
Comparative Transcriptome Analysis of Agrobacterium tumefaciens Reveals the Molecular Basis for the Recalcitrant Genetic Transformation of Camellia sinensis L.



 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
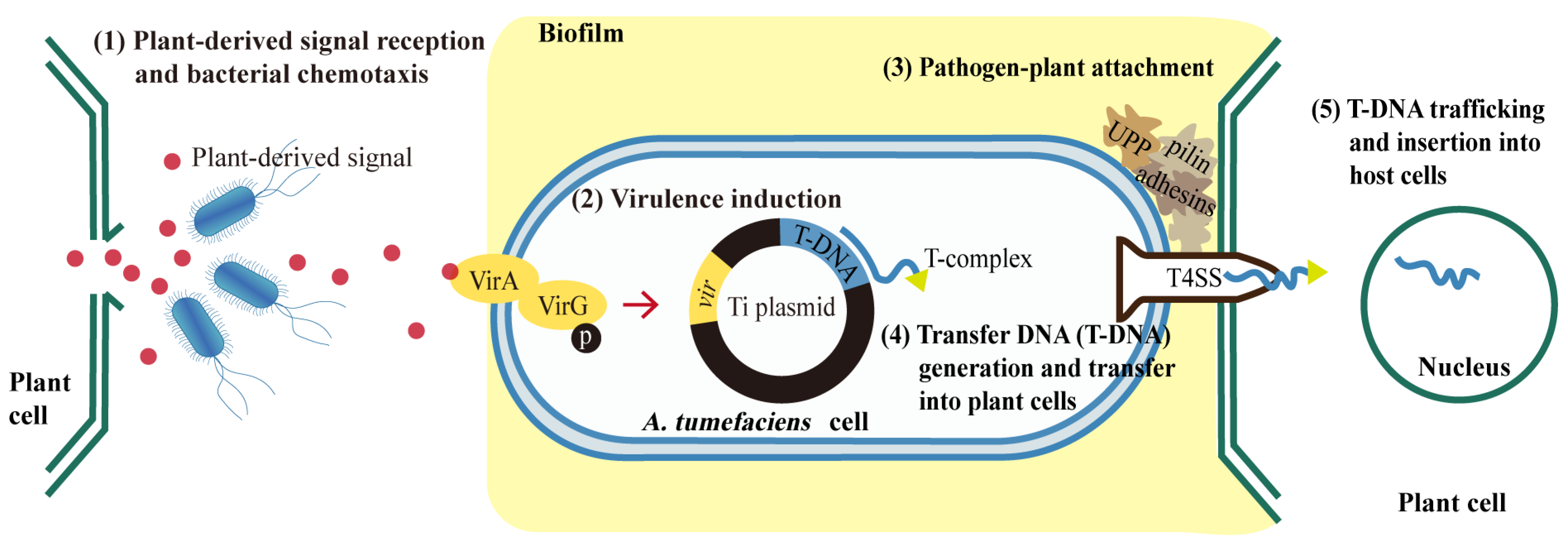
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Bacterial Culture
2.3. Agrobacterium-Mediated Transformation (AMT)
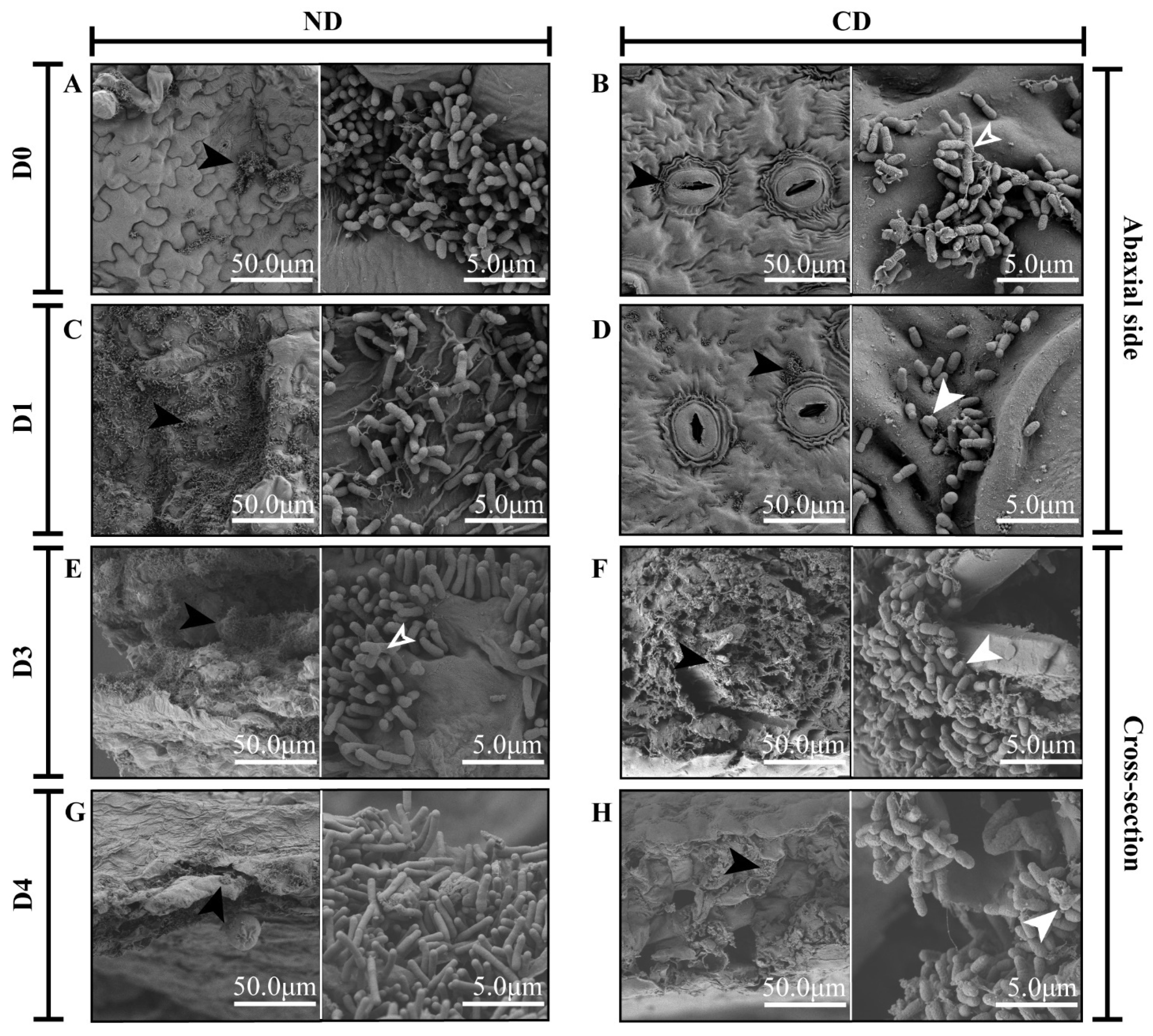
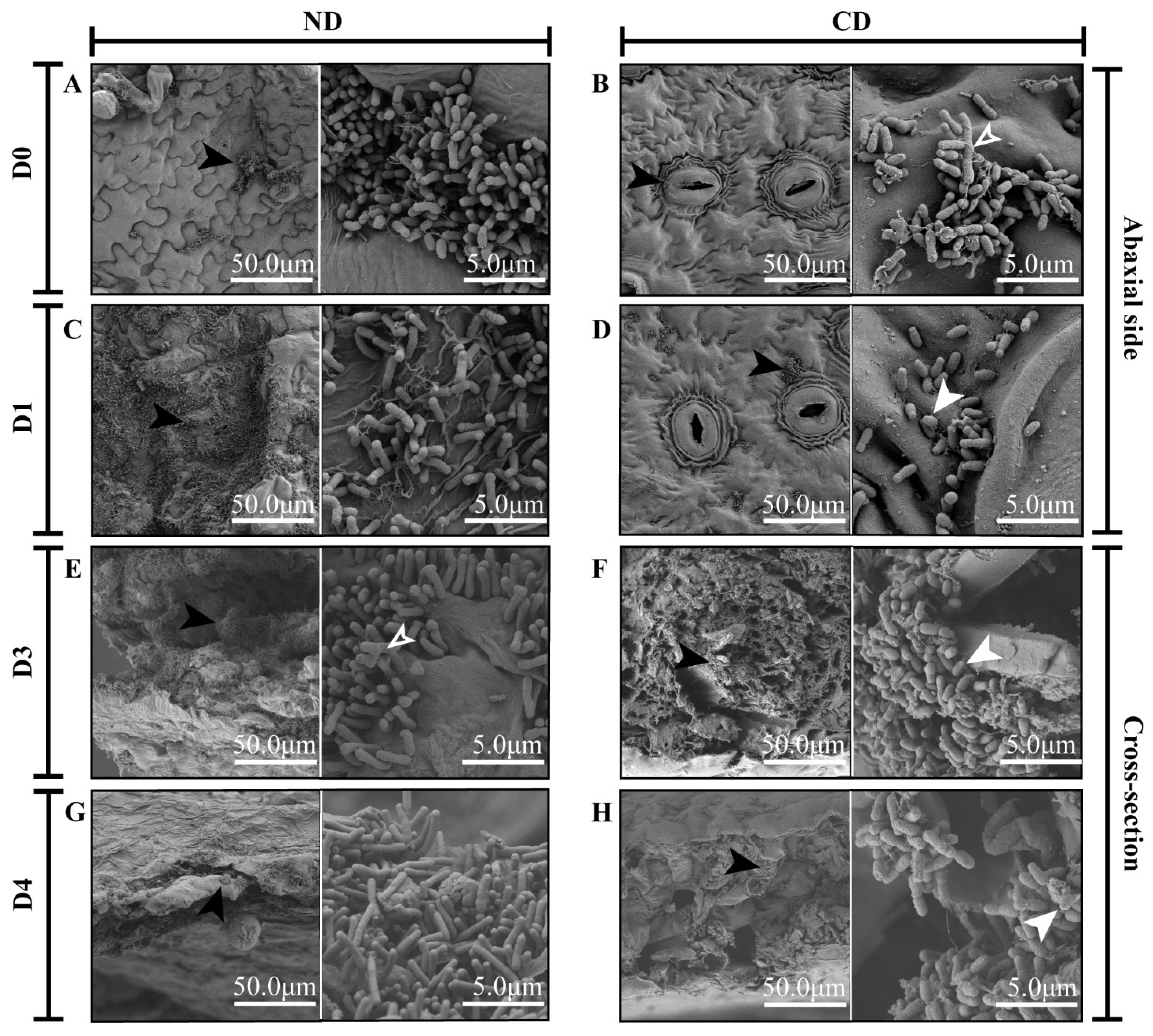
2.4. Scanning Electron Microscopy Observations
2.5. Transcriptomic Sequencing and Analysis
2.6. Quantitative Reverse Transcription-PCR (qRT-PCR) Verification
2.7. Statistical Analysis
3. Results
3.1. Growth and Morphological Changes in Agrobacterium during Transformation
3.2. Transcriptomic Analysis of Agrobacterium during Genetic Transformation
3.3. Gene Ontology Enrichment Analysis
3.4. Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analysis
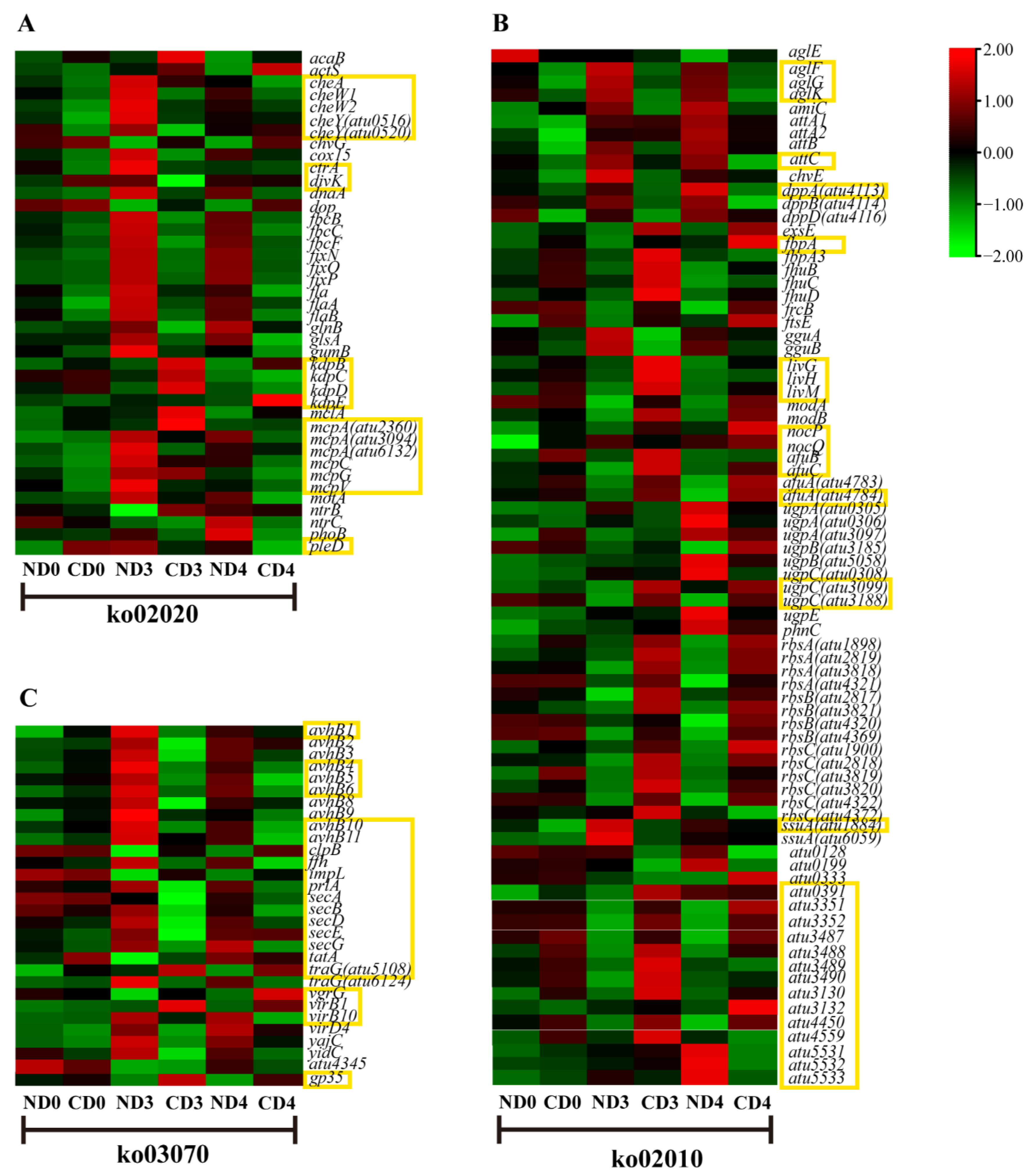
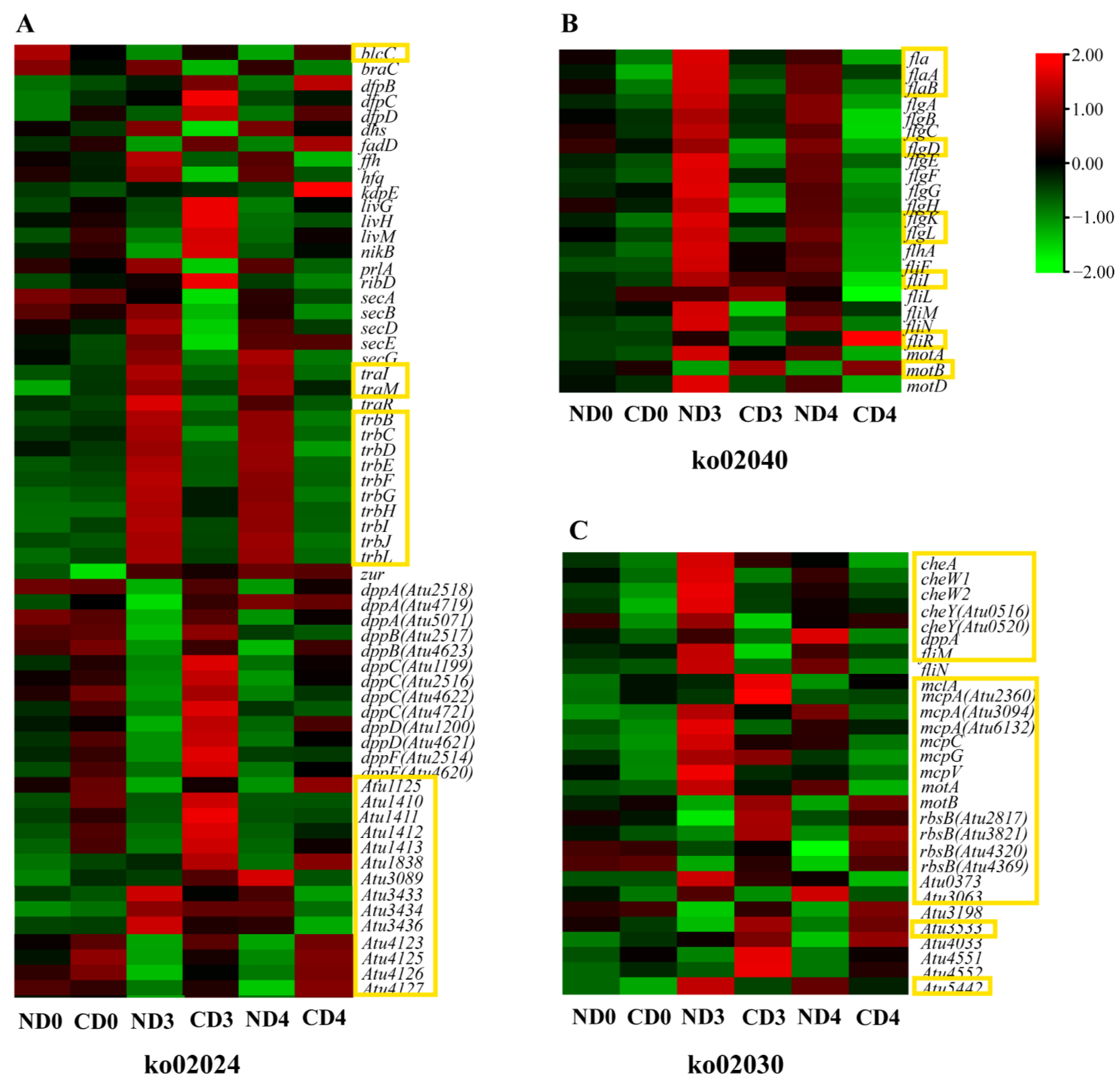
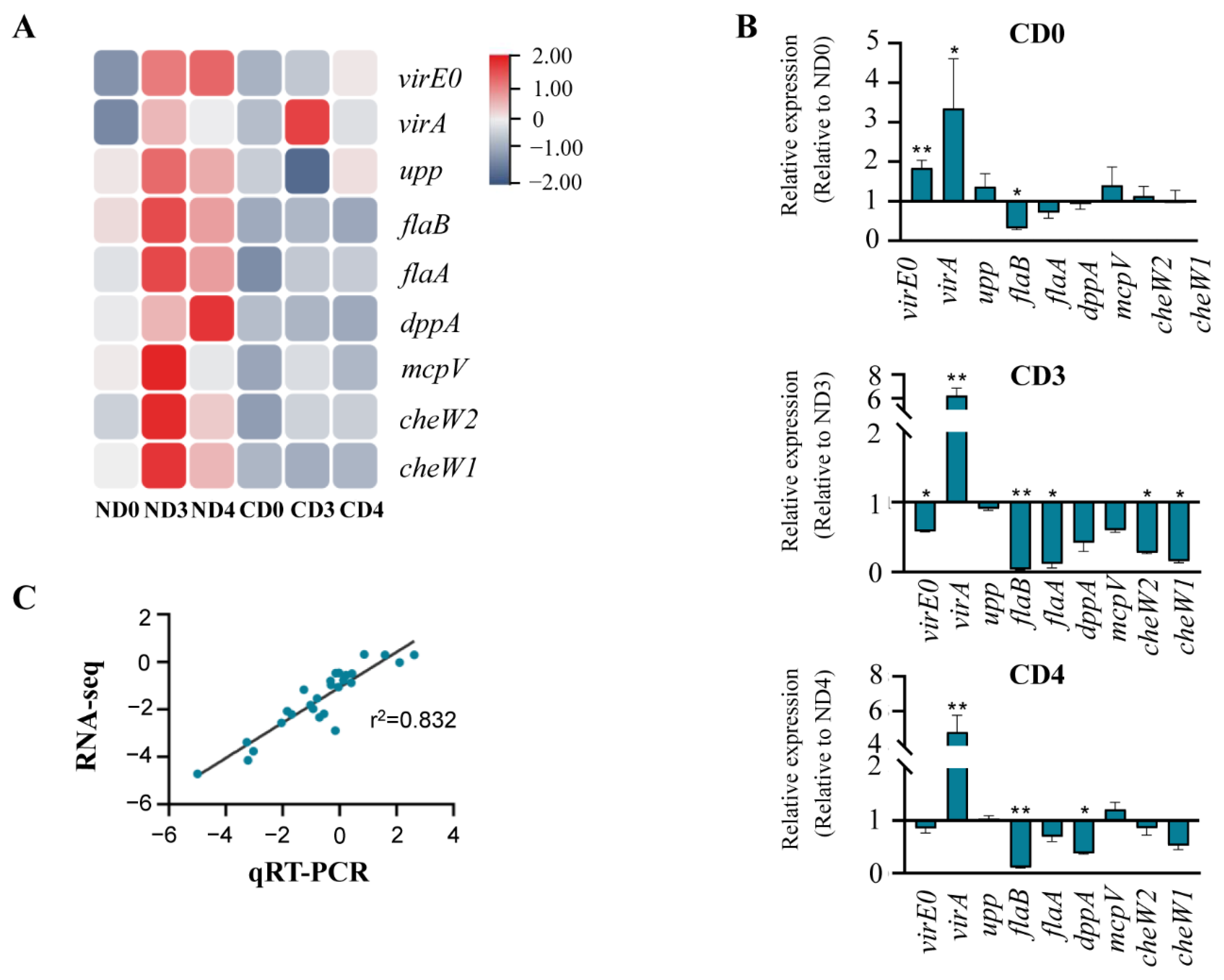
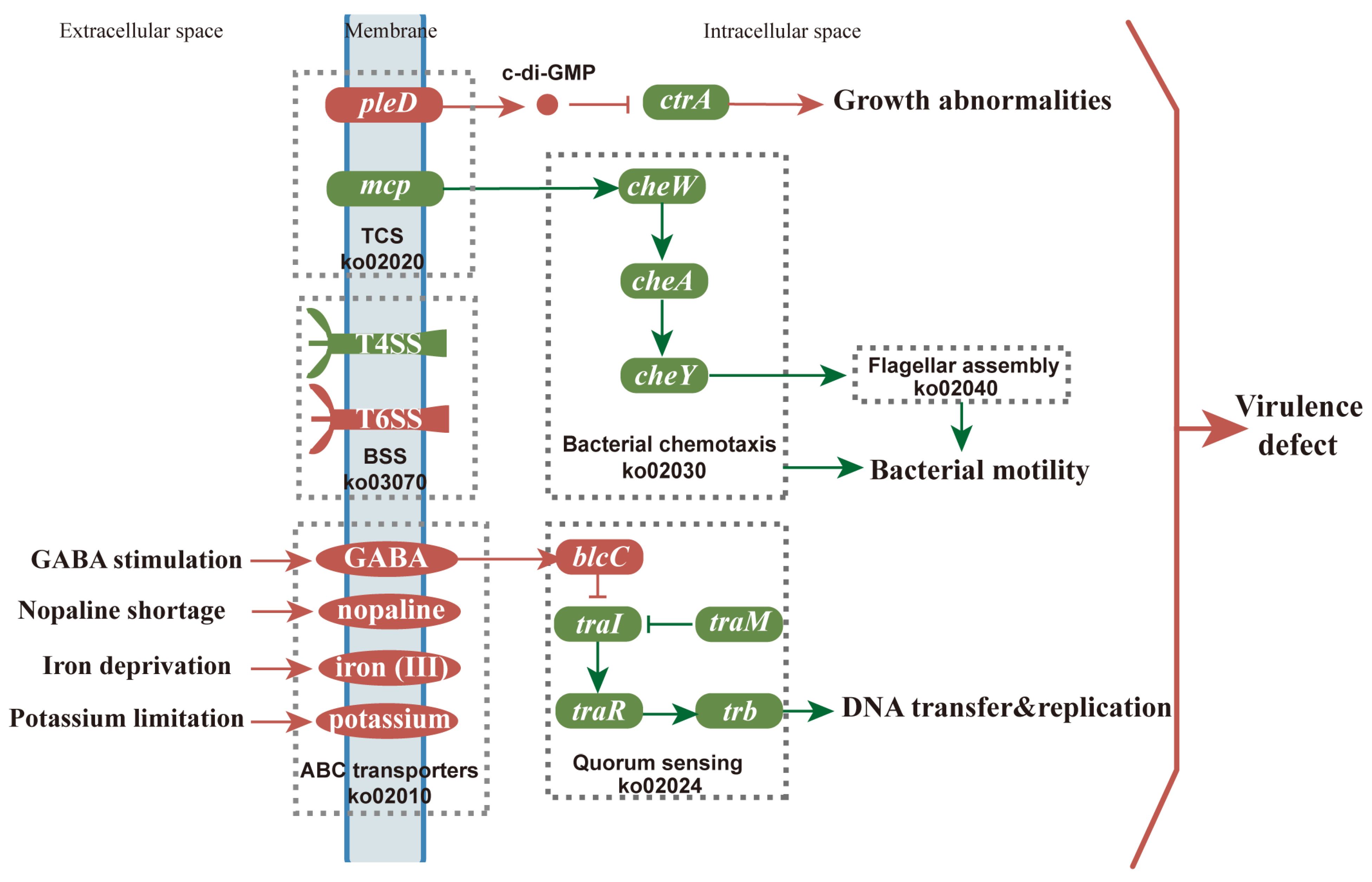
3.5. Transcriptional Changes of Genes Related to Agrobacterium-Mediated Transformation (AMT)
3.5.1. Expression Pattern Analysis of Genes Related to Environmental Information Processing
3.5.2. Expression Pattern Analysis of Genes Related to Cellular Processes
3.6. Quantitative Reverse Transcription-PCR (qRT-PCR) Verification
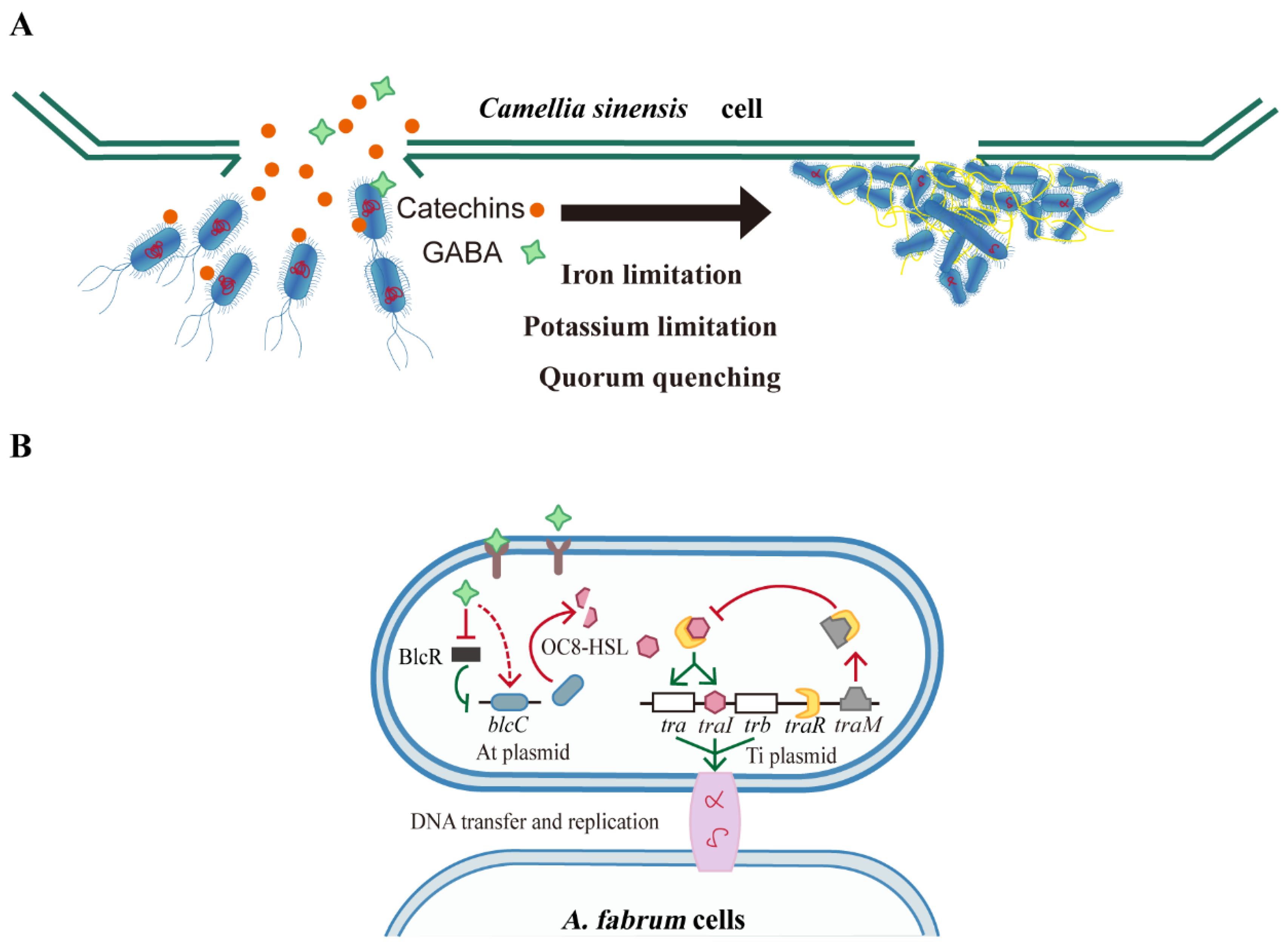
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Prasanth, M.I.; Sivamaruthi, B.S.; Chaiyasut, C.; Tencomnao, T. A review of the role of green tea (Camellia sinensis) in antiphotoaging, stress resistance, neuroprotection, and autophagy. Nutrients 2019, 11, 474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondal, T.K.; Bhattacharya, A.; Laxmikumaran, M.; Singh Ahuja, P. Recent advances of tea (Camellia sinensis) biotechnology. Plant Cell Tissue Organ Cult. 2004, 76, 195–254. [Google Scholar] [CrossRef]
- Butelli, E.; Titta, L.; Giorgio, M.; Mock, H.-P.; Matros, A.; Peterek, S.; Schijlen, E.G.W.M.; Hall, R.D.; Bovy, A.G.; Luo, J.; et al. Enrichment of tomato fruit with health-promoting anthocyanins by expression of select transcription factors. Nat. Biotechnol. 2008, 26, 1301–1308. [Google Scholar] [CrossRef] [PubMed]
- Chilcoat, D.; Liu, Z.-B.; Sander, J. Chapter Two-Use of CRISPR/Cas9 for Crop Improvement in Maize and Soybean. In Progress in Molecular Biology and Translational Science; Weeks, D.P., Yang, B., Eds.; Academic Press: Cambridge, MA, USA, 2017; Volume 149, pp. 27–46. [Google Scholar] [CrossRef]
- Mondal, T.; Bhattacharya, A.; Ahuja, P.; Chand, P. Transgenic tea [Camellia sinensis (L.) O. Kuntze cv. Kangra Jat] plants obtained by Agrobacterium-mediated transformation of somatic embryos. Plant Cell Rep. 2001, 20, 712–720. [Google Scholar] [CrossRef]
- Singh, H.R.; Bhattacharyya, N.; Agarwala, N.; Bhagawati, P.; Deka, M.; Das, S. Exogenous gene transfer in Assam tea [Camellia assamica (Masters)] by Agrobacterium-mediated transformation using somatic embryo. Eur. J. Exp. Biol. 2014, 4, 166–175. [Google Scholar]
- Singh, H.R.; Hazarika, P.; Agarwala, N.; Bhattacharyya, N.; Bhagawati, P.; Gohain, B.; Bandyopadhyay, T.; Bharalee, R.; Gupta, S.; Deka, M.; et al. Transgenic tea over-expressing Solanum tuberosum endo-1,3-beta-d-glucanase gene conferred resistance against blister blight disease. Plant Mol. Biol. Rep. 2018, 36, 107–122. [Google Scholar] [CrossRef]
- Sandal, I.; Saini, U.; Lacroix, B.; Bhattacharya, A.; Ahuja, P.S.; Citovsky, V. Agrobacterium-mediated genetic transformation of tea leaf explants: Effects of counteracting bactericidity of leaf polyphenols without loss of bacterial virulence. Plant Cell Rep. 2007, 26, 169–176. [Google Scholar] [CrossRef]
- Xia, E.-H.; Zhang, H.-B.; Sheng, J.; Li, K.; Zhang, Q.-J.; Kim, C.; Zhang, Y.; Liu, Y.; Zhu, T.; Li, W.; et al. The tea tree genome provides insights into tea flavor and independent evolution of caffeine biosynthesis. Mol. Plant 2017, 10, 866–877. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, M.; Mondal, T.; Chand, P. Biotechnological advances in tea (Camellia sinensis [L.] O. Kuntze): A review. Plant Cell Rep. 2015, 35, 255–287. [Google Scholar] [CrossRef]
- Mohanpuria, P.; Kumar, V.; Ahuja, P.S.; Yadav, S.K. Agrobacterium-mediated silencing of caffeine synthesis through root transformation in Camellia sinensis L. Mol. Biotechnol. 2011, 48, 235–243. [Google Scholar] [CrossRef]
- Matsumoto, S.; Fukui, M. Effect of acetosyringone application on Agrobacterium-mediated gene transfer in tea plant (Camellia sinensis). Bull. Natl. Res. Inst. Veg. Ornam. Plants Tea Jpn. 1999, 14, 9–15. [Google Scholar]
- Lopez, S.J.; Kumar, R.R.; Pius, P.K.; Muraleedharan, N. Agrobacterium tumefaciens-Mediated genetic transformation in tea (Camellia sinensis [L.] O. Kuntze). Plant Mol. Biol. Rep. 2004, 22, 201–202. [Google Scholar] [CrossRef]
- Gelvin, S.B. Agrobacterium-mediated plant transformation: The biology behind the “gene-jockeying” tool. Microbiol. Mol. Biol. Rev. 2003, 67, 16–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, M.G.; Moore, W.M.; Hummel, N.F.C.; Pearson, A.N.; Barnum, C.R.; Scheller, H.V.; Shih, P.M. Agrobacterium tumefaciens: A bacterium primed for synthetic biology. BioDes. Res. 2020, 2020, 8189219. [Google Scholar] [CrossRef]
- Guo, M.; Huang, Z.; Yang, J. Is there any crosstalk between the chemotaxis and virulence induction signaling in Agrobacterium tumefaciens? Biotechnol. Adv. 2017, 35, 505–511. [Google Scholar] [CrossRef]
- Matthysse, A.G. Attachment of Agrobacterium to plant surfaces. Front. Plant Sci. 2014, 5, 252. [Google Scholar] [CrossRef] [Green Version]
- Dazzo, F.; Truchet, G.; Sherwood, J.; Hrabak, E.; Abe, M.; Pankratz, S. Specific phases of root hair attachment in the Rhizobium trifolii-clover symbiosis. Appl. Environ. Microbiol. 1984, 48, 1140–1150. [Google Scholar] [CrossRef] [Green Version]
- Heindl, J.E.; Wang, Y.; Heckel, B.C.; Mohari, B.; Feirer, N.; Fuqua, C. Mechanisms and regulation of surface interactions and biofilm formation in Agrobacterium. Front. Plant Sci. 2014, 5, 176. [Google Scholar] [CrossRef] [Green Version]
- Bernal, P.; Llamas, M.A.; Filloux, A. Type VI secretion systems in plant-associated bacteria. Environ. Microbiol. 2018, 20, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Wang, Y.-C.; Huang, C.-J.; Ma, L.-S.; Lai, E.-M. Agrobacterium tumefaciens deploys a versatile antibacterial strategy to increase its competitiveness. J. Bacteriol. 2021, 203, e00490-20. [Google Scholar] [CrossRef]
- Dong, T.G.; Dong, S.; Catalano, C.; Moore, R.; Liang, X.; Mekalanos, J.J. Generation of reactive oxygen species by lethal attacks from competing microbes. Proc. Natl. Acad. Sci. USA 2015, 112, 2181–2186. [Google Scholar] [CrossRef] [Green Version]
- Silva, L.N.; Zimmer, K.R.; Macedo, A.J.; Trentin, D.S. Plant natural products targeting bacterial virulence factors. Chem. Rev. 2016, 116, 9162–9236. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Bassler, B.L. Bacterial quorum sensing in complex and dynamically changing environments. Nat. Rev. Microbiol. 2019, 17, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Murphy, P.J.; Kerr, A.; Tate, M.E. Agrobacterium conjugation and gene regulation by N-acyl-L-homoserine lactones. Nature 1993, 362, 446–448. [Google Scholar] [CrossRef] [PubMed]
- Grandclément, C.; Tannières, M.; Moréra, S.; Dessaux, Y.; Faure, D. Quorum quenching: Role in nature and applied developments. FEMS Microbiol. Rev. 2015, 40, 86–116. [Google Scholar] [CrossRef] [PubMed]
- White, C.E.; Winans, S.C. Identification of amino acid residues of the Agrobacterium tumefaciens quorum-sensing regulator TraR that are critical for positive control of transcription. Mol. Microbiol. 2005, 55, 1473–1486. [Google Scholar] [CrossRef] [PubMed]
- Dessaux, Y.; Faure, D. Quorum sensing and quorum quenching in Agrobacterium: A “Go/No Go System”? Genes 2018, 9, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.-W.; Xuan, C.-G.; Lu, C.-H.; Guo, S.; Yu, J.-F.; Asif, M.; Jiang, W.-J.; Zhou, Z.-G.; Luo, Z.-Q.; Zhang, L.-Q. AidB, a novel thermostable N-acylhomoserine lactonase from the bacterium Bosea sp. Appl. Environ. Microbiol. 2019, 85, e02065-19. [Google Scholar] [CrossRef]
- Haudecoeur, E.; Faure, D. A fine control of quorum-sensing communication in Agrobacterium tumefaciens. Commun. Integr. Biol. 2010, 3, 84–88. [Google Scholar] [CrossRef]
- Haudecoeur, E.; Planamente, S.; Cirou, A.; Tannières, M.; Shelp, B.J.; Moréra, S.; Faure, D. Proline antagonizes GABA-induced quenching of quorum-sensing in Agrobacterium tumefaciens. Proc. Natl. Acad. Sci. USA 2009, 106, 14587–14592. [Google Scholar] [CrossRef] [Green Version]
- Planamente, S.; Vigouroux, A.; Mondy, S.; Aumont-Nicaise, M.; Faure, D.; Moréra, S. A conserved mechanism of GABA binding and antagonism is revealed by structure-function analysis of the periplasmic binding protein Atu2422 in Agrobacterium tumefaciens. J. Biol. Chem. 2010, 285, 30294–30303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engström, P.; Zambryski, P.; Van Montagu, M.; Stachel, S. Characterization of Agrobacterium tumefaciens virulence proteins induced by the plant factor acetosyringone. J. Mol. Biol. 1987, 197, 635–645. [Google Scholar] [CrossRef]
- Bourras, S.; Rouxel, T.; Meyer, M. Agrobacterium tumefaciens gene transfer: How a plant pathogen hacks the nuclei of plant and nonplant organisms. Phytopathology 2015, 105, 1288–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathi, K.M.; Tula, S.; Tuteja, N. High frequency regeneration via direct somatic embryogenesis and efficient Agrobacterium-mediated genetic transformation of tobacco. Plant Signal. Behav. 2013, 8, e24354. [Google Scholar] [CrossRef] [Green Version]
- Maher, M.F.; Nasti, R.A.; Vollbrecht, M.; Starker, C.G.; Clark, M.D.; Voytas, D.F. Plant gene editing through de novo induction of meristems. Nat. Biotechnol. 2020, 38, 84–89. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [Green Version]
- Wood, D.W.; Setubal, J.C.; Kaul, R.; Monks, D.E.; Kitajima, J.P.; Okura, V.K.; Zhou, Y.; Chen, L.; Wood, G.E.; Almeida, N.F.; et al. The genome of the natural genetic engineer Agrobacterium tumefaciens C58. Science 2001, 294, 2317–2323. [Google Scholar] [CrossRef] [Green Version]
- Matthysse, A.G. Exopolysaccharides of Agrobacterium tumefaciens. In Agrobacterium Biology: From Basic Science to Biotechnology; Gelvin, S.B., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 111–141. [Google Scholar] [CrossRef]
- Flores, S.A.; Howell, M.; Daniel, J.J.; Piccolo, R.; Brown, P.J.B. Absence of the Min system does not cause major cell division defects in Agrobacterium tumefaciens. Front. Microbiol. 2018, 9, 681. [Google Scholar] [CrossRef]
- Kaczmarczyk, A.; Hempel, A.M.; von Arx, C.; Böhm, R.; Dubey, B.N.; Nesper, J.; Schirmer, T.; Hiller, S.; Jenal, U. Precise timing of transcription by c-di-GMP coordinates cell cycle and morphogenesis in Caulobacter. Nat. Commun. 2020, 11, 816. [Google Scholar] [CrossRef] [Green Version]
- Paul, R.; Jaeger, T.; Abel, S.; Wiederkehr, I.; Folcher, M.; Biondi, E.G.; Laub, M.T.; Jenal, U. Allosteric regulation of histidine kinases by their cognate response regulator determines cell fate. Cell 2008, 133, 452–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrera-Calderon, R.G.; Pallegar, P.; Westbye, A.B.; Wiesmann, C.; Lang, A.S.; Beatty, J.T. The CckA-ChpT-CtrA phosphorelay controlling Rhodobacter capsulatus gene transfer agent production is bidirectional and regulated by cyclic di-GMP. J. Bacteriol. 2021, 203, e00525-20. [Google Scholar] [CrossRef] [PubMed]
- Wolinsky, L.E.; Sote, E.O. Isolation of natural plaque-inhibiting substances from ‘Nigerian chewing sticks’. Caries Res. 1984, 18, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Matthysse, A.G.; White, S.; Lightfoot, R. Genes required for cellulose synthesis in Agrobacterium tumefaciens. J. Bacteriol. 1995, 177, 1069–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnhart, D.M.; Su, S.; Baccaro, B.E.; Banta, L.M.; Farrand, S.K. CelR, an ortholog of the diguanylate cyclase PleD of Caulobacter, regulates cellulose synthesis in Agrobacterium tumefaciens. Appl. Environ. Microbiol. 2013, 79, 7188–7202. [Google Scholar] [CrossRef] [Green Version]
- Pandey, S.S.; Patnana, P.K.; Lomada, S.K.; Tomar, A.; Chatterjee, S. Co-regulation of iron metabolism and virulence associated functions by iron and XibR, a novel iron binding transcription factor, in the plant pathogen Xanthomonas. PLoS Pathog. 2016, 12, e1006019. [Google Scholar] [CrossRef]
- Heindl, J.E.; Hibbing, M.E.; Xu, J.; Natarajan, R.; Buechlein, A.M.; Fuqua, C. Discrete responses to limitation for iron and manganese in Agrobacterium tumefaciens: Influence on attachment and biofilm formation. J. Bacteriol. 2015, 198, 816–829. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Pan, X.; Xu, Y.; Li, Y.; Xu, N.; Huang, Z.; Ye, J.; Gao, D.; Guo, M. Agrobacterium tumefaciens ferritins play an important role in full virulence through regulating iron homeostasis and oxidative stress survival. Mol. Plant Pathol. 2020, 21, 1167–1178. [Google Scholar] [CrossRef]
- Renzetti, A.; Betts, J.W.; Fukumoto, K.; Rutherford, R.N. Antibacterial green tea catechins from a molecular perspective: Mechanisms of action and structure–activity relationships. Food Funct. 2020, 11, 9370–9396. [Google Scholar] [CrossRef]
- Freeman, Z.N.; Dorus, S.; Waterfield, N.R. The KdpD/KdpE two-component system: Integrating K+ homeostasis and virulence. PLoS Pathog. 2013, 9, e1003201. [Google Scholar] [CrossRef] [Green Version]
- Zanker, H.; von Lintig, J.; Schröder, J. Opine transport genes in the octopine (occ) and nopaline (noc) catabolic regions in Ti plasmids of Agrobacterium tumefaciens. J. Bacteriol. 1992, 174, 841–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosie, A.H.F.; Poole, P.S. Bacterial ABC transporters of amino acids. Res. Microbiol. 2001, 152, 259–270. [Google Scholar] [CrossRef]
- Irino, N.; Nakayama, K.; Nakayama, H. The recQ gene of Escherichia coli K12: Primary structure and evidence for SOS regulation. Mol. Genet. Genom. 1986, 205, 298–304. [Google Scholar] [CrossRef]
- Baharoglu, Z.; Mazel, D. SOS, the formidable strategy of bacteria against aggressions. FEMS Microbiol. Rev. 2014, 38, 1126–1145. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Shen, B.; Du, P.; Wang, N.; Wang, J.; Li, J.; Sun, A. Transcriptomic analysis of the response of Pseudomonas fluorescens to epigallocatechin gallate by RNA-seq. PLoS ONE 2017, 12, e0177938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, D.; Feng, L.; Rana, M.; Gao, M.; Wei, S. Effects of catechins on Agrobacterium-mediated genetic transformation of Camellia sinensis. Plant Cell Tissue Organ Cult. 2014, 119, 27–37. [Google Scholar] [CrossRef]
- Lv, Q.; Chen, C.; Xu, Y.; Hu, S.; Wang, L.; Sun, K.; Chen, X.; Li, X. Optimization of Agrobacterium tumefaciens-Mediated Transformation Systems in Tea Plant (Camellia sinensis). Hortic. Plant J. 2017, 3, 105–109. [Google Scholar] [CrossRef]
- Attai, H.; Rimbey, J.; Smith, G.P.; Brown, P.J.B. Expression of a peptidoglycan hydrolase from lytic bacteriophages Atu_ph02 and Atu_ph03 triggers lysis of Agrobacterium tumefaciens. Appl. Environ. Microbiol. 2017, 83, e01498-17. [Google Scholar] [CrossRef] [Green Version]
- Tannières, M.; Lang, J.; Barnier, C.; Shykoff, J.A.; Faure, D. Quorum-quenching limits quorum-sensing exploitation by signal-negative invaders. Sci. Rep. 2017, 7, 40126. [Google Scholar] [CrossRef] [Green Version]
- Mei, X.; Chen, Y.; Zhang, L.; Fu, X.; Wei, Q.; Grierson, D.; Zhou, Y.; Huang, Y.; Dong, F.; Yang, Z. Dual mechanisms regulating glutamate decarboxylases and accumulation of gamma-aminobutyric acid in tea (Camellia sinensis) leaves exposed to multiple stresses. Sci. Rep. 2016, 6, 23685. [Google Scholar] [CrossRef] [Green Version]
- Kumar, N.; Gulati, A.; Bhattacharya, A. L-glutamine and l-glutamic acid facilitate successful Agrobacterium Infection of recalcitrant tea cultivars. Appl. Biochem. Biotechnol. 2013, 170, 1649–1664. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, S.; Someya, T.; Zhou, S.; Takayama, M.; Nakamura, K.; Ezura, H. An Agrobacterium tumefaciens strain with gamma-aminobutyric acid transaminase activity shows an enhanced genetic transformation ability in plants. Sci. Rep. 2017, 7, 42649. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, K.; Tian, N.; da Silva Ferreira, J.F.; Sandhu, D.; Xiao, L.; Gu, M.; Luo, Y.; Zhang, X.; Liu, G.; Liu, Z.; et al. Comparative Transcriptome Analysis of Agrobacterium tumefaciens Reveals the Molecular Basis for the Recalcitrant Genetic Transformation of Camellia sinensis L. Biomolecules 2022, 12, 688. https://doi.org/10.3390/biom12050688
Jin K, Tian N, da Silva Ferreira JF, Sandhu D, Xiao L, Gu M, Luo Y, Zhang X, Liu G, Liu Z, et al. Comparative Transcriptome Analysis of Agrobacterium tumefaciens Reveals the Molecular Basis for the Recalcitrant Genetic Transformation of Camellia sinensis L. Biomolecules. 2022; 12(5):688. https://doi.org/10.3390/biom12050688
Chicago/Turabian StyleJin, Ke, Na Tian, Jorge Freire da Silva Ferreira, Devinder Sandhu, Lizheng Xiao, Meiyi Gu, Yiping Luo, Xiangqin Zhang, Guizhi Liu, Zhonghua Liu, and et al. 2022. "Comparative Transcriptome Analysis of Agrobacterium tumefaciens Reveals the Molecular Basis for the Recalcitrant Genetic Transformation of Camellia sinensis L." Biomolecules 12, no. 5: 688. https://doi.org/10.3390/biom12050688
APA StyleJin, K., Tian, N., da Silva Ferreira, J. F., Sandhu, D., Xiao, L., Gu, M., Luo, Y., Zhang, X., Liu, G., Liu, Z., Huang, J., & Liu, S. (2022). Comparative Transcriptome Analysis of Agrobacterium tumefaciens Reveals the Molecular Basis for the Recalcitrant Genetic Transformation of Camellia sinensis L. Biomolecules, 12(5), 688. https://doi.org/10.3390/biom12050688