
ADP-Induced Conformational Transition of Human Adenylate Kinase 1 Is Triggered by Suppressing Internal Motion of α3α4 and α7α8 Fragments on the ps-ns Timescale

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Recombinant hAK1 and Its Mutants
2.2. Structure Calculation
2.3. NMR Chemical Shift Perturbation Experiments
2.4. Enzymatic Activity Assay
2.5. Backbone Relaxation Measurements
2.6. Model-Free Analysis
2.7. Reduced Spectral Density Mapping
3. Results
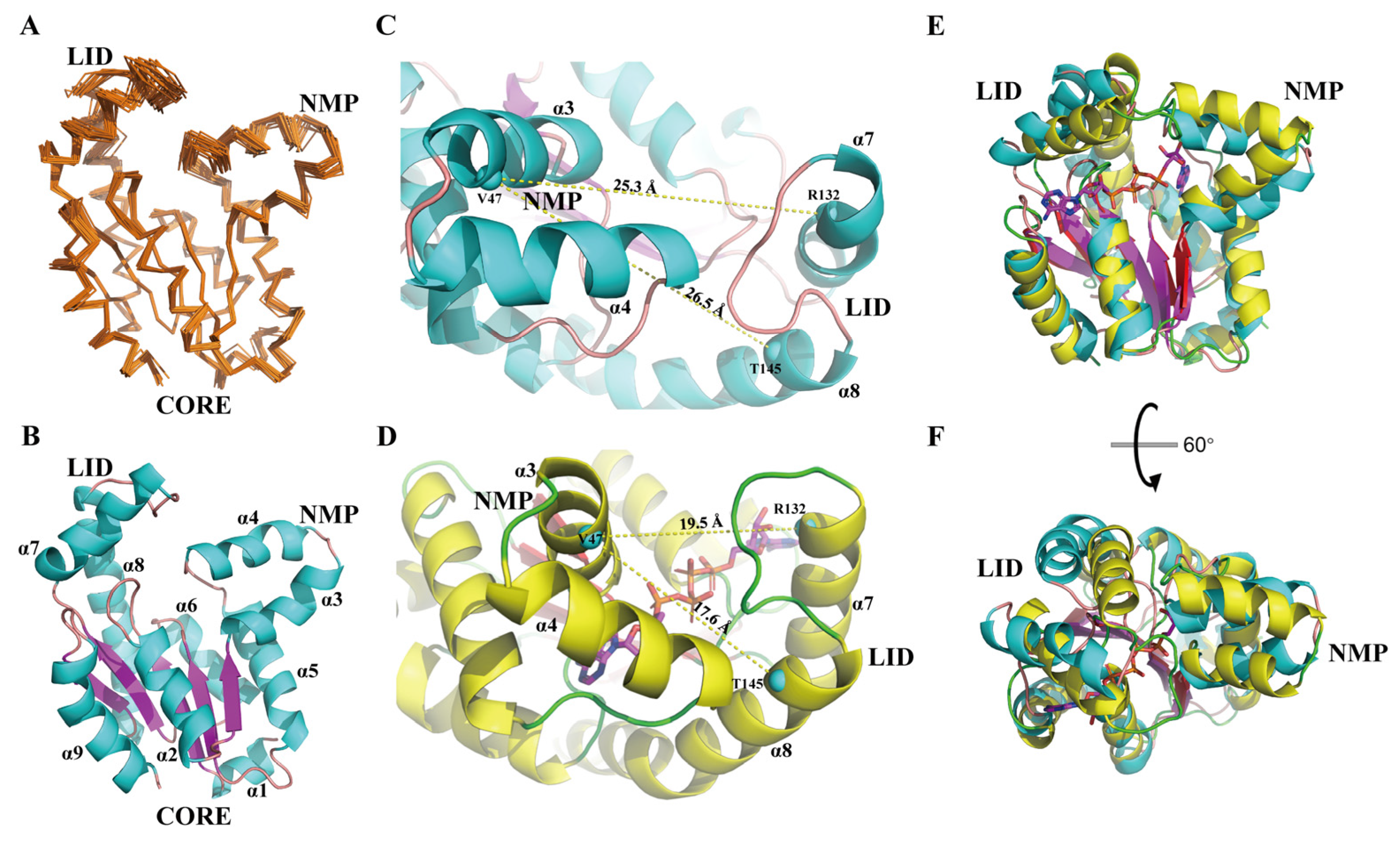
3.1. NMR Solution Structure of hAK1
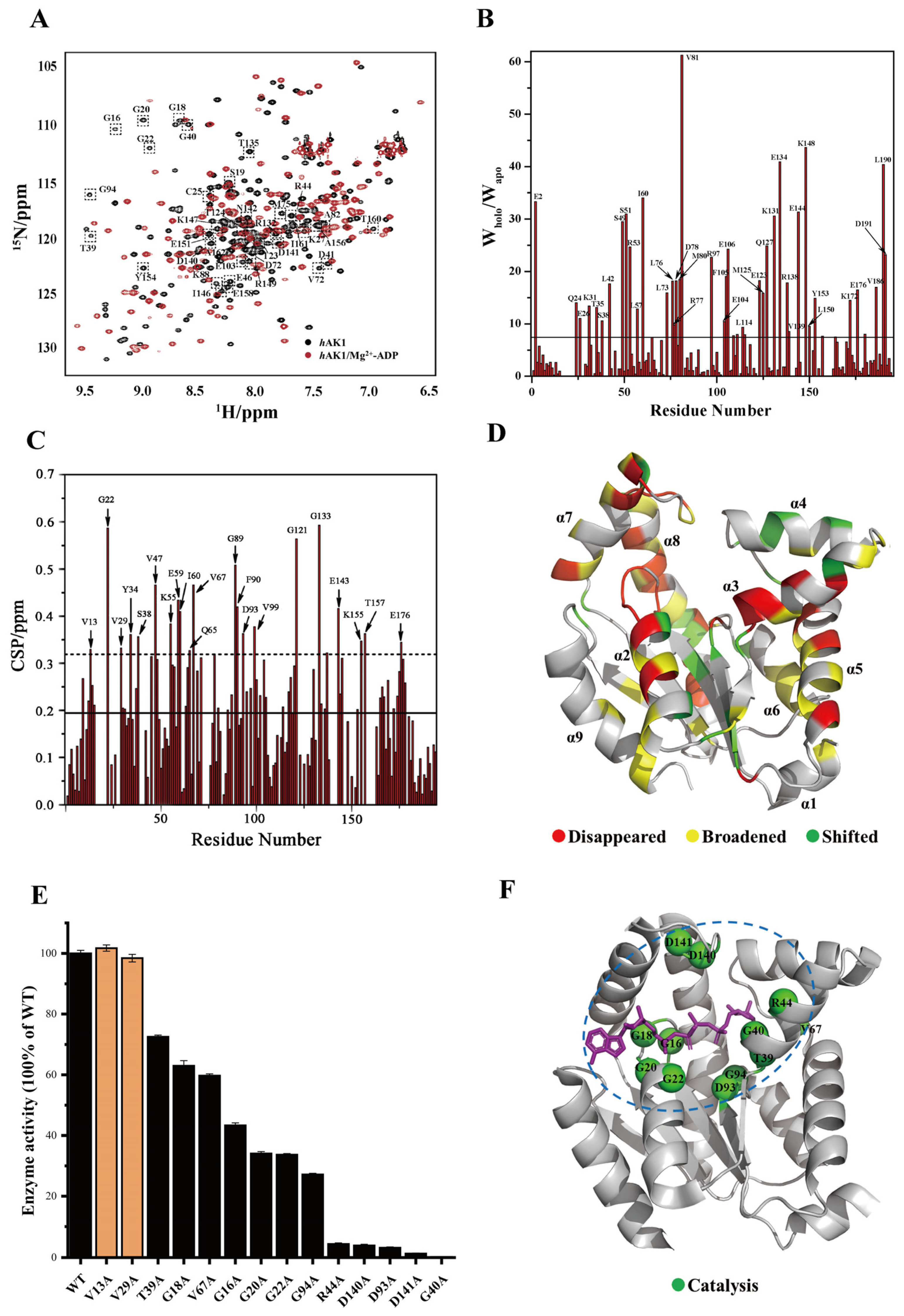
3.2. Key Residues for hAK1 Catalyzing ADP
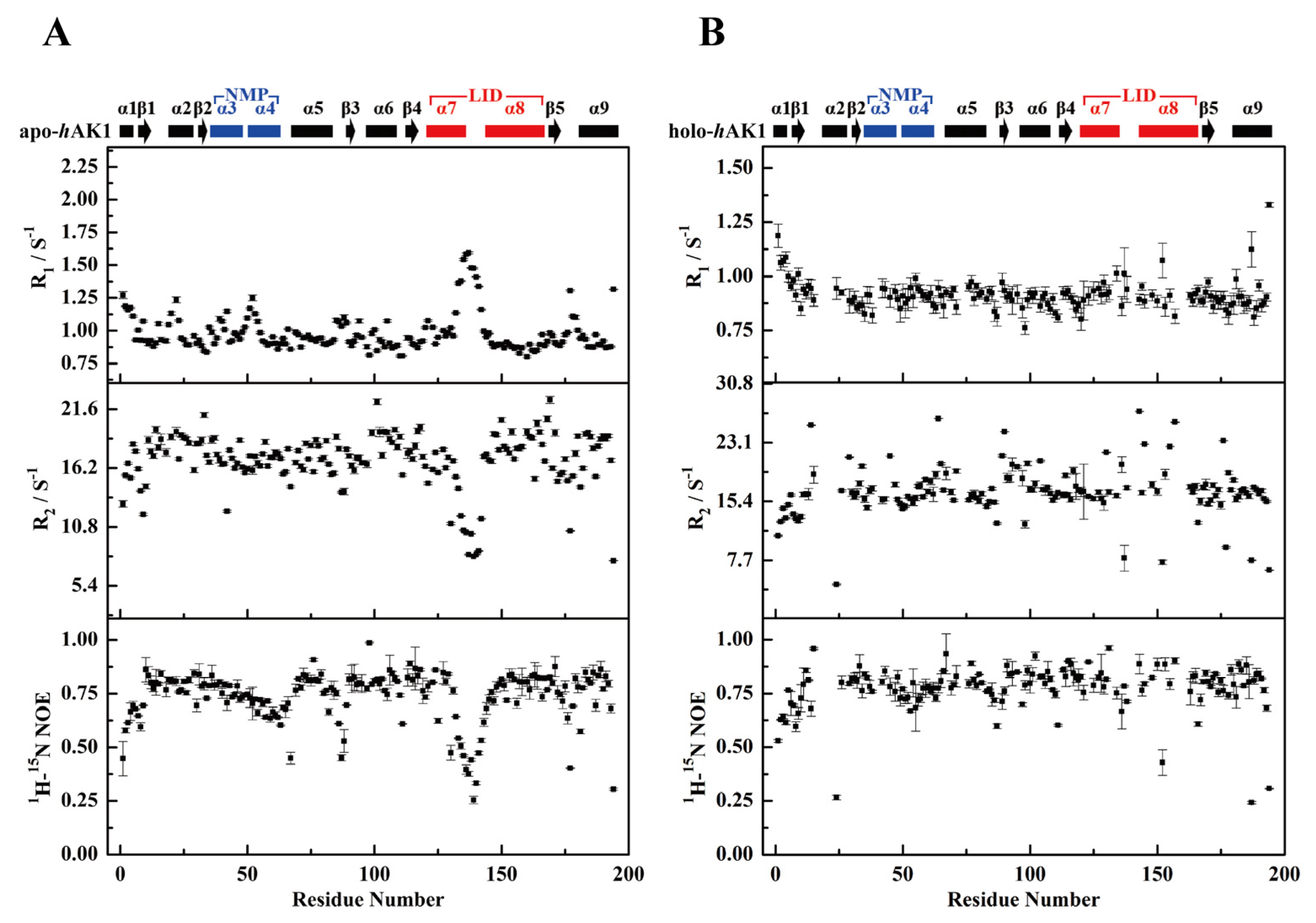
3.3. Comparison of Relaxation Data between apo-hAK1 and holo-hAK1
3.4. Model-Free Analysis
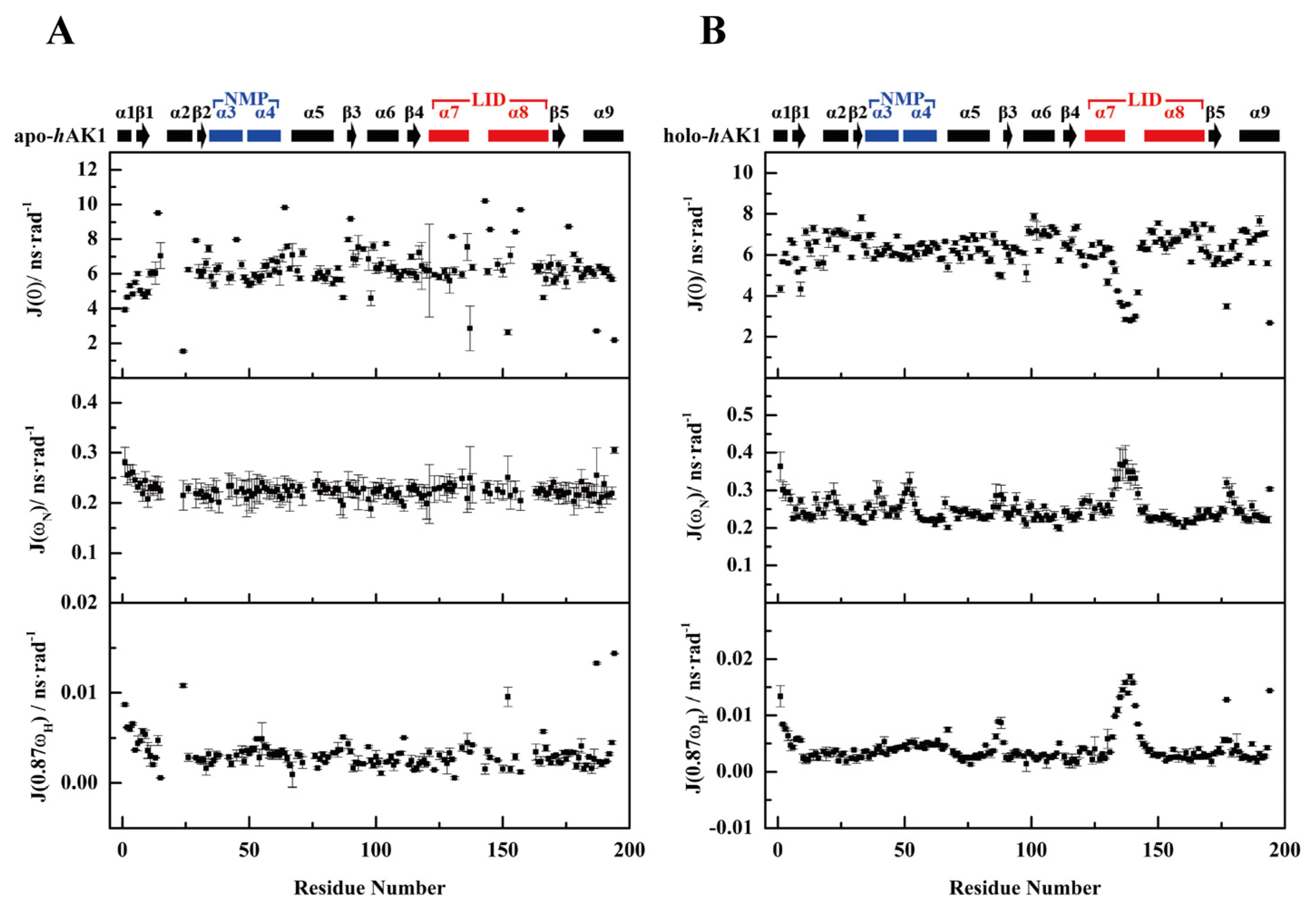
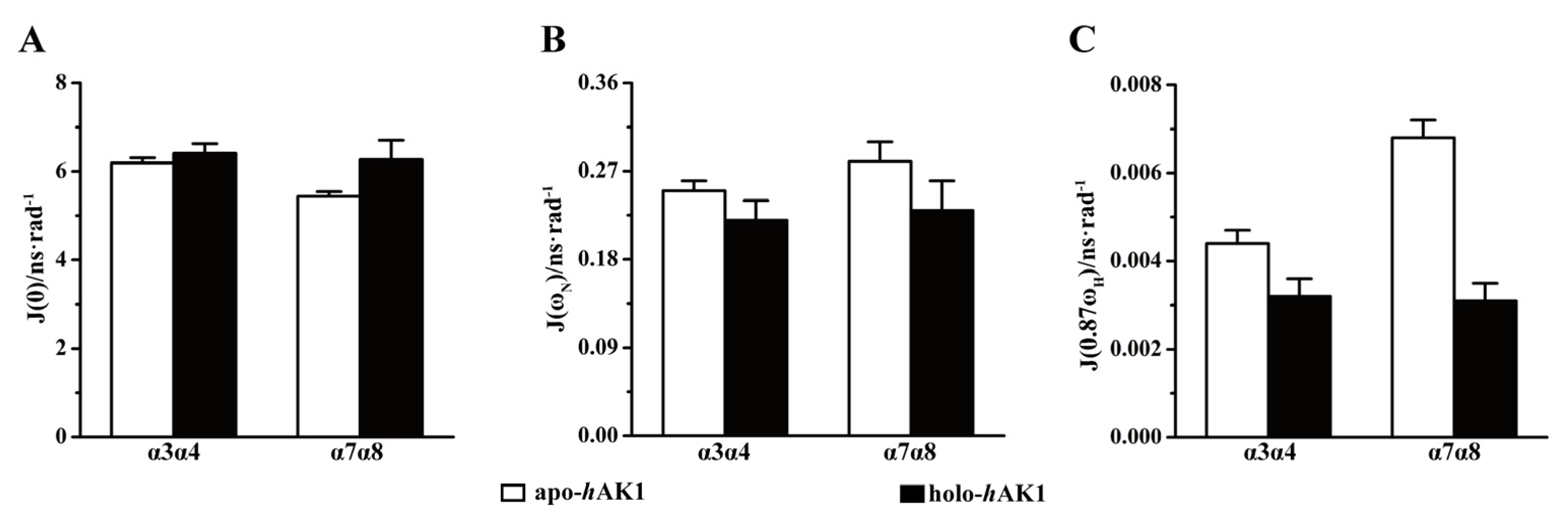
3.5. Reduced Spectral Density Mapping
4. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Downs, S.M.; Hudson, E.R.; Hardie, D.G. A Potential Role for AMP-Activated Protein Kinase in Meiotic Induction in Mouse Oocytes. Dev. Biol. 2002, 245, 200–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Koh, H.; Kim, M.; Kim, Y.; Lee, S.Y.; Karess, R.E.; Lee, S.-H.; Shong, M.; Kim, J.-M.; Kim, J.; et al. Energy-Dependent Regulation of Cell Structure by AMP-Activated Protein Kinase. Nature 2007, 447, 1017–1020. [Google Scholar] [CrossRef] [PubMed]
- Brenman, J.E. AMPK/LKB1 Signaling in Epithelial Cell Polarity and Cell Division. Cell Cycle 2007, 6, 2755–2759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zwerschke, W.; Mazurek, S.; Stöckl, P.; Hütter, E.; Eigenbrodt, E.; Jansen-Dürr, P. Metabolic Analysis of Senescent Human Fibroblasts Reveals a Role for AMP in Cellular Senescence. Biochem. J. 2003, 376, 403–411. [Google Scholar] [CrossRef] [Green Version]
- Panayiotou, C.; Solaroli, N.; Karlsson, A. The Many Isoforms of Human Adenylate Kinases. Int. J. Biochem. Cell. B 2014, 49, 75–83. [Google Scholar] [CrossRef]
- Dzeja, P.P.; Zeleznikar, R.J.; Goldberg, N.D. Adenylate Kinase: Kinetic Behavior in Intact Cells Indicates It Is Integral to Multiple Cellular Processes. Mol. Cell. Biochem. 1998, 184, 169–182. [Google Scholar] [CrossRef]
- Shapiro, Y.E.; Meirovitch, E. Activation Energy of Catalysis-Related Domain Motion in E. Coli Adenylate Kinase. J. Phys. Chem. B 2006, 110, 11519–11524. [Google Scholar] [CrossRef]
- Reinstein, J.; Vetter, I.R.; Schlichting, I.; Roesch, P.; Wittinghofer, A.; Goody, R.S. Fluorescence and NMR Investigations on the Ligand Binding Properties of Adenylate Kinases. Biochemistry 1990, 29, 7440–7450. [Google Scholar] [CrossRef]
- Müller, C.W.; Schulz, G.E. Structure of the Complex between Adenylate Kinase from Escherichia Coli and the Inhibitor Ap5A Refined at 1.9 Å Resolution: A Model for a Catalytic Transition State. J. Mol. Biol. 1992, 224, 159–177. [Google Scholar] [CrossRef]
- Müller, C.; Schlauderer, G.; Reinstein, J.; Schulz, G. Adenylate Kinase Motions during Catalysis: An Energetic Counterweight Balancing Substrate Binding. Structure 1996, 4, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Pelz, B.; Žoldák, G.; Zeller, F.; Zacharias, M.; Rief, M. Subnanometre Enzyme Mechanics Probed by Single-Molecule Force Spectroscopy. Nat. Commun. 2016, 7, 10848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miron, S.; Munier-Lehmann, H.; Craescu, C.T. Structural and Dynamic Studies on Ligand-Free Adenylate Kinase from Mycobacterium Tuberculosis Revealed a Closed Conformation that Can Be Related to the Reduced Catalytic Activity. Biochemistry 2004, 43, 67–77. [Google Scholar] [CrossRef]
- Bellinzoni, M.; Haouz, A.; Graña, M.; Munier-Lehmann, H.; Shepard, W.; Alzari, P.M. The Crystal Structure of Mycobacterium Tuberculosis Adenylate Kinase in Complex with Two Molecules of ADP and Mg2+ Supports an Associative Mechanism for Phosphoryl Transfer. Protein Sci. 2006, 15, 1489–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, C.; Peng, Y.; Liao, X.; Guo, C.; Lin, D. 1H, 13C, 15N Backbone and Side-Chain Resonance Assignments of the Human Adenylate Kinase 1 in Apo Form. Biomol. NMR Assign. 2013, 7, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Rieping, W.; Habeck, M.; Bardiaux, B.; Bernard, A.; Malliavin, T.E.; Nilges, M. ARIA2: Automated NOE Assignment and Data Integration in NMR Structure Calculation. Bioinformatics 2007, 23, 381–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Bax, A. Protein Backbone and Sidechain Torsion Angles Predicted from NMR Chemical Shifts Using Artificial Neural Networks. J. Biomol. NMR 2013, 56, 227–241. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A Program to Check the Stereochemical Quality of Protein Structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Borglund, E.; Brolin, S.E.; Ågren, A. Fluorometric Microassays of Adenylate Kinase, an Enzyme Important in Energy Metabolism. Ups. J. Med. Sci. 1978, 83, 81–84. [Google Scholar] [CrossRef] [Green Version]
- Lipari, G.; Szabo, A. Model-Free Approach to the Interpretation of Nuclear Magnetic Resonance Relaxation in Macromolecules. 1. Theory and Range of Validity. J. Am. Chem. Soc. 1982, 104, 4546–4559. [Google Scholar] [CrossRef]
- Lipari, G.; Szabo, A. Model-Free Approach to the Interpretation of Nuclear Magnetic Resonance Relaxation in Macromolecules. 2. Analysis of Experimental Results. J. Am. Chem. Soc. 1982, 104, 4559–4570. [Google Scholar] [CrossRef]
- Dosset, P.; Hus, J.-C.; Blackledge, M.; Marion, D. Efficient Analysis of Macromolecular Rotational Diffusion from Heteronuclear Relaxation Data. J. Biomol. NMR 2000, 16, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Cole, R.; Loria, J.P. FAST-Modelfree: A Program for Rapid Automated Analysis of Solution NMR Spin-Relaxation Data. J. Biomol. NMR 2003, 26, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Farrow, N.A.; Zhang, O.; Szabo, A.; Torchia, D.A.; Kay, L.E. Spectral Density Function Mapping Using 15N Relaxation Data Exclusively. J. Biomol. NMR 1995, 6, 153–162. [Google Scholar] [CrossRef]
- Spyracopoulos, L. A Suite of Mathematica Notebooks for the Analysis of Protein Main Chain 15N NMR Relaxation Data. J. Biomol. NMR 2006, 36, 215. [Google Scholar] [CrossRef]
- Cui, D.-C.; Ren, W.-T.; Li, W.-F.; Wang, W. Metadynamics Simulations of Mg2+ Transfer in the Late Stage of the Adenylate Kinase Catalytic Cycle. Acta Phys.-Chim. Sin. 2016, 32, 429–435. [Google Scholar] [CrossRef]
- Onuk, E.; Badger, J.; Wang, Y.J.; Bardhan, J.; Chishti, Y.; Akcakaya, M.; Brooks, D.H.; Erdogmus, D.; Minh, D.D.L.; Makowski, L. Effects of Catalytic Action and Ligand Binding on Conformational Ensembles of Adenylate Kinase. Biochemistry 2017, 56, 4559–4567. [Google Scholar] [CrossRef]
- Lescop, E.; Lu, Z.; Liu, Q.; Xu, H.; Li, G.; Xia, B.; Yan, H.; Jin, C. Dynamics of the Conformational Transitions in the Assembling of the Michaelis Complex of a Bisubstrate Enzyme: A 15N Relaxation Study of Escherichia Coli 6-Hydroxymethyl-7,8-Dihydropterin Pyrophosphokinase. Biochemistry 2009, 48, 302–312. [Google Scholar] [CrossRef]
- Song, H.D.; Zhu, F. Conformational Dynamics of a Ligand-Free Adenylate Kinase. PLoS ONE 2013, 8, e68023. [Google Scholar] [CrossRef] [Green Version]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [Green Version]
- Pawnikar, S.; Bhattarai, A.; Wang, J.; Miao, Y. Binding Analysis Using Accelerated Molecular Dynamics Simulations and Future Perspectives. AABC 2022, 15, 1–19. [Google Scholar] [CrossRef]
- Kerns, S.J.; Agafonov, R.V.; Cho, Y.-J.; Pontiggia, F.; Otten, R.; Pachov, D.V.; Kutter, S.; Phung, L.A.; Murphy, P.N.; Thai, V.; et al. The Energy Landscape of Adenylate Kinase during Catalysis. Nat. Struct. Mol. Biol. 2015, 22, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Henzler-Wildman, K.A.; Thai, V.; Lei, M.; Ott, M.; Wolf-Watz, M.; Fenn, T.; Pozharski, E.; Wilson, M.A.; Petsko, G.A.; Karplus, M.; et al. Intrinsic Motions along an Enzymatic Reaction Trajectory. Nature 2007, 450, 838–844. [Google Scholar] [CrossRef] [PubMed]
- Henzler-Wildman, K.A.; Lei, M.; Thai, V.; Kerns, S.J.; Karplus, M.; Kern, D. A Hierarchy of Timescales in Protein Dynamics Is Linked to Enzyme Catalysis. Nature 2007, 450, 913–916. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NMR Distance and Dihedral Constraints | Values |
|---|---|
| Total unambiguous distance restraints | 2603 |
| Intra-residual | 884 |
| Sequential (|i–j| = 1) | 728 |
| Short-range (2 ≤ |i–j| ≤ 3) | 388 |
| Medium-range (4 ≤ |i–j| ≤ 5) | 144 |
| Long-range (|i–j| > 5) | 459 |
| Total ambiguous distance restraints | 402 |
| Total dihedral angle restraints | 350 |
| φ (backbone dihedral angle Ci-1–Ni–Ci,α–Ci) | 175 |
| ψ (backbone dihedral angle Ni–Ci,α–Ci–Ni+1) | 175 |
| Structural statistics | |
| Mean restraint violations | |
| Distance restraint violations (>0.5 Å) | 0.90 |
| Dihedral restraint violations (>5°) | 1.35 |
| Average RMSD (Å) to mean structure (all residues) | |
| Backbone RMSD | 0.57 ± 0.09 |
| Heavy atoms RMSD | 0.84 ± 0.06 |
| Ramachandran plot statistics 1 | |
| Residues in favored regions | 89.2% ± 1.5% |
| Residues in allowed regions | 10.6% ± 1.7% |
| Residues in generously allowed regions | 0.2% ± 0.3% |
| Residues in disallowed regions | 0% ± 0% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, C.; Zhang, H.; Lin, W.; Chen, H.; Chang, T.; Wu, Z.; Yu, J.; Lin, D. ADP-Induced Conformational Transition of Human Adenylate Kinase 1 Is Triggered by Suppressing Internal Motion of α3α4 and α7α8 Fragments on the ps-ns Timescale. Biomolecules 2022, 12, 671. https://doi.org/10.3390/biom12050671
Guo C, Zhang H, Lin W, Chen H, Chang T, Wu Z, Yu J, Lin D. ADP-Induced Conformational Transition of Human Adenylate Kinase 1 Is Triggered by Suppressing Internal Motion of α3α4 and α7α8 Fragments on the ps-ns Timescale. Biomolecules. 2022; 12(5):671. https://doi.org/10.3390/biom12050671
Chicago/Turabian StyleGuo, Chenyun, Haoran Zhang, Weiliang Lin, Hanyu Chen, Ting Chang, Zhihua Wu, Jiaxin Yu, and Donghai Lin. 2022. "ADP-Induced Conformational Transition of Human Adenylate Kinase 1 Is Triggered by Suppressing Internal Motion of α3α4 and α7α8 Fragments on the ps-ns Timescale" Biomolecules 12, no. 5: 671. https://doi.org/10.3390/biom12050671