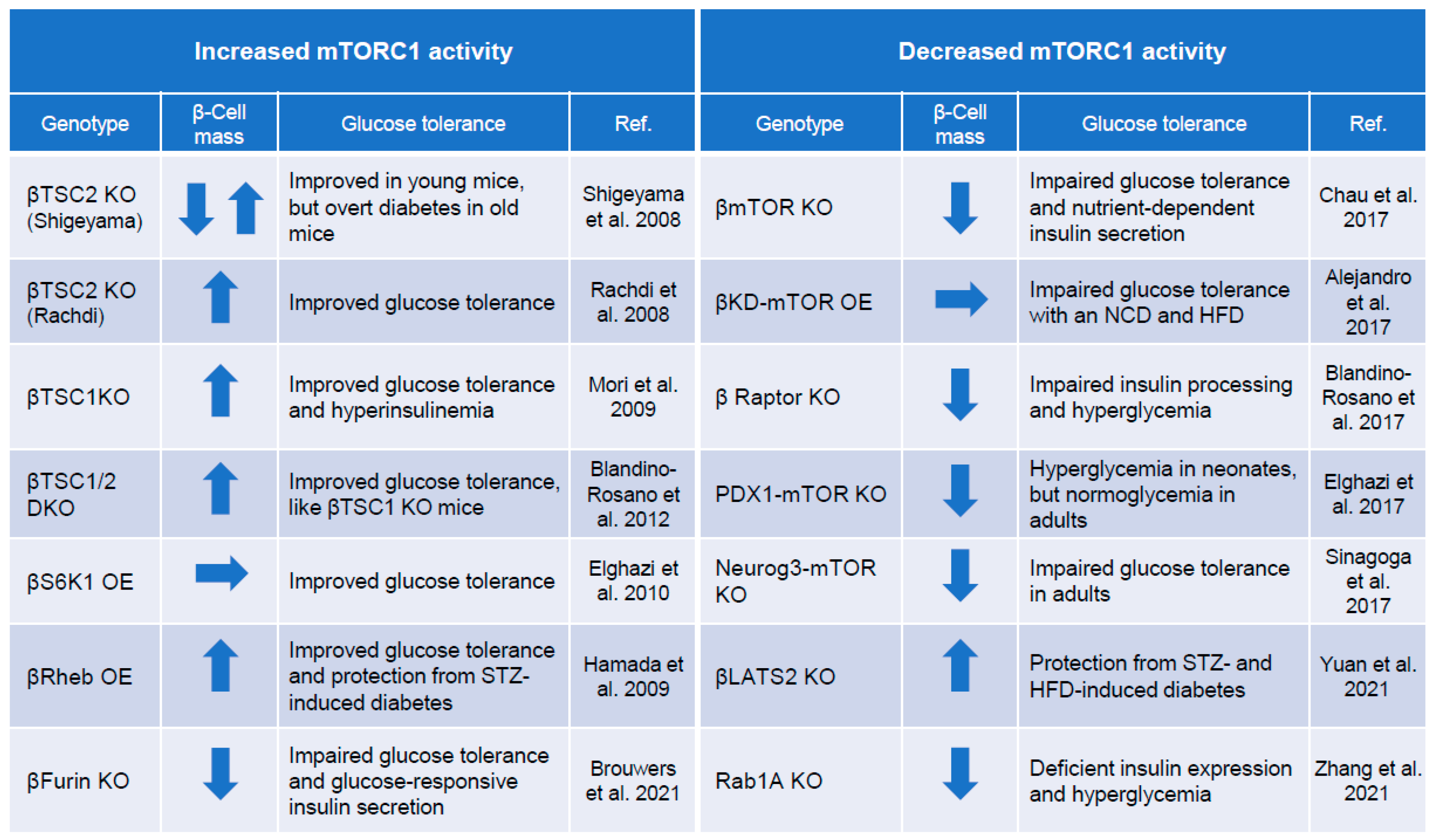
4. mTORC1 and Regulation of Pancreatic β-Cell Mass
5. Autophagy
6. ER Stress
7. mTORC2 and β-Cell Growth and Function
8. Is mTOR “Good” or “Bad” for Pancreatic β-Cells?
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- IDF Diabetes Atlas, 10th ed.; IDF Executive Office: Brussels, Belgium, 2021.
- Tchobroutsky, G. Relation of diabetic control to development of microvascular complications. Diabetologia 1978, 15, 143–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quality of life in type 2 diabetic patients is affected by complications but not by intensive policies to improve blood glucose or blood pressure control (UKPDS 37). U.K. Prospective Diabetes Study Group. Diabetes Care 1999, 22, 1125–1136.
- Ali, M.K.; Pearson-Stuttard, J.; Selvin, E.; Gregg, E.W. Interpreting global trends in type 2 diabetes complications and mortality. Diabetologia 2022, 65, 3–13. [Google Scholar] [CrossRef]
- Weyer, C.; Bogardus, C.; Mott, D.M.; Pratley, R.E. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J. Clin. Investig. 1999, 104, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Kasuga, M.; Zick, Y.; Blithe, D.L.; Crettaz, M.; Kahn, C.R. Insulin stimulates tyrosine phosphorylation of the insulin receptor in a cell-free system. Nature 1982, 298, 667–669. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Croniger, C.; Arizmendi, C.; Harada-Shiba, M.; Ren, J.; Poli, V.; Hanson, R.W.; Friedman, J.E. Hypoglycemia and impaired hepatic glucose production in mice with a deletion of the C/EBPbeta gene. J. Clin. Investig. 1999, 103, 207–213. [Google Scholar] [CrossRef] [Green Version]
- Lane, M.D.; Flores-Riveros, J.R.; Hresko, R.C.; Kaestner, K.H.; Liao, K.; Janicot, M.; Hoffman, R.D.; McLenithan, J.C.; Kastelic, T.; Christy, R.J. Insulin-receptor tyrosine kinase and glucose transport. Diabetes Care 1990, 13, 565–575. [Google Scholar] [CrossRef]
- Mulder, H.; Yang, S.; Winzell, M.S.; Holm, C.; Ahrén, B. Inhibition of lipase activity and lipolysis in rat islets reduces insulin secretion. Diabetes 2004, 53, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, N.; Kido, Y.; Uchida, T.; Asahara, S.; Shigeyama, Y.; Matsuda, T.; Takeda, A.; Tsuchihashi, D.; Nishizawa, A.; Ogawa, W.; et al. Ablation of PDK1 in pancreatic beta cells induces diabetes as a result of loss of beta cell mass. Nat. Genet. 2006, 38, 589–593. [Google Scholar] [CrossRef]
- Ueki, K.; Okada, T.; Hu, J.; Liew, C.W.; Assmann, A.; Dahlgren, G.M.; Peters, J.L.; Shackman, J.G.; Zhang, M.; Artner, I.; et al. Total insulin and IGF-I resistance in pancreatic beta cells causes overt diabetes. Nat. Genet. 2006, 38, 583–588. [Google Scholar] [CrossRef]
- Kubota, N.; Terauchi, Y.; Tobe, K.; Yano, W.; Suzuki, R.; Ueki, K.; Takamoto, I.; Satoh, H.; Maki, T.; Kubota, T.; et al. Insulin receptor substrate 2 plays a crucial role in beta cells and the hypothalamus. J. Clin. Investig. 2004, 114, 917–927. [Google Scholar] [CrossRef] [Green Version]
- Tanabe, K.; Liu, Z.; Patel, S.; Doble, B.W.; Li, L.; Cras-Méneur, C.; Martinez, S.C.; Welling, C.M.; White, M.F.; Bernal-Mizrachi, E.; et al. Genetic deficiency of glycogen synthase kinase-3beta corrects diabetes in mouse models of insulin resistance. PLoS Biol. 2008, 6, e37. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, K.; Ueki, K.; Takahashi, N.; Hashimoto, S.; Okamoto, M.; Awazawa, M.; Okazaki, Y.; Ohsugi, M.; Inabe, K.; Umehara, T.; et al. Class IA Phosphatidylinositol 3-Kinase in Pancreatic β Cells Controls Insulin Secretion by Multiple Mechanisms. Cell Metab. 2010, 12, 619–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kido, Y. Gene–environment interaction in type 2 diabetes. Diabetol. Int. 2016, 8, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Fu, X.; Chin, R.M.; Vergnes, L.; Hwang, H.; Deng, G.; Xing, Y.; Pai, M.Y.; Li, S.; Ta, L.; Fazlollahi, F.; et al. 2-Hydroxyglutarate Inhibits ATP Synthase and mTOR Signaling. Cell Metab. 2015, 22, 508–515. [Google Scholar] [CrossRef] [Green Version]
- Aoki, M.; Blazek, E.; Vogt, P.K. A role of the kinase mTOR in cellular transformation induced by the oncoproteins P3k and Akt. Proc. Natl. Acad. Sci. USA 2001, 98, 136–141. [Google Scholar] [CrossRef]
- Sparks, C.A.; Guertin, D.A. Targeting mTOR: Prospects for mTOR complex 2 inhibitors in cancer therapy. Oncogene 2010, 29, 3733–3744. [Google Scholar] [CrossRef]
- Jacobs, B.L.; McNally, R.M.; Kim, K.J.; Blanco, R.; Privett, R.E.; You, J.S.; Hornberger, T.A. Identification of mechanically regulated phosphorylation sites on tuberin (TSC2) that control mechanistic target of rapamycin (mTOR) signaling. J. Biol. Chem. 2017, 292, 6987–6997. [Google Scholar] [CrossRef] [Green Version]
- Tomita, I.; Kume, S.; Sugahara, S.; Osawa, N.; Yamahara, K.; Yasuda-Yamahara, M.; Takeda, N.; Chin-Kanasaki, M.; Kaneko, T.; Mayoux, E.; et al. SGLT2 Inhibition Mediates Protection from Diabetic Kidney Disease by Promoting Ketone Body-Induced mTORC1 Inhibition. Cell Metab. 2020, 32, 404–419.e6. [Google Scholar] [CrossRef] [PubMed]
- Umemura, A.; Park, E.J.; Taniguchi, K.; Lee, J.H.; Shalapour, S.; Valasek, M.; Aghajan, M.; Nakagawa, H.; Seki, E.; Hall, M.N.; et al. Liver Damage, Inflammation, and Enhanced Tumorigenesis after Persistent mTORC1 Inhibition. Cell Metab. 2014, 20, 133–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blandino-Rosano, M.; Chen, A.Y.; Scheys, J.O.; Alejandro, E.U.; Gould, A.P.; Taranukha, T.; Elghazi, L.; Cras-Méneur, C.; Bernal-Mizrachi, E. mTORC1 signaling and regulation of pancreatic β-cell mass. Cell Cycle 2012, 11, 1892–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, R.N.; Mizrachi, E.B.; Ocana, A.G.; Stewart, A.F. Human β-cell proliferation and intracellular signaling: Driving in the dark without a road map. Diabetes 2012, 61, 2205–2213. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Herbert, T.P. The role of mammalian target of rapamycin (mTOR) in the regulation of pancreatic β-cell mass: Implications in the development of type-2 diabetes. Cell Mol. Life Sci. 2012, 69, 1289–1304. [Google Scholar] [CrossRef]
- Nie, J.; Liu, X.; Lilley, B.N.; Zhang, H.; Pan, Y.A.; Kimball, S.R.; Zhang, J.; Zhang, W.; Wang, L.; Jefferson, L.S.; et al. SAD-A kinase controls islet β-cell size and function as a mediator of mTORC1 signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 13857–13862. [Google Scholar] [CrossRef] [Green Version]
- Le Bacquer, O.; Queniat, G.; Gmyr, V.; Kerr-Conte, J.; Lefebvre, B.; Pattou, F. mTORC1 and mTORC2 regulate insulin secretion through Akt in INS-1 cells. J. Endocrinol. 2013, 216, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Ardestani, A.; Lupse, B.; Kido, Y.; Leibowitz, G.; Maedler, K. mTORC1 Signaling: A Double-Edged Sword in Diabetic β Cells. Cell Metab. 2018, 27, 314–331. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.G.; Buel, G.R.; Blenis, J. Nutrient regulation of the mTOR complex 1 signaling pathway. Mol. Cells 2013, 35, 463–473. [Google Scholar] [CrossRef] [Green Version]
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef] [Green Version]
- Choo, A.Y.; Yoon, S.O.; Kim, S.G.; Roux, P.P.; Blenis, J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc. Natl. Acad. Sci. USA 2008, 105, 17414–17419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.C.; Guan, K.-L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, C.H.; Jun, C.B.; Ro, S.-H.; Kim, Y.-M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.-H. ULK-Atg13-FIP200 Complexes Mediate mTOR Signaling to the Autophagy Machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamming, D.; Sabatini, D.M. A Central Role for mTOR in Lipid Homeostasis. Cell Metab. 2013, 18, 465–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyanagi, M.; Asahara, S.-I.; Matsuda, T.; Hashimoto, N.; Shigeyama, Y.; Shibutani, Y.; Kanno, A.; Fuchita, M.; Mikami, T.; Hosooka, T.; et al. Ablation of TSC2 Enhances Insulin Secretion by Increasing the Number of Mitochondria through Activation of mTORC1. PLoS ONE 2011, 6, e23238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dibble, C.C.; Manning, B.D. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nature 2013, 15, 555–564. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag Complex Targets mTORC1 to the Lysosomal Surface and Is Necessary for Its Activation by Amino Acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [Green Version]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 Senses Lysosomal Amino Acids Through an Inside-Out Mechanism That Requires the Vacuolar H+-ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef] [Green Version]
- Kishton, R.J.; Barnes, C.E.; Nichols, A.G.; Cohen, S.; Gerriets, V.; Siska, P.J.; Macintyre, A.; Goraksha-Hicks, P.; de Cubas, A.A.; Liu, T.; et al. AMPK Is Essential to Balance Glycolysis and Mitochondrial Metabolism to Control T-ALL Cell Stress and Survival. Cell Metab. 2016, 23, 649–662. [Google Scholar] [CrossRef] [Green Version]
- Tzatsos, A.; Tsichlis, P.N. Energy Depletion Inhibits Phosphatidylinositol 3-Kinase/Akt Signaling and Induces Apoptosis via AMP-activated Protein Kinase-dependent Phosphorylation of IRS-1 at Ser-794. J. Biol. Chem. 2007, 282, 18069–18082. [Google Scholar] [CrossRef] [Green Version]
- Choo, A.Y.; Kim, S.G.; Vander Heiden, M.G.; Mahoney, S.J.; Vu, H.; Yoon, S.O.; Cantley, L.C.; Blenis, J. Glucose addiction of TSC null cells is caused by failed mTORC1-dependent balancing of metabolic demand with supply. Mol. Cell 2010, 38, 487–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, M.; Gravel, S.-P.; Chénard, V.; Sikström, K.; Zheng, L.; Alain, T.; Gandin, V.; Avizonis, D.; Arguello, M.; Zakaria, C.; et al. mTORC1 Controls Mitochondrial Activity and Biogenesis through 4E-BP-Dependent Translational Regulation. Cell Metab. 2013, 18, 698–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, H.; Inoki, K.; Opland, D.; Münzberg, H.; Villanueva, E.C.; Faouzi, M.; Ikenoue, T.; Kwiatkowski, D.J.; Macdougald, O.A.; Myers, M.G., Jr.; et al. Critical roles for the TSC-mTOR pathway in β-cell function. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1013–E1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallace, D.C. Mitochondrial Diseases in Man and Mouse. Science 1999, 283, 1482–1488. [Google Scholar] [CrossRef] [Green Version]
- Velho, G.; Byrne, M.M.; Clément, K.; Sturis, J.; Pueyo, M.E.; Blanché, H.; Vionnet, N.; Fiet, J.; Passa, P.; Robert, J.J.; et al. Clinical phenotypes, insulin secretion, and insulin sensitivity in kindreds with maternally inherited diabetes and deafness due to mitochondrial tRNALeu (UUR) gene mutation. Diabetes 1996, 45, 478–487. [Google Scholar] [CrossRef]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms Controlling Mitochondrial Biogenesis and Respiration through the Thermogenic Coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A Cold-Inducible Coactivator of Nuclear Receptors Linked to Adaptive Thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Falkenberg, M.; Larsson, N.-G.; Gustafsson, C.M. DNA Replication and Transcription in Mammalian Mitochondria. Annu. Rev. Biochem. 2007, 76, 679–699. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Transcriptional Paradigms in Mammalian Mitochondrial Biogenesis and Function. Physiol. Rev. 2008, 88, 611–638. [Google Scholar] [CrossRef] [Green Version]
- Duncan, J.G.; Fong, J.L.; Medeiros, D.M.; Finck, B.N.; Kelly, D.P. Insulin-resistant heart exhibits a mitochondrial biogenic response driven by the peroxisome proliferator-activated receptor-alpha/PGC-1alpha gene regulatory pathway. Circulation 2007, 115, 909–917. [Google Scholar] [CrossRef] [Green Version]
- Silva, J.P.; Köhler, M.; Graff, C.; Oldfors, A.; Magnuson, M.A.; Berggren, P.O.; Larsson, N.G. Impaired insulin secretion and beta-cell loss in tissue-specific knockout mice with mitochondrial diabetes. Nat. Genet. 2000, 26, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Rutter, G.; Leclerc, I. The AMP-regulated kinase family: Enigmatic targets for diabetes therapy. Mol. Cell. Endocrinol. 2009, 297, 41–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahn, B.B.; Alquier, T.; Carling, D.; Hardie, D.G. AMP-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005, 1, 15–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergeron, R.; Ren, J.M.; Cadman, K.S.; Moore, I.K.; Perret, P.; Pypaert, M.; Young, L.H.; Semenkovich, C.F.; Shulman, G.I. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am. J. Physiol. Endocrinol. Metab. 2001, 281, E1340–E1346. [Google Scholar] [CrossRef]
- Winder, W.W.; Holmes, B.F.; Rubink, D.S.; Jensen, E.B.; Chen, M.; Holloszy, J.O. Activation of AMP-activated protein kinase increases mitochondrial enzymes in skeletal muscle. J. Appl. Physiol. 2000, 88, 2219–2226. [Google Scholar] [CrossRef] [Green Version]
- Schieke, S.M.; Finkel, T. Mitochondrial signaling, TOR, and life span. Biol. Chem. 2006, 387, 1357–1361. [Google Scholar] [CrossRef]
- Cunningham, J.T.; Rodgers, J.T.; Arlow, D.H.; Vazquez, F.; Mootha, V.K.; Puigserver, P. mTOR controls mitochondrial oxidative function through a YY1–PGC-1α transcriptional complex. Nature 2007, 450, 736–740. [Google Scholar] [CrossRef]
- Morino, K.; Petersen, K.F.; Dufour, S.; Befroy, D.; Frattini, J.; Shatzkes, N.; Neschen, S.; White, M.F.; Bilz, S.; Sono, S.; et al. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J. Clin. Investig. 2005, 115, 3587–3593. [Google Scholar] [CrossRef] [Green Version]
- Iwabu, M.; Yamauchi, T.; Okada-Iwabu, M.; Sato, K.; Nakagawa, T.; Funata, M.; Yamaguchi, M.; Namiki, S.; Nakayama, R.; Tabata, M.; et al. Adiponectin and AdipoR1 regulate PGC-1α and mitochondria by Ca2+ and AMPK/SIRT1. Nature 2010, 464, 1313–1319. [Google Scholar] [CrossRef]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Jäger, S.; Handschin, C.; St.-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Z.; Hu, W.; de Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: Stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007, 67, 3043–3053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn-Windgassen, A.; Nogueira, V.; Chen, C.-C.; Skeen, J.E.; Sonenberg, N.; Hay, N. Akt Activates the Mammalian Target of Rapamycin by Regulating Cellular ATP Level and AMPK Activity. J. Biol. Chem. 2005, 280, 32081–32089. [Google Scholar] [CrossRef] [Green Version]
- Quan, Y.; Xin, Y.; Tian, G.; Zhou, J.; Liu, X. Mitochondrial ROS-Modulated mtDNA: A Potential Target for Cardiac Aging. Oxid. Med. Cell. Longev. 2020, 2020, 9423593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sattar, N.; Preiss, D.; Murray, H.M.; Welsh, P.; Buckley, B.M.; de Craen, A.J.; Seshasai, S.R.; McMurray, J.J.; Freeman, D.J.; Jukema, J.W.; et al. Statins and risk of incident diabetes: A collaborative meta-analysis of randomised statin trials. Lancet 2010, 375, 735–742. [Google Scholar] [CrossRef]
- Preiss, D.; Seshasai, S.R.; Welsh, P.; Murphy, S.A.; Ho, J.E.; Waters, D.D.; DeMicco, D.A.; Barter, P.; Cannon, C.P.; Sabatine, M.S.; et al. Risk of incident diabetes with intensive-dose compared with moderate-dose statin therapy: A meta-analysis. JAMA 2011, 305, 2556–2564. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Gu, Y.; Qiu, Y.; Cheng, T.; Nie, A.; Cui, C.; Fu, C.; Li, T.; Li, X.; Fu, L.; et al. Atorvastatin Targets the Islet Mevalonate Pathway to Dysregulate mTOR Signaling and Reduce β-Cell Functional Mass. Diabetes 2019, 69, 48–59. [Google Scholar] [CrossRef]
- Pascoe, J.; Hollern, D.; Stamateris, R.; Abbasi, M.; Romano, L.C.; Zou, B.; O’Donnell, C.P.; Garcia-Ocana, A.; Alonso, L.C. Free fatty acids block glucose-induced b-cell proliferation in mice by inducing cell cycle inhibitors p16 and p18. Diabetes 2012, 61, 632–641. [Google Scholar] [CrossRef] [Green Version]
- Fontés, G.; Zarrouki, B.; Hagman, D.K.; Latour, M.G.; Semache, M.; Roskens, V.; Moore, P.C.; Prentki, M.; Rhodes, C.J.; Jetton, T.L.; et al. Glucolipotoxicity age-dependently impairs beta cell function in rats despite a marked increase in beta cell mass. Diabetologia 2010, 53, 2369–2379. [Google Scholar] [CrossRef] [Green Version]
- Poitout, V.; Amyot, J.; Semache, M.; Zarrouki, B.; Hagman, D.; Fontés, G. Glucolipotoxicity of the pancreatic beta cell. Biochim. Biophys. Acta 2010, 1801, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Hatanaka, M.; Maier, B.; Sims, E.K.; Templin, A.T.; Kulkarni, R.N.; Evans-Molina, C.; Mirmira, R.G. Palmitate Induces mRNA Translation and Increases ER Protein Load in Islet β-Cells via Activation of the Mammalian Target of Rapamycin Pathway. Diabetes 2014, 63, 3404–3415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigeyama, Y.; Kobayashi, T.; Kido, Y.; Hashimoto, N.; Asahara, S.; Matsuda, T.; Takeda, A.; Inoue, T.; Shibutani, Y.; Koyanagi, M.; et al. Biphasic response of pancreatic beta-cell mass to ablation of tuberous sclerosis complex 2 in mice. Mol. Cell Biol. 2008, 28, 2971–2979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rachdi, L.; Balcazar, N.; Osorio-Duque, F.; Elghazi, L.; Weiss, A.; Gould, A.; Chang-Chen, K.J.; Gambello, M.J.; Bernal-Mizrachi, E. Disruption of Tsc2 in pancreatic beta cells induces beta cell mass expansion and improved glucose tolerance in a TORC1-dependent manner. Proc. Natl. Acad. Sci. USA 2008, 105, 9250–9255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werneck-De-Castro, J.P.; Peçanha, F.L.M.; Silvestre, D.H.; Bernal-Mizrachi, E. The RNA-binding protein LARP1 is dispensable for pancreatic β-cell function and mass. Sci. Rep. 2021, 11, 2079. [Google Scholar] [CrossRef] [PubMed]
- Elghazi, L.; Balcazar, N.; Blandino-Rosano, M.; Cras-Méneur, C.; Fatrai, S.; Gould, A.P.; Chi, M.M.; Moley, K.H.; Bernal-Mizrachi, E. Decreased IRS signaling impairs beta-cell cycle progression and survival in transgenic mice overexpressing S6K in beta-cells. Diabetes 2010, 59, 2390–2399. [Google Scholar] [CrossRef] [Green Version]
- Blandino-Rosano, M.; Barbaresso, R.; Jimenez-Palomares, M.; Bozadjieva, N.; Werneck-De-Castro, J.P.; Hatanaka, M.; Mirmira, R.G.; Sonenberg, N.; Liu, M.; Rüegg, M.A.; et al. Loss of mTORC1 signalling impairs β-cell homeostasis and insulin processing. Nat. Commun. 2017, 8, 16014. [Google Scholar] [CrossRef]
- Sinagoga, K.L.; Stone, W.; Schiesser, J.V.; Schweitzer, J.I.; Sampson, L.; Zheng, Y.; Wells, J.M. Distinct roles for the mTOR pathway in postnatal morphogenesis, maturation and function of pancreatic islets. Development 2017, 144, 2402–2414. [Google Scholar] [CrossRef] [Green Version]
- Hamada, S.; Hara, K.; Hamada, T.; Yasuda, H.; Moriyama, H.; Nakayama, R.; Nagata, M.; Yokono, K. Upregulation of the mammalian target of rapamycin complex 1 pathway by Ras homolog enriched in brain in pancreatic beta-cells leads to increased beta-cell mass and prevention of hyperglycemia. Diabetes 2009, 58, 1321–1332. [Google Scholar] [CrossRef] [Green Version]
- Chau, G.C.; Im, D.U.; Kang, T.M.; Bae, J.M.; Kim, W.; Pyo, S.; Moon, E.Y.; Um, S.H. mTOR controls ChREBP transcriptional activity and pancreatic β cell survival under diabetic stress. J. Cell Biol. 2017, 216, 2091–2105. [Google Scholar] [CrossRef] [Green Version]
- Maedler, K.; Ardestani, A. mTORC in β cells: More Than Only Recognizing Comestibles. J. Cell Biol. 2017, 216, 1883–1885. [Google Scholar] [CrossRef] [Green Version]
- Ni, Q.; Gu, Y.; Xie, Y.; Yin, Q.; Zhang, H.; Nie, A.; Li, W.; Wang, Y.; Ning, G.; Wang, W.; et al. Raptor regulates functional maturation of murine beta cells. Nat. Commun. 2017, 8, 15755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alejandro, E.U.; Bozadjieva, N.; Blandino-Rosano, M.; Wasan, M.A.; Elghazi, L.; Vadrevu, S.; Satin, L.; Bernal-Mizrachi, E. Overexpression of Kinase-Dead mTOR Impairs Glucose Homeostasis by Regulating Insulin Secretion and Not β-Cell Mass. Diabetes 2017, 66, 2150–2162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouwers, B.; Coppola, I.; Vints, K.; Dislich, B.; Jouvet, N.; Van Lommel, L.; Segers, C.; Gounko, N.V.; Thorrez, L.; Schuit, F.; et al. Loss of Furin in β-Cells Induces an mTORC1-ATF4 Anabolic Pathway That Leads to β-Cell Dysfunction. Diabetes 2021, 70, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Elghazi, L.; Blandino-Rosano, M.; Alejandro, E.; Cras-Méneur, C.; Bernal-Mizrachi, E. Role of nutrients and mTOR signaling in the regulation of pancreatic progenitors development. Mol. Metab. 2017, 6, 560–573. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Annamalai, K.; Naik, S.; Lupse, B.; Geravandi, S.; Pal, A.; Dobrowolski, A.; Ghawali, J.; Ruhlandt, M.; Gorrepati, K.D.D.; et al. The Hippo kinase LATS2 impairs pancreatic β-cell survival in diabetes through the mTORC1-autophagy axis. Nat. Commun. 2021, 12, 4928. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, X.; Yuan, Z.; Radford, S.J.; Liu, C.; Libutti, S.K.; Zheng, X.F.S. Amino acids-Rab1A-mTORC1 signaling controls whole-body glucose homeostasis. Cell Rep. 2021, 34, 108830. [Google Scholar] [CrossRef]
- Alejandro, E.; Gregg, B.; Wallen, T.; Kumusoglu, D.; Meister, D.; Chen, A.; Merrins, M.J.; Satin, L.S.; Liu, M.; Arvan, P.; et al. Maternal diet–induced microRNAs and mTOR underlie β cell dysfunction in offspring. J. Clin. Investig. 2014, 124, 4395–4410. [Google Scholar] [CrossRef] [Green Version]
- Um, S.H.; Sticker-Jantscheff, M.; Chau, G.C.; Vintersten, K.; Mueller, M.; Gangloff, Y.-G.; Adams, R.H.; Spetz, J.-F.; Elghazi, L.; Pfluger, P.T.; et al. S6K1 controls pancreatic β cell size independently of intrauterine growth restriction. J. Clin. Investig. 2015, 125, 2736–2747. [Google Scholar] [CrossRef] [Green Version]
- Roos, S.; Jansson, N.; Palmberg, I.; Säljö, K.; Powell, T.; Jansson, T. Mammalian target of rapamycin in the human placenta regulates leucine transport and is down-regulated in restricted fetal growth. J. Physiol. 2007, 582, 449–459. [Google Scholar] [CrossRef]
- Sati, L.; Soygur, B.; Celik-Ozenci, C. Expression of Mammalian Target of Rapamycin and Downstream Targets in Normal and Gestational Diabetic Human Term Placenta. Reprod. Sci. 2015, 23, 324–332. [Google Scholar] [CrossRef]
- Jaafar, R.; Tran, S.; Shah, A.N.; Sun, G.; Valdearcos, M.; Marchetti, P.; Masini, M.; Swisa, A.; Giacometti, S.; Bernal-Mizrachi, E.; et al. mTORC1-to-AMPK switching underlies β cell metabolic plasticity during maturation and diabetes. J. Clin. Investig. 2019, 129, 4124–4137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masini, M.; Bugliani, M.; Lupi, R.; del Guerra, S.; Boggi, U.; Filipponi, F.; Marselli, L.; Masiello, P.; Marchetti, P. Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia 2009, 52, 1083–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mir, S.U.; George, N.M.; Zahoor, L.; Harms, R.; Guinn, Z.; Sarvetnick, N.E. Inhibition of autophagic turnover in b-cells by fatty acids and glucose leads to apoptotic cell death. J. Biol. Chem. 2015, 290, 6071–6085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebato, C.; Uchida, T.; Arakawa, M.; Komatsu, M.; Ueno, T.; Komiya, K.; Azuma, K.; Hirose, T.; Tanaka, K.; Kominami, E.; et al. Autophagy Is Important in Islet Homeostasis and Compensatory Increase of Beta Cell Mass in Response to High-Fat Diet. Cell Metab. 2008, 8, 325–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, H.S.; Chung, K.W.; Won Kim, J.; Kim, J.; Komatsu, M.; Tanaka, K.; Nguyen, Y.H.; Kang, T.M.; Yoon, K.H.; Kim, J.W.; et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 2008, 8, 318–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riahi, Y.; Wikstrom, J.D.; Bachar-Wikstrom, E.; Polin, N.; Zucker, H.; Lee, M.-S.; Quan, W.; Haataja, L.; Liu, M.; Arvan, P.; et al. Autophagy is a major regulator of beta cell insulin homeostasis. Diabetologia 2016, 59, 1480–1491. [Google Scholar] [CrossRef] [Green Version]
- Stienstra, R.; Haim, Y.; Riahi, Y.; Netea, M.; Rudich, A.; Leibowitz, G. Autophagy in adipose tissue and the beta cell: Implications for obesity and diabetes. Diabetologia 2014, 57, 1505–1516. [Google Scholar] [CrossRef] [Green Version]
- Bartolomé, A.; Kimura-Koyanagi, M.; Asahara, S.-I.; Guillén, C.; Inoue, H.; Teruyama, K.; Shimizu, S.; Kanno, A.; García-Aguilar, A.; Koike, M.; et al. Pancreatic β-Cell Failure Mediated by mTORC1 Hyperactivity and Autophagic Impairment. Diabetes 2014, 63, 2996–3008. [Google Scholar] [CrossRef] [Green Version]
- Bachar-Wikstrom, E.; Wikstrom, J.D.; Ariav, Y.; Tirosh, B.; Kaiser, N.; Cerasi, E.; Leibowitz, G. Stimulation of autophagy improves endoplasmic reticulum stress-induced diabetes. Diabetes 2013, 62, 1227–1237. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Pasquier, A.; Vivot, K.; Erbs, E.; Spiegelhalter, C.; Zhang, Z.; Aubert, V.; Liu, Z.; Senkara, M.; Maillard, E.; Pinget, M.; et al. Lysosomal degradation of newly formed insulin granules contributes to β cell failure in diabetes. Nat. Commun. 2019, 10, 3312. [Google Scholar] [CrossRef] [PubMed]
- Back, S.H.; Kaufman, R.J. Endoplasmic Reticulum Stress and Type 2 Diabetes. Annu. Rev. Biochem. 2012, 81, 767–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biden, T.J.; Boslem, E.; Chu, K.Y.; Sue, N. Lipotoxic endoplasmic reticulum stress, β cell failure, and type 2 diabetes mellitus. Trends Endocrinol. Metab. 2014, 25, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, S.G.; Gromada, J.; Urano, F. Endoplasmic reticulum stress and pancreatic β-cell death. Trends Endocrinol. Metab. 2011, 22, 266–274. [Google Scholar] [CrossRef] [Green Version]
- Oyadomari, S.; Koizumi, A.; Takeda, K.; Gotoh, T.; Akira, S.; Araki, E.; Mori, M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J. Clin. Investig. 2002, 109, 525–532. [Google Scholar] [CrossRef]
- Appenzeller-Herzog, C.; Hall, M.N. Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends Cell Biol. 2012, 22, 274–282. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Yang, X.; Zhang, J. Bridges between mitochondrial oxidative stress, ER stress and mTOR signaling in pancreatic β cells. Cell Signal. 2016, 28, 1099–1104. [Google Scholar] [CrossRef]
- Ozcan, U.; Ozcan, L.; Yilmaz, E.; Duvel, K.; Sahin, M.; Manning, B.D.; Hotamisligil, G.S. Loss of the tuberous sclerosis complex tumor suppressors triggers the unfolded protein response to regulate insulin signaling and apoptosis. Mol. Cell 2008, 29, 541–551. [Google Scholar] [CrossRef] [Green Version]
- Bachar, E.; Ariav, Y.; Ketzinel-Gilad, M.; Cerasi, E.; Kaiser, N.; Leibowitz, G. Glucose Amplifies Fatty Acid-Induced Endoplasmic Reticulum Stress in Pancreatic β-Cells via Activation of mTORC1. PLoS ONE 2009, 4, e4954. [Google Scholar] [CrossRef] [Green Version]
- Riahi, Y.; Israeli, T.; Yeroslaviz, R.; Chimenez, S.; Avrahami, D.; Stolovich-Rain, M.; Alter, I.; Sebag, M.; Polin, N.; Bernal-Mizrachi, E.; et al. Inhibition of mTORC1 by ER stress impairs neonatal β-cell expansion and predisposes to diabetes in the Akita mouse. eLife 2018, 7, e38472. [Google Scholar] [CrossRef]
- Guan, B.J.; Krokowski, D.; Majumder, M.; Schmotzer, C.L.; Kimball, S.R.; Merrick, W.C.; Koromilas, A.E.; Hatzoglou, M. Translational control during endoplasmic reticulum stress beyond phosphorylation of the translation initiation factor eIF2α. J. Biol. Chem. 2014, 289, 12593–12611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krokowski, D.; Han, J.; Saikia, M.; Majumder, M.; Yuan, C.L.; Guan, B.J.; Bevilacqua, E.; Bussolati, O.; Bröer, S.; Arvan, P.; et al. A self-defeating anabolic program leads to b-cell apoptosis in endoplasmic reticulum stress induced diabetes via regulation of amino acid flux. J. Biol. Chem. 2013, 288, 17202–17213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.; Reyna-Neyra, A.; Philippe, L.; Thoreen, C.C. mTORC1 Balances Cellular Amino Acid Supply with Demand for Protein Synthesis through Post-transcriptional Control of ATF4. Cell Rep. 2017, 19, 1083–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Y.; Lindner, J.; Kumar, A.; Yuan, W.; Magnuson, M.A. Rictor/mTORC2 is essential for maintaining a balance between beta-cell proliferation and cell size. Diabetes 2011, 60, 827–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Cui, C.; Nie, A.; Wang, Y.; Ni, Q.; Liu, Y.; Yin, Q.; Zhang, H.; Li, Y.; Wang, Q.; et al. The mTORC2/PKC pathway sustains compensatory insulin secretion of pancreatic β cells in response to metabolic stress. Biochim Biophys Acta Gen Subj. 2017, 1861, 2039–2047. [Google Scholar] [CrossRef]
- Yin, Q.; Ni, Q.; Wang, Y.; Zhang, H.; Li, W.; Nie, A.; Wang, S.; Gu, Y.; Wang, Q.; Ning, G. Raptor determines β-cell identity and plasticity independent of hyperglycemia in mice. Nat. Commun. 2020, 11, 2538. [Google Scholar] [CrossRef]
- Lupse, B.; Annamalai, K.; Ibrahim, H.; Kaur, S.; Geravandi, S.; Sarma, B.; Pal, A.; Awal, S.; Joshi, A.; Rafizadeh, S.; et al. Inhibition of PHLPP1/2 phosphatases rescues pancreatic β-cells in diabetes. Cell Rep. 2021, 36, 109490. [Google Scholar] [CrossRef]
- Yuan, T.; Rafizadeh, S.; Gorrepati, K.D.D.; Lupse, B.; Oberholzer, J.; Maedler, K.; Ardestani, A. Reciprocal regulation of mTOR complexes in pancreatic islets from humans with type 2 diabetes. Diabetologia 2016, 60, 668–678. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asahara, S.-i.; Inoue, H.; Watanabe, H.; Kido, Y. Roles of mTOR in the Regulation of Pancreatic β-Cell Mass and Insulin Secretion. Biomolecules 2022, 12, 614. https://doi.org/10.3390/biom12050614
Asahara S-i, Inoue H, Watanabe H, Kido Y. Roles of mTOR in the Regulation of Pancreatic β-Cell Mass and Insulin Secretion. Biomolecules. 2022; 12(5):614. https://doi.org/10.3390/biom12050614
Chicago/Turabian StyleAsahara, Shun-ichiro, Hiroyuki Inoue, Hitoshi Watanabe, and Yoshiaki Kido. 2022. "Roles of mTOR in the Regulation of Pancreatic β-Cell Mass and Insulin Secretion" Biomolecules 12, no. 5: 614. https://doi.org/10.3390/biom12050614