
Structural Insights into the Intrinsically Disordered GPCR C-Terminal Region, Major Actor in Arrestin-GPCR Interaction

 , , ,
, , , 
Abstract
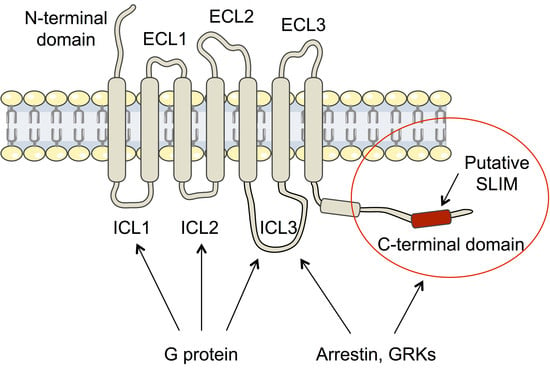
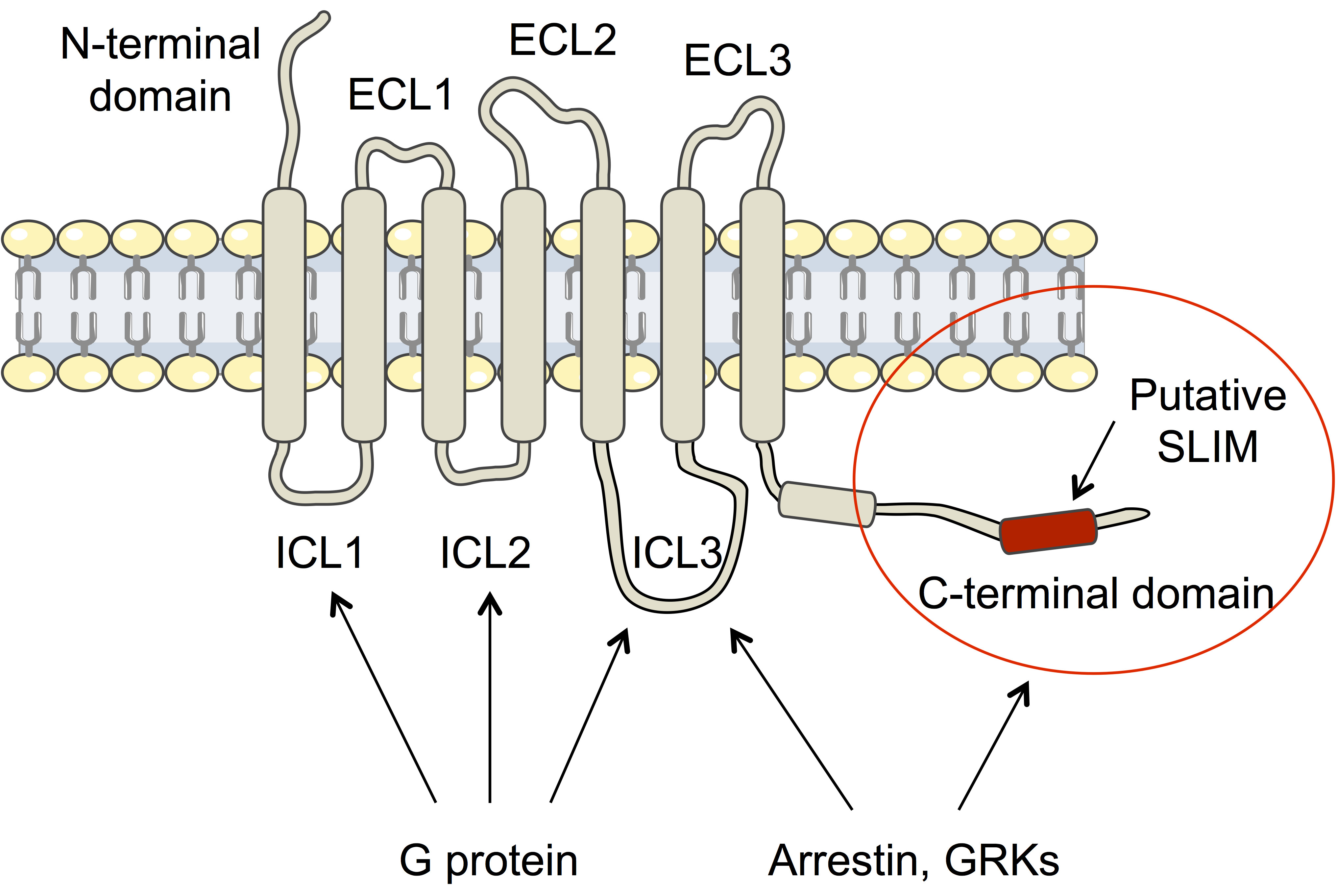
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. Bioinformatic Analyses
2.2.1. Disorder Prediction
2.2.2. Secondary Structure Prediction
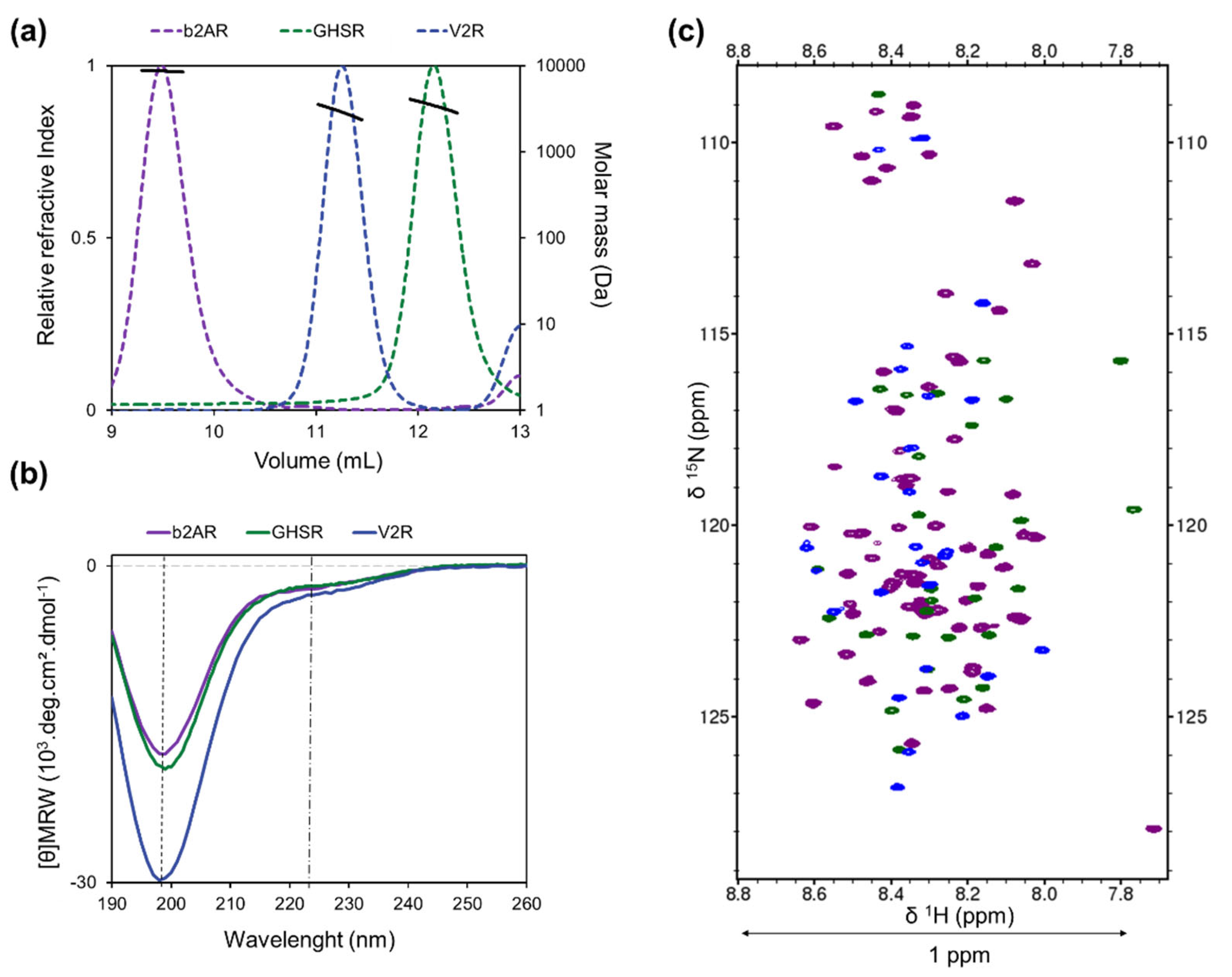
2.3. Size Exclusion Chromatography-Multi-Angle Light Scattering (SEC-MALS)
2.4. Circular Dichroism (CD)
2.5. Small-Angle X-ray Scattering (SAXS) Measurement and Analysis
2.6. Nuclear Magnetic Resonance (NMR) Spectroscopy
2.6.1. Backbone Assignment
2.6.2. Secondary Chemical Shift (SCS)
2.6.3. 3JHNHA Coupling
2.6.4. 1H-15N Residual Dipolar Couplings (RDCs)
2.6.5. 15N Relaxation Experiments
2.6.6. Paramagnetic Relaxation Enhancement (PRE)
2.6.7. Ensemble Calculations
3. Results
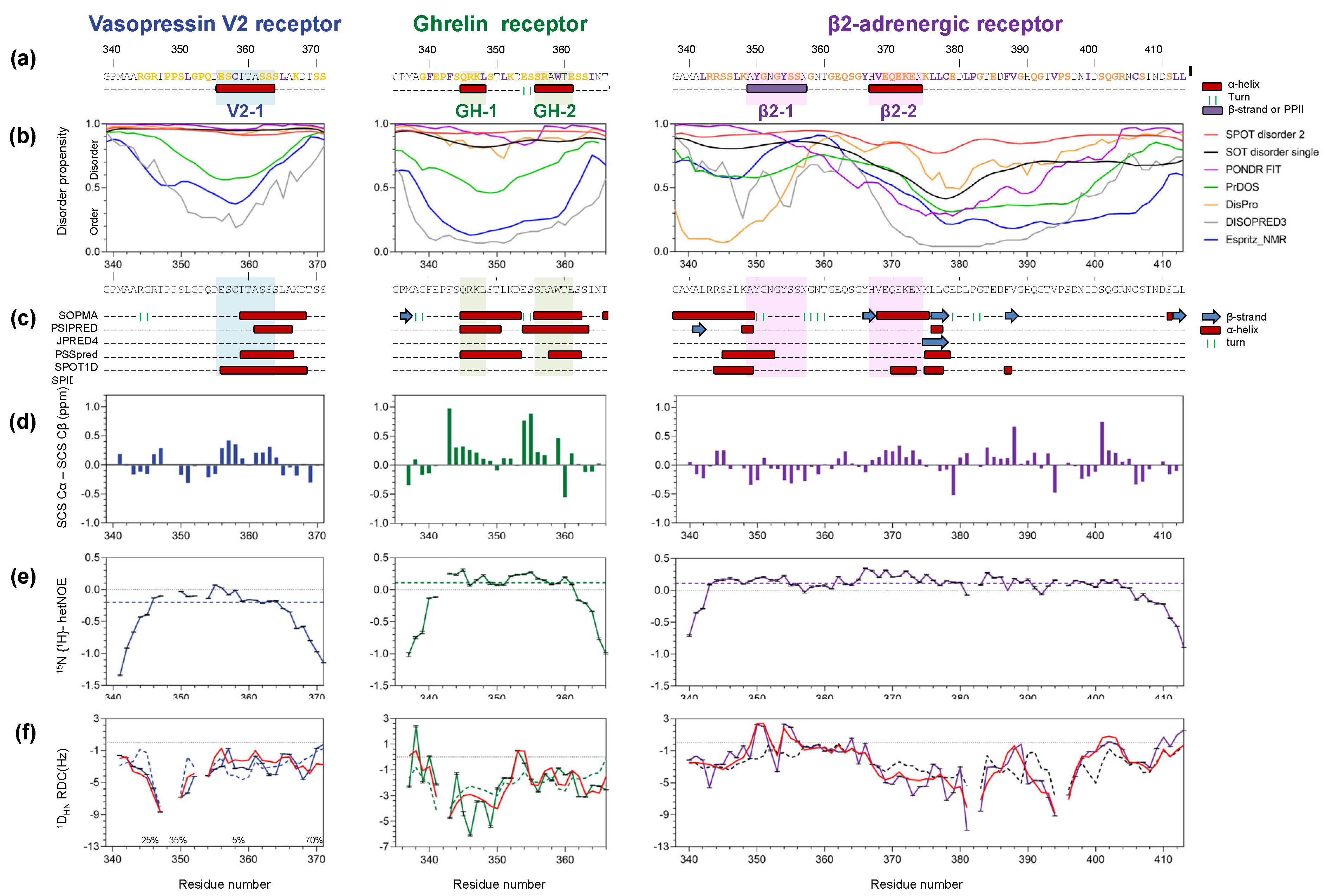
3.1. The Disordered C-Termini of V2R, GHSR, and β2AR Contain Transient Secondary Structures
3.2. Location of the Transient Secondary Structures in V2R, GHSR, and β2AR C-Termini
3.2.1. Vasopressin V2 Receptor C-Terminal Domain (V2R-Cter)
3.2.2. Ghrelin Receptor C-Terminal Domain (GHSR-Cter)
3.2.3. β2-Adrenergic Receptor C-Terminal Domain (β2AR-Cter)
3.2.4. Conformational Ensemble of GPCR-Cters
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bockaert, J.; Philippe Pin, J. Molecular Tinkering of G Protein–Coupled Receptors: An Evolutionary Success. EMBO J. 1999, 18, 1723–1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schöneberg, T.; Schulz, A.; Biebermann, H.; Hermsdorf, T.; Römpler, H.; Sangkuhl, K. Mutant G-Protein-Coupled Receptors as a Cause of Human Diseases. Pharmacol. Ther. 2004, 104, 173–206. [Google Scholar] [CrossRef] [PubMed]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR Drug Discovery: New Agents, Targets and Indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, R. The G-Protein-Coupled Receptors in the Human Genome Form Five Main Families. Phylogenetic Analysis, Paralogon Groups, and Fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef] [Green Version]
- Lagerström, M.C.; Schiöth, H.B. Structural Diversity of G Protein-Coupled Receptors and Significance for Drug Discovery. Nat. Rev. Drug Discov. 2008, 7, 339–357. [Google Scholar] [CrossRef]
- Fonin, A.V.; Darling, A.L.; Kuznetsova, I.M.; Turoverov, K.K.; Uversky, V.N. Multi-Functionality of Proteins Involved in GPCR and G Protein Signaling: Making Sense of Structure-Function Continuum with Intrinsic Disorder-Based Proteoforms. Cell Mol. Life Sci. 2019, 76, 4461–4492. [Google Scholar] [CrossRef]
- Gurevich, V.V.; Gurevich, E.V. GPCR Signaling Regulation: The Role of GRKs and Arrestins. Front. Pharmacol. 2019, 10, 125. [Google Scholar] [CrossRef] [Green Version]
- Ren, X.-R.; Reiter, E.; Ahn, S.; Kim, J.; Chen, W.; Lefkowitz, R.J. Different G Protein-Coupled Receptor Kinases Govern G Protein and Beta-Arrestin-Mediated Signaling of V2 Vasopressin Receptor. Proc. Natl. Acad. Sci. USA 2005, 102, 1448–1453. [Google Scholar] [CrossRef] [Green Version]
- Nobles, K.N.; Xiao, K.; Ahn, S.; Shukla, A.K.; Lam, C.M.; Rajagopal, S.; Strachan, R.T.; Huang, T.-Y.; Bressler, E.A.; Hara, M.R.; et al. Distinct Phosphorylation Sites on the 2-Adrenergic Receptor Establish a Barcode That Encodes Differential Functions of -Arrestin. Sci. Signal. 2011, 4, ra51. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, V.V.; Gurevich, E.V. The Structural Basis of the Arrestin Binding to GPCRs. Mol. Cell. Endocrinol. 2019, 484, 34–41. [Google Scholar] [CrossRef]
- Venkatakrishnan, A.; Flock, T.; Prado, D.E.; Oates, M.E.; Gough, J.; Madan Babu, M. Structured and Disordered Facets of the GPCR Fold. Curr. Opin. Struct. Biol. 2014, 27, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Jaakola, V.-P.; Prilusky, J.; Sussman, J.L.; Goldman, A. G Protein-Coupled Receptors Show Unusual Patterns of Intrinsic Unfolding. Protein Eng. Des. Sel. 2005, 18, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. What Does It Mean to Be Natively Unfolded? Eur. J. Biochem. 2002, 269, 2–12. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically Disordered Proteins in Cellular Signalling and Regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M.; Simon, I.; Friedrich, P.; Tompa, P. Preformed Structural Elements Feature in Partner Recognition by Intrinsically Unstructured Proteins. J. Mol. Biol. 2004, 338, 1015–1026. [Google Scholar] [CrossRef]
- Oakley, R.H.; Laporte, S.A.; Holt, J.A.; Caron, M.G.; Barak, L.S. Differential Affinities of Visual Arrestin, ΒArrestin1, and ΒArrestin2 for G Protein-Coupled Receptors Delineate Two Major Classes of Receptors. J. Biol. Chem. 2000, 275, 17201–17210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyson, H.J.; Wright, P.E. Unfolded Proteins and Protein Folding Studied by NMR. Chem. Rev. 2004, 104, 3607–3622. [Google Scholar] [CrossRef] [PubMed]
- Sibille, N.; Bernadó, P. Structural Characterization of Intrinsically Disordered Proteins by the Combined Use of NMR and SAXS. Biochem. Soc. Trans. 2012, 40, 955–962. [Google Scholar] [CrossRef] [Green Version]
- Delaforge, E.; Cordeiro, T.N.; Bernadó, P.; Sibille, N. Conformational Characterization of Intrinsically Disordered Proteins and Its Biological Significance. In Modern Magnetic Resonance; Webb, G.A., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 381–399. ISBN 978-3-319-28388-3. [Google Scholar]
- Milles, S.; Salvi, N.; Blackledge, M.; Jensen, M.R. Characterization of Intrinsically Disordered Proteins and Their Dynamic Complexes: From in Vitro to Cell-like Environments. Prog. Nucl. Magn. Reson. Spectrosc. 2018, 109, 79–100. [Google Scholar] [CrossRef]
- Shukla, A.K.; Manglik, A.; Kruse, A.C.; Xiao, K.; Reis, R.I.; Tseng, W.-C.; Staus, D.P.; Hilger, D.; Uysal, S.; Huang, L.-Y.; et al. Structure of Active β-Arrestin1 Bound to a G Protein-Coupled Receptor Phosphopeptide. Nature 2013, 497, 137–141. [Google Scholar] [CrossRef] [Green Version]
- Staus, D.P.; Hu, H.; Robertson, M.J.; Kleinhenz, A.L.W.; Wingler, L.M.; Capel, W.D.; Latorraca, N.R.; Lefkowitz, R.J.; Skiniotis, G. Structure of the M2 Muscarinic Receptor–β-Arrestin Complex in a Lipid Nanodisc. Nature 2020, 579, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.H.; Thomsen, A.R.B.; Cahill, T.J.; Huang, R.; Huang, L.-Y.; Marcink, T.; Clarke, O.B.; Heissel, S.; Masoudi, A.; Ben-Hail, D.; et al. Structure of an Endosomal Signaling GPCR–G Protein–β-Arrestin Megacomplex. Nat. Struct. Mol. Biol. 2019, 26, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Obradovic, Z.; Li, X.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence Complexity of Disordered Protein. Proteins 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. PONDR-FIT: A Meta-Predictor of Intrinsically Disordered Amino Acids. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2010, 1804, 996–1010. [Google Scholar] [CrossRef] [Green Version]
- Ishida, T.; Kinoshita, K. PrDOS: Prediction of Disordered Protein Regions from Amino Acid Sequence. Nucleic Acids Res. 2007, 35, W460–W464. [Google Scholar] [CrossRef]
- Hanson, J.; Paliwal, K.; Zhou, Y. Accurate Single-Sequence Prediction of Protein Intrinsic Disorder by an Ensemble of Deep Recurrent and Convolutional Architectures. J. Chem. Inf. Model. 2018, 58, 2369–2376. [Google Scholar] [CrossRef] [Green Version]
- Hanson, K.; Paliwal, K.; Litfin, T.; Zhou, Y. SPOT-Disorder2: Improved Protein Intrinsic Disorder Prediction by Ensembled Deep Learning—ScienceDirect. Available online: https://www.sciencedirect.com/science/article/pii/S1672022920300243 (accessed on 29 June 2021).
- Walsh, I.; Martin, A.J.M.; Di Domenico, T.; Tosatto, S.C.E. ESpritz: Accurate and Fast Prediction of Protein Disorder. Bioinformatics 2012, 28, 503–509. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Sweredoski, M.J.; Baldi, P. Accurate Prediction of Protein Disordered Regions by Mining Protein Structure Data. Data Min. Knowl. Disc. 2005, 11, 213–222. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T.; Cozzetto, D. DISOPRED3: Precise Disordered Region Predictions with Annotated Protein-Binding Activity. Bioinformatics 2015, 31, 857–863. [Google Scholar] [CrossRef]
- Geourjon, C.; Deléage, G. SOPMA: Significant Improvements in Protein Secondary Structure Prediction by Consensus Prediction from Multiple Alignments. Bioinformatics 1995, 11, 681–684. [Google Scholar] [CrossRef]
- Buchan, D.W.A.; Jones, D.T. The PSIPRED Protein Analysis Workbench: 20 Years On. Nucleic Acids Res. 2019, 47, W402–W407. [Google Scholar] [CrossRef] [Green Version]
- Drozdetskiy, A.; Cole, C.; Procter, J.; Barton, G.J. JPred4: A Protein Secondary Structure Prediction Server. Nucleic Acids Res. 2015, 43, W389–W394. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Xu, D.; Yang, J.; Walker, S.; Zhang, Y. A Comparative Assessment and Analysis of 20 Representative Sequence Alignment Methods for Protein Structure Prediction. Sci. Rep. 2013, 3, 2619. [Google Scholar] [CrossRef] [Green Version]
- Hanson, J.; Paliwal, K.; Litfin, T.; Yang, Y.; Zhou, Y. Improving Prediction of Protein Secondary Structure, Backbone Angles, Solvent Accessibility and Contact Numbers by Using Predicted Contact Maps and an Ensemble of Recurrent and Residual Convolutional Neural Networks. Bioinformatics 2019, 35, 2403–2410. [Google Scholar] [CrossRef] [PubMed]
- Heffernan, R.; Yang, Y.; Paliwal, K.; Zhou, Y. Capturing Non-Local Interactions by Long Short-Term Memory Bidirectional Recurrent Neural Networks for Improving Prediction of Protein Secondary Structure, Backbone Angles, Contact Numbers and Solvent Accessibility. Bioinformatics 2017, 33, 2842–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chemes, L.B.; Alonso, L.G.; Noval, M.G.; de Prat-Gay, G. Circular Dichroism Techniques for the Analysis of Intrinsically Disordered Proteins and Domains. In Intrinsically Disordered Protein Analysis: Volume 1, Methods and Experimental Tools; Uversky, V.N., Dunker, A.K., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2012; pp. 387–404. ISBN 978-1-61779-927-3. [Google Scholar]
- Thureau, A.; Roblin, P.; Pérez, J. BioSAXS on the SWING Beamline at Synchrotron SOLEIL. J. Appl. Crystallogr. 2021, 54, 1698–1710. [Google Scholar] [CrossRef]
- Blanchet, C.E.; Svergun, D.I. Small-Angle X-ray Scattering on Biological Macromolecules and Nanocomposites in Solution. Annu. Rev. Phys. Chem. 2013, 64, 37–54. [Google Scholar] [CrossRef]
- Manalastas-Cantos, K.; Konarev, P.V.; Hajizadeh, N.R.; Kikhney, A.G.; Petoukhov, M.V.; Molodenskiy, D.S.; Panjkovich, A.; Mertens, H.D.T.; Gruzinov, A.; Borges, C.; et al. ATSAS 3.0: Expanded Functionality and New Tools for Small-Angle Scattering Data Analysis. J. Appl. Crystallogr. 2021, 54, 343–355. [Google Scholar] [CrossRef]
- Wishart, D.S.; Sykes, B.D. [12] Chemical Shifts as a Tool for Structure Determination. In Methods in Enzymology; Elsevier: New York, NY, USA, 1994; Volume 239, pp. 363–392. ISBN 978-0-12-182140-1. [Google Scholar]
- Lee, W.; Tonelli, M.; Markley, J.L. NMRFAM-SPARKY: Enhanced Software for Biomolecular NMR Spectroscopy. Bioinformatics 2015, 31, 1325–1327. [Google Scholar] [CrossRef] [Green Version]
- Tamiola, K.; Acar, B.; Mulder, F.A.A. Sequence-Specific Random Coil Chemical Shifts of Intrinsically Disordered Proteins. J. Am. Chem. Soc. 2010, 132, 18000–18003. [Google Scholar] [CrossRef]
- Nielsen, J.T.; Mulder, F.A.A. POTENCI: Prediction of Temperature, Neighbor and PH-Corrected Chemical Shifts for Intrinsically Disordered Proteins. J. Biomol. NMR 2018, 70, 141–165. [Google Scholar] [CrossRef] [PubMed]
- Vuister, G.W.; Bax, A. Quantitative J Correlation: A New Approach for Measuring Homonuclear Three-Bond J(HNH.Alpha.) Coupling Constants in 15N-Enriched Proteins. J. Am. Chem. Soc. 1993, 115, 7772–7777. [Google Scholar] [CrossRef]
- Shen, Y.; Roche, J.; Grishaev, A.; Bax, A. Prediction of Nearest Neighbor Effects on Backbone Torsion Angles and NMR Scalar Coupling Constants in Disordered Proteins. Protein Sci. 2018, 27, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Ottiger, M.; Delaglio, F.; Bax, A. Measurement OfJand Dipolar Couplings from Simplified Two-Dimensional NMR Spectra. J. Magn. Reson. 1998, 131, 373–378. [Google Scholar] [CrossRef] [Green Version]
- Rückert, M.; Otting, G. Alignment of Biological Macromolecules in Novel Nonionic Liquid Crystalline Media for NMR Experiments. J. Am. Chem. Soc. 2000, 122, 7793–7797. [Google Scholar] [CrossRef]
- Hansen, M.R.; Mueller, L.; Pardi, A. Tunable Alignment of Macromolecules by Filamentous Phage Yields Dipolar Coupling Interactions. Nat. Struct. Biol. 1998, 5, 1065–1074. [Google Scholar] [CrossRef]
- Teilum, K.; Kragelund, B.B.; Poulsen, F.M. Transient Structure Formation in Unfolded Acyl-Coenzyme A-Binding Protein Observed by Site-Directed Spin Labelling. J. Mol. Biol. 2002, 324, 349–357. [Google Scholar] [CrossRef]
- Ozenne, V.; Bauer, F.; Salmon, L.; Huang, J.R.; Jensen, M.R.; Segard, S.; Bernado, P.; Charavay, C.; Blackledge, M. Flexible-meccano: A Tool for the Generation of Explicit Ensemble Descriptions of Intrinsically Disordered Proteins and Their Associated Experimental Observables. Bioinformatics 2012, 28, 1463–1470. [Google Scholar] [CrossRef]
- Senicourt, L.; Le Maire, A.; Allemand, F.; Carvalho, J.E.; Guee, L.; Germain, P.; Schubert, M.; Bernadó, P.; Bourguet, W.; Sibille, N. Structural Insights into the Interaction of the Intrinsically Disordered Co-Activator TIF2 with Retinoic Acid Receptor Heterodimer (RXR/RAR). J. Mol. Biol. 2021, 433, 166899. [Google Scholar] [CrossRef]
- Tovo-Rodrigues, L.; Roux, A.; Hutz, M.H.; Rohde, L.A.; Woods, A.S. Functional Characterization of G-Protein-Coupled Receptors: A Bioinformatics Approach. Neuroscience 2014, 277, 764–779. [Google Scholar] [CrossRef] [Green Version]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically Disordered Protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why Are “Natively Unfolded” Proteins Unstructured under Physiologic Conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Uversky, V.N. Size-Exclusion Chromatography in Structural Analysis of Intrinsically Disordered Proteins. In Intrinsically Disordered Protein Analysis: Volume 2, Methods and Experimental Tools; Uversky, V.N., Dunker, A.K., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2012; pp. 179–194. ISBN 978-1-4614-3704-8. [Google Scholar]
- Woody, R.W. Circular Dichroism of Intrinsically Disordered Proteins. In Instrumental Analysis of Intrinsically Disordered Proteins; John Wiley & Sons, Ltd.: New York, NY, USA, 2010; pp. 303–321. ISBN 978-0-470-60261-4. [Google Scholar]
- Woody, R.W. Theory of Circular Dichroism of Proteins. In Circular Dichroism and the Conformational Analysis of Biomolecules; Fasman, G.D., Ed.; Springer: Boston, MA, USA, 1996; pp. 25–67. ISBN 978-1-4757-2508-7. [Google Scholar]
- Bernadó, P.; Svergun, D.I. Structural Analysis of Intrinsically Disordered Proteins by Small-Angle X-Ray Scattering. Mol. BioSyst. 2011, 8, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, T.N.; Herranz-Trillo, F.; Urbanek, A.; Estaña, A.; Cortés, J.; Sibille, N.; Bernadó, P. Small-Angle Scattering Studies of Intrinsically Disordered Proteins and Their Complexes. Curr. Opin. Struct. Biol. 2017, 42, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, I.; Ueda, T.; Kofuku, Y.; Eddy, M.T.; Wüthrich, K. GPCR Drug Discovery: Integrating Solution NMR Data with Crystal and Cryo-EM Structures. Nat. Rev. Drug Discov. 2019, 18, 59–82. [Google Scholar] [CrossRef]
- Wishart, D.S.; Sykes, B.D. The 13C Chemical-Shift Index: A Simple Method for the Identification of Protein Secondary Structure Using 13C Chemical-Shift Data. J. Biomol. NMR 1994, 4, 171–180. [Google Scholar] [CrossRef]
- Bax, A.; Kontaxis, G.; Tjandra, N. Dipolar Couplings in Macromolecular Structure Determination. In Methods in Enzymology; Elsevier: New York, NY, USA, 2001; Volume 339, pp. 127–174. ISBN 978-0-12-182240-8. [Google Scholar]
- Bernadó, P.; Blanchard, L.; Timmins, P.; Marion, D.; Ruigrok, R.W.H.; Blackledge, M. A Structural Model for Unfolded Proteins from Residual Dipolar Couplings and Small-Angle X-ray Scattering. Proc. Natl. Acad. Sci. USA 2005, 102, 17002–17007. [Google Scholar] [CrossRef] [Green Version]
- Bernadó, P.; Bertoncini, C.W.; Griesinger, C.; Zweckstetter, M.; Blackledge, M. Defining Long-Range Order and Local Disorder in Native α-Synuclein Using Residual Dipolar Couplings. J. Am. Chem. Soc. 2005, 127, 17968–17969. [Google Scholar] [CrossRef]
- Salmon, L.; Nodet, G.; Ozenne, V.; Yin, G.; Jensen, M.R.; Zweckstetter, M.; Blackledge, M. NMR Characterization of Long-Range Order in Intrinsically Disordered Proteins. J. Am. Chem. Soc. 2010, 132, 8407–8418. [Google Scholar] [CrossRef] [Green Version]
- Guillien, M.; le Maire, A.; Mouhand, A.; Bernadó, P.; Bourguet, W.; Banères, J.-L.; Sibille, N. IDPs and Their Complexes in GPCR and Nuclear Receptor Signaling. In Progress in Molecular Biology and Translational Science; Elsevier: New York, NY, USA, 2020; Volume 174, pp. 105–155. ISBN 978-0-12-822615-5. [Google Scholar]
- Appadurai, R.; Uversky, V.N.; Srivastava, A. The Structural and Functional Diversity of Intrinsically Disordered Regions in Transmembrane Proteins. J. Membr. Biol. 2019, 252, 273–292. [Google Scholar] [CrossRef]
- Ahmad, R.; Lahuna, O.; Sidibe, A.; Daulat, A.; Zhang, Q.; Luka, M.; Guillaume, J.-L.; Gallet, S.; Guillonneau, F.; Hamroune, J.; et al. GPR50-Ctail Cleavage and Nuclear Translocation: A New Signal Transduction Mode for G Protein-Coupled Receptors. Cell. Mol. Life Sci. 2020, 77, 5189–5205. [Google Scholar] [CrossRef] [PubMed]
- Dufourny, L.; Levasseur, A.; Migaud, M.; Callebaut, I.; Pontarotti, P.; Malpaux, B.; Monget, P. GPR50 Is the Mammalian Ortholog of Mel1c: Evidence of Rapid Evolution in Mammals. BMC Evol. Biol. 2008, 8, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouzo-Lorenzo, M.; Santo-Zas, I.; Lodeiro, M.; Nogueiras, R.; Casanueva, F.F.; Castro, M.; Pazos, Y.; Tobin, A.B.; Butcher, A.J.; Camiña, J.P. Distinct Phosphorylation Sites on the Ghrelin Receptor, GHSR1a, Establish a Code That Determines the Functions of ß-Arrestins. Sci. Rep. 2016, 6, 22495. [Google Scholar] [CrossRef] [PubMed]
- Nobles, K.N.; Guan, Z.; Xiao, K.; Oas, T.G.; Lefkowitz, R.J. The Active Conformation of Beta-Arrestin1: Direct Evidence for the Phosphate Sensor in the N-Domain and Conformational Differences in the Active States of Beta-Arrestins1 and -2. J. Biol. Chem. 2007, 282, 21370–21381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Warne, T.; Nehmé, R.; Pandey, S.; Dwivedi-Agnihotri, H.; Chaturvedi, M.; Edwards, P.C.; García-Nafría, J.; Leslie, A.G.W.; Shukla, A.K.; et al. Molecular Basis of β-Arrestin Coupling to Formoterol-Bound β 1-Adrenoceptor. Nature 2020, 583, 862–866. [Google Scholar] [CrossRef] [PubMed]
- Tompa, P.; Schad, E.; Tantos, A.; Kalmar, L. Intrinsically Disordered Proteins: Emerging Interaction Specialists. Curr. Opin. Struct. Biol. 2015, 35, 49–59. [Google Scholar] [CrossRef]
- Mayer, D.; Damberger, F.F.; Samarasimhareddy, M.; Feldmueller, M.; Vuckovic, Z.; Flock, T.; Bauer, B.; Mutt, E.; Zosel, F.; Allain, F.H.T.; et al. Distinct G Protein-Coupled Receptor Phosphorylation Motifs Modulate Arrestin Affinity and Activation and Global Conformation. Nat. Commun. 2019, 10, 1261. [Google Scholar] [CrossRef]
- Bah, A.; Vernon, R.M.; Siddiqui, Z.; Krzeminski, M.; Muhandiram, R.; Zhao, C.; Sonenberg, N.; Kay, L.E.; Forman-Kay, J.D. Folding of an Intrinsically Disordered Protein by Phosphorylation as a Regulatory Switch. Nature 2015, 519, 106–109. [Google Scholar] [CrossRef]
- Cordeiro, T.N.; Herranz-Trillo, F.; Urbanek, A.; Estaña, A.; Cortés, J.; Sibille, N.; Bernadó, P. Structural Characterization of Highly Flexible Proteins by Small-Angle Scattering. Adv. Exp. Med. Biol. 2017, 1009, 107–129. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guillien, M.; Mouhand, A.; Fournet, A.; Gontier, A.; Martí Navia, A.; Cordeiro, T.N.; Allemand, F.; Thureau, A.; Banères, J.-L.; Bernadó, P.; et al. Structural Insights into the Intrinsically Disordered GPCR C-Terminal Region, Major Actor in Arrestin-GPCR Interaction. Biomolecules 2022, 12, 617. https://doi.org/10.3390/biom12050617
Guillien M, Mouhand A, Fournet A, Gontier A, Martí Navia A, Cordeiro TN, Allemand F, Thureau A, Banères J-L, Bernadó P, et al. Structural Insights into the Intrinsically Disordered GPCR C-Terminal Region, Major Actor in Arrestin-GPCR Interaction. Biomolecules. 2022; 12(5):617. https://doi.org/10.3390/biom12050617
Chicago/Turabian StyleGuillien, Myriam, Assia Mouhand, Aurélie Fournet, Amandine Gontier, Aleix Martí Navia, Tiago N. Cordeiro, Frédéric Allemand, Aurélien Thureau, Jean-Louis Banères, Pau Bernadó, and et al. 2022. "Structural Insights into the Intrinsically Disordered GPCR C-Terminal Region, Major Actor in Arrestin-GPCR Interaction" Biomolecules 12, no. 5: 617. https://doi.org/10.3390/biom12050617