
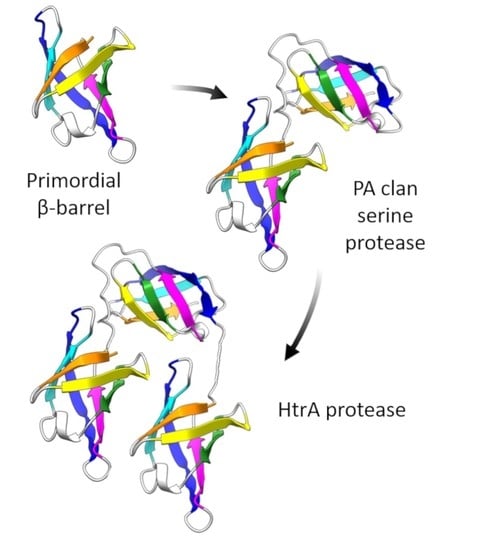
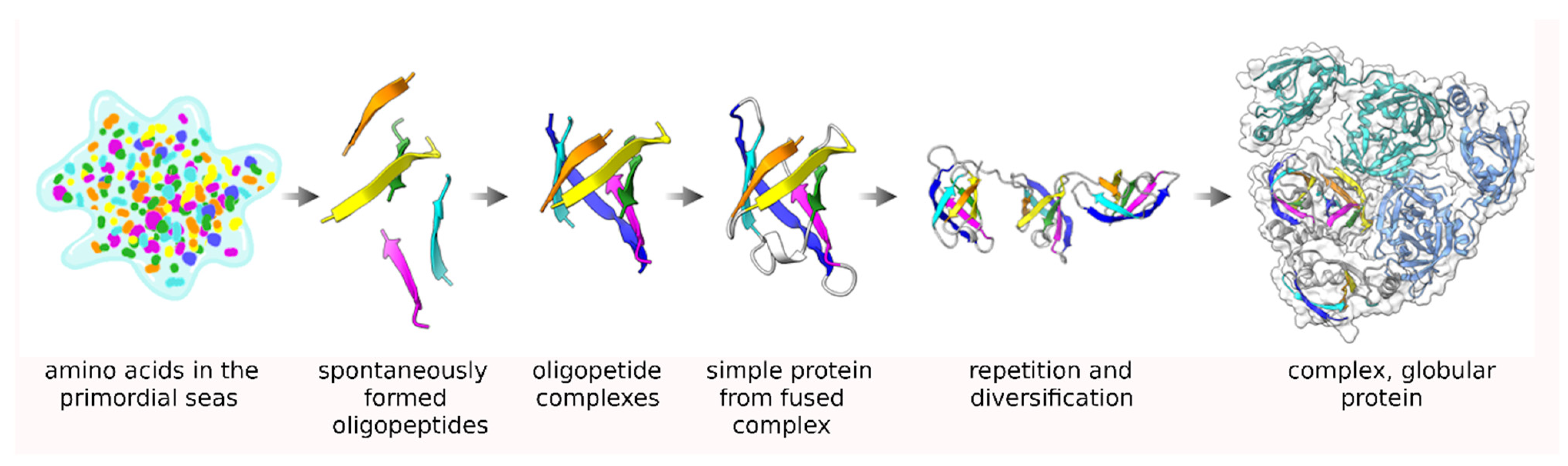
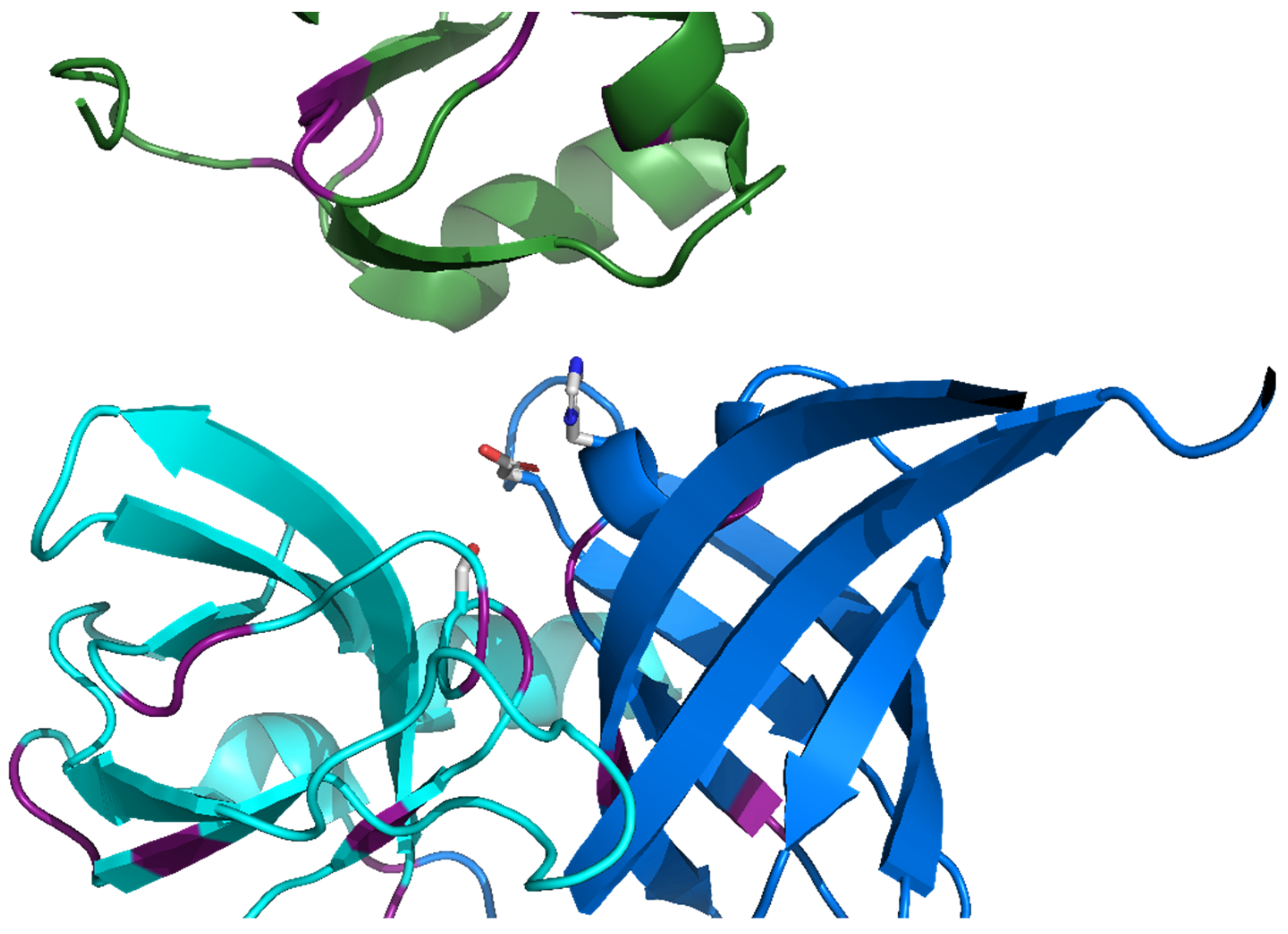
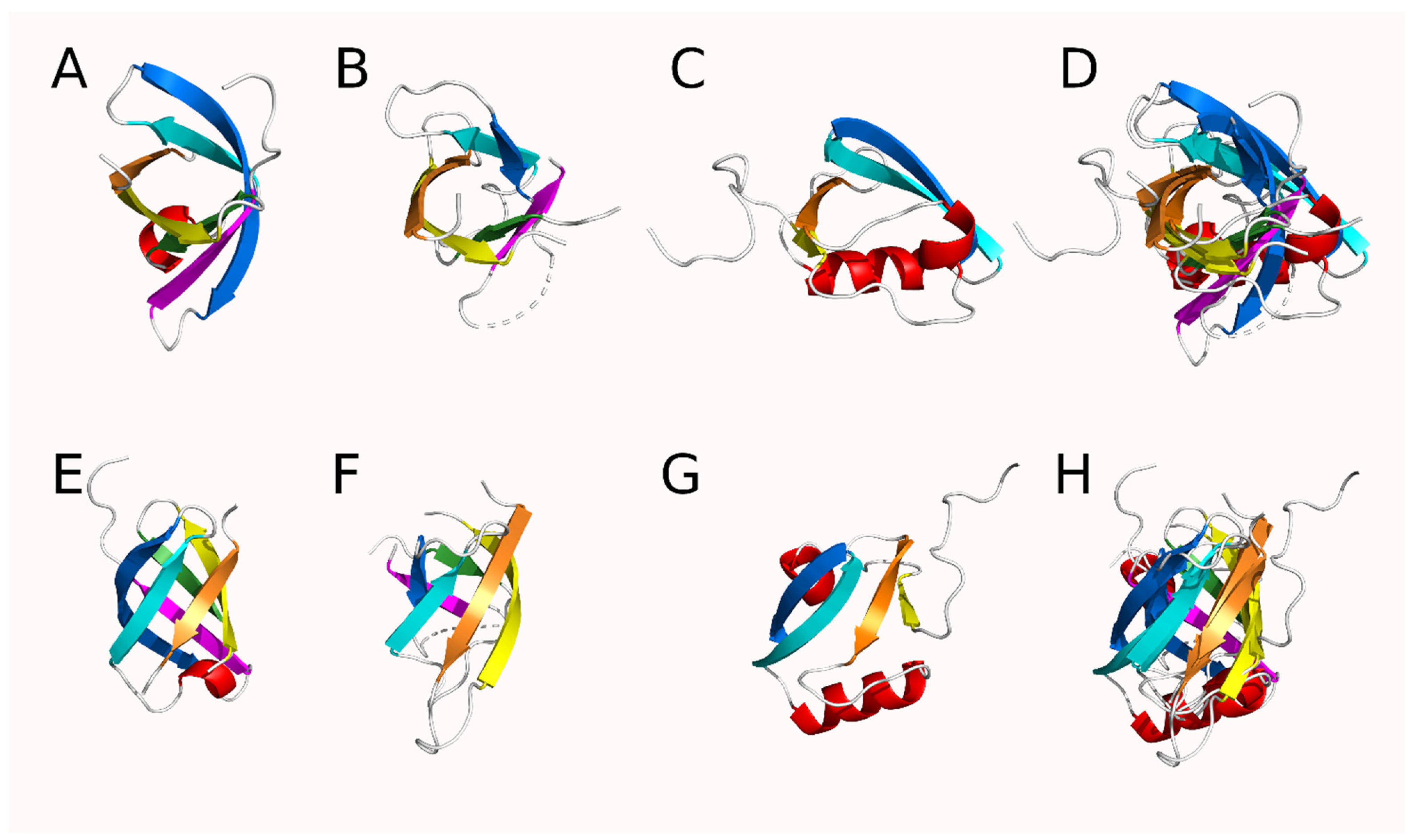
The Repeating, Modular Architecture of the HtrA Proteases


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Radó-Trilla, N.; Albà, M. Dissecting the role of low-complexity regions in the evolution of vertebrate proteins. BMC Evol. Biol. 2012, 12, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrade, M.A.; Perez-Iratxeta, C.; Ponting, C.P. Protein repeats: Structures, functions, and evolution. J. Struct. Biol. 2001, 134, 117–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espada, R.; Parra, R.G.; Sippl, M.J.; Mora, T.; Walczak, A.M.; Ferreiro, D.U. Repeat proteins challenge the concept of structural domains. Biochem. Soc. Trans. 2015, 43, 844–849. [Google Scholar] [CrossRef] [Green Version]
- Kajava, A.V. Tandem repeats in proteins: From sequence to structure. J. Struct. Biol. 2011, 179, 279–288. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.X.; Chen, L.; Di Costanzo, L.; Christie, C.; Dalenberg, K.; Duarte, J.M.; Dutta, S.; et al. RCSB Protein Data Bank: Biological macromolecular structures enabling research and education in fundamental bi-ology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019, 47, D464–D474. [Google Scholar] [CrossRef] [Green Version]
- Illergård, K.; Ardell, D.H.; Elofsson, A. Structure is three to ten times more conserved than sequence-A study of structural response in protein cores. Proteins: Struct. Funct. Bioinform. 2009, 77, 499–508. [Google Scholar] [CrossRef]
- Alva, V.; Söding, J.; Lupas, A.N. A vocabulary of ancient peptides at the origin of folded proteins. eLife 2015, 4, e09410. [Google Scholar] [CrossRef]
- Ranea, J.A.G.; Sillero, A.; Thornton, J.M.; Orengo, C.A. Protein superfamily evolution and the last universal common an-cestor (LUCA). J. Mol. Evol 2006, 63, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Eck, R.V.; Dayhoff, M.O. Evolution of the Structure of Ferredoxin Based on Living Relics of Primitive Amino Acid Sequences. Science 1966, 152, 363–366. [Google Scholar] [CrossRef]
- Alva, V.; Lupas, A.N. From ancestral peptides to designed proteins. Curr. Opin. Struct. Biol. 2018, 48, 103–109. [Google Scholar] [CrossRef]
- Broom, A.; Doxey, A.C.; Lobsanov, Y.D.; Berthin, L.G.; Rose, D.R.; Howell, P.L.; McConkey, B.J.; Meiering, E.M. Modu-lar Evolution and the Origins of Symmetry: Reconstruction of a Three-Fold Symmetric Globular Protein. Structure 2012, 20, 161–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, V.A.; Busby, J.N.; Janssen, B.; Lott, S.; Seiradake, E. Teneurin Structures Are Composed of Ancient Bacterial Protein Domains. Front. Neurosci. 2019, 13, 183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wieczorek, R. On Prebiotic Ecology, Supramolecular Selection and Autopoiesis. Orig. Life Evol. Biosphere 2012, 42, 445–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Söding, J.; Lupas, A.N. More than the sum of their parts: On the evolution of proteins from peptides. BioEssays 2003, 25, 837–846. [Google Scholar] [CrossRef]
- Lupas, A.N.; Ponting, C.; Russell, R.B. On the Evolution of Protein Folds: Are Similar Motifs in Different Protein Folds the Result of Convergence, Insertion, or Relics of an Ancient Peptide World? J. Struct. Biol. 2001, 134, 191–203. [Google Scholar] [CrossRef]
- Marcotte, E.; Pellegrini, M.; Yeates, T.; Eisenberg, D. A census of protein repeats. J. Mol. Biol. 1999, 293, 151–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, M.L.R.; Rabin, A.; Tawfik, D.S. Functional Proteins from Short Peptides: Dayhoff’s Hypothesis Turns 50. Angew. Chem. Int. Ed. 2016, 55, 15966–15971. [Google Scholar] [CrossRef]
- Laurino, P.; Tóth-Petróczy, A.; Meana-Pañeda, R.; Lin, W.; Truhlar, D.; Tawfik, D.S. An Ancient Fingerprint Indicates the Common Ancestry of Rossmann-Fold Enzymes Utilizing Different Ribose-Based Cofactors. PLOS Biol. 2016, 14, e1002396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galpern, E.A.; Freiberger, M.I.; Ferreiro, D.U. Large Ankyrin repeat proteins are formed with similar and energetically fa-vorable units. PLoS ONE 2020, 15, e0233865. [Google Scholar] [CrossRef]
- Clausen, T.; Kaiser, M.; Huber, R.; Ehrmann, M. HTRA proteases: Regulated proteolysis in protein quality control. Nat. Rev. Mol. Cell Biol. 2011, 12, 152–162. [Google Scholar] [CrossRef]
- Ortega, J.; Iwanczyk, J.; Jomaa, A. Escherichia coli DegP: A Structure-Driven Functional Model. J. Bacteriol. 2009, 191, 4705–4713. [Google Scholar] [CrossRef] [Green Version]
- Sohn, J.; Grant, R.A.; Sauer, R.T. Allostery Is an Intrinsic Property of the Protease Domain of DegS: Implications for Enzyme Function and Evolution. J. Biol. Chem. 2010, 285, 34039–34047. [Google Scholar] [CrossRef] [Green Version]
- Kley, J.; Schmidt, B.; Boyanov, B.; Stolt-Bergner, P.C.; Kirk, R.; Ehrmann, M.; Knopf, R.R.; Naveh, L.; Adam, Z.; Clausen, T. Structural adaptation of the plant protease Deg1 to repair photosystem II during light exposure. Nat. Struct. Mol. Biol. 2011, 18, 728–731. [Google Scholar] [CrossRef] [PubMed]
- Hegde, R.; Srinivasula, S.M.; Zhang, Z.J.; Wassell, R.; Mukattash, R.; Cilenti, L.; DuBois, G.; Lazebnik, Y.; Zervos, A.S.; Fernandes-Alnemri, T.; et al. Identification of Omi/HtrA-2 as a mitochondrial apoptotic serine protease that dis-rupts inhibitor of apoptosis protein-caspase interaction. J. Biol. Chem. 2002, 277, 432–438. [Google Scholar] [CrossRef] [Green Version]
- Li, W.Y.; Srinivasula, S.M.; Chai, J.J.; Li, P.W.; Wu, J.W.; Zhang, Z.J.; Alnemri, E.S.; Shi, Y.G. Structural insights into the pro-apoptotic function of mitochondrial serine protease HtrA2/Omi. Nat. Struct. Biol. 2002, 9, 436–441. [Google Scholar] [CrossRef]
- Merski, M.; Moreira, C.; Abreu, R.M.; Ramos, M.J.; Fernandes, P.; Martins, L.M.; Pereira, P.; Macedo-Ribeiro, S. Molecular motion regulates the activity of the Mitochondrial Serine Protease HtrA2. Cell Death Dis. 2017, 8, e3119. [Google Scholar] [CrossRef] [Green Version]
- Zurawa-Janicka, D.; Jarzab, M.; Polit, A.; Skorko-Glonek, J.; Lesner, A.; Gitlin, A.; Gieldon, A.; Ciarkowski, J.; Glaza, P.; Lubomska, A.; et al. Temperature-induced changes of HtrA2(Omi) protease activity and structure. Cell Stress Chaperones 2012, 18, 35–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Cera, E. Serine Proteases. Iubmb Life 2009, 61, 510–515. [Google Scholar] [CrossRef]
- Krojer, T.; Garrido-Franco, M.; Huber, R.; Ehrmann, M.; Clausen, T. Crystal structure of DegP (HtrA) reveals a new prote-ase-chaperone machine. Nature 2002, 416, 455–459. [Google Scholar] [CrossRef]
- Lee, H.-J.; Zheng, J.J. PDZ domains and their binding partners: Structure, specificity, and modification. Cell Commun. Signal. 2010, 8, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, L.M.; Turk, B.E.; Cowling, V.; Borg, A.; Jarrell, E.T.; Cantley, L.C.; Downward, J. Binding specificity and regula-tion of the serine protease and PDZ domains of HtrA2/Omi. J. Biol. Chem. 2003, 278, 49417–49427. [Google Scholar] [CrossRef] [Green Version]
- Toyama, Y.; Harkness, R.W.; Kay, L.E. Structural basis of protein substrate processing by human mitochondrial high-temperature requirement A2 protease. Proc. Natl. Acad. Sci. USA 2022, 119, e2203172119. [Google Scholar] [CrossRef] [PubMed]
- Merski, M.; Młynarczyk, K.; Ludwiczak, J.; Skrzeczkowski, J.; Dunin-Horkawicz, S.; Górna, M.W. Self-analysis of repeat proteins reveals evolutionarily conserved patterns. BMC Bioinform. 2020, 21, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Sonnhammer, E.L.L.; Durbin, R. A dot-matrix program with dynamic threshold control suited for genomic DNA and pro-tein sequence analysis (Reprinted from Gene Combis, vol 167, pg GC1-GC10, 1996). Gene 1995, 167, Gc1–Gc10. [Google Scholar] [CrossRef] [Green Version]
- Bateman, A.; Martin, M.J.; Orchard, S.; Magrane, M.; Alpi, E.; Bely, B.; Bingley, M.; Britto, R.; Bursteinas, B.; Busiello, G.; et al. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar]
- Huang, Y.; Niu, B.F.; Gao, Y.; Fu, L.M.; Li, W.Z. CD-HIT Suite: A web server for clustering and comparing biological se-quences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [Green Version]
- Stothard, P. The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA se-quences. Biotechniques 2000, 28, 1102–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Open-Source PyMOL 1.6.0.0; Schrodinger LLC: New York, NY, USA, 2013.
- Zhang, Y.; Skolnick, J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef]
- Clausen, T.; Southan, C.; Ehrmann, M. The HtrA Family of Proteases: Implications for Protein Composition and Cell Fate. Mol. Cell 2002, 10, 443–455. [Google Scholar] [CrossRef]
- Schubert, A.; Wrase, R.; Hilgenfeld, R.; Hansen, G. Structures of DegQ from Legionella pneumophila Define Distinct ON and OFF States. J. Mol. Biol. 2015, 427, 2840–2851. [Google Scholar] [CrossRef]
- Teyra, J.; Ernst, A.; Singer, A.; Sicheri, F.; Sidhu, S.S. Comprehensive analysis of all evolutionary paths between two diver-gent PDZ domain specificities. Protein Sci. 2020, 29, 433–442. [Google Scholar] [CrossRef]
- Jorda, J.; Xue, B.; Uversky, V.N.; Kajava, A.V. Protein tandem repeats—The more perfect, the less structured. FEBS J. 2010, 277, 2673–2682. [Google Scholar] [CrossRef] [PubMed]
- Skelton, N.J.; Koehler, M.F.T.; Zobel, K.; Wong, W.L.; Yeh, S.; Pisabarro, M.T.; Yin, J.P.; Lasky, L.A.; Sidhu, S.S. Origins of PDZ domain ligand specificity—Structure determination and mutagenesis of the erbin PDZ domain. J. Biol. Chem. 2003, 278, 7645–7654. [Google Scholar] [CrossRef] [Green Version]
- Krojer, T.; Sawa, J.; Schafer, E.; Saibil, H.R.; Ehrmann, M.; Clausen, T. Structural basis for the regulated protease and chap-erone function of DegP. Nature 2008, 453, 885-U31. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Fan, H.; Gao, F.; Lin, Y.; Zhang, L.; Gong, W.; Liu, L. Crystal Structure of Arabidopsis Deg2 Protein Reveals an Internal PDZ Ligand Locking the Hexameric Resting State. J. Biol. Chem. 2012, 287, 37564–37569. [Google Scholar] [CrossRef] [Green Version]
- Burroughs, A.M.; Allen, K.N.; Dunaway-Mariano, D.; Aravind, L. Evolutionary genomics of the HAD superfamily: Under-standing the structural adaptations and catalytic diversity in a superfamily of phosphoesterases and allied enzymes. J. Mol. Bi-ol. 2006, 361, 1003–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vos, M.; Hesselman, M.C.; te Beek, T.A.; van Passel, M.W.J.; Eyre-Walker, A. Rates of Lateral Gene Transfer in Prokary-otes: High but Why? Trends Microbiol. 2015, 23, 598–605. [Google Scholar] [CrossRef]
- Schaper, E.; Kajava, A.V.; Hauser, A.; Anisimova, M. Repeat or not repeat?—Statistical validation of tandem repeat predic-tion in genomic sequences. Nucleic Acids Res. 2012, 40, 10005–10017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marold, J.D.; Kavran, J.M.; Bowman, G.D.; Barrick, D. A Naturally Occurring Repeat Protein with High Internal Sequence Identity Defines a New Class of TPR-like Proteins. Structure 2015, 23, 2055–2065. [Google Scholar] [CrossRef] [Green Version]
- Gul, I.S.; Hulpiau, P.; Saeys, Y.; van Roy, F. Metazoan evolution of the armadillo repeat superfamily. Cell. Mol. Life Sci. 2016, 74, 525–541. [Google Scholar] [CrossRef] [PubMed]
- Renault, L.; Nassar, N.; Vetter, I.; Becker, J.; Klebe, C.; Roth, M.; Wittinghofer, A. The 1.7 angstrom crystal structure of the regulator of chromosome condensation (RCC1) reveals a seven-bladed propeller. Nature 1998, 392, 97–101. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, L.D.; Regan, L. TPR proteins: The versatile helix. Trends Biochem. Sci. 2003, 28, 655–662. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merski, M.; Macedo-Ribeiro, S.; Wieczorek, R.M.; Górna, M.W. The Repeating, Modular Architecture of the HtrA Proteases. Biomolecules 2022, 12, 793. https://doi.org/10.3390/biom12060793
Merski M, Macedo-Ribeiro S, Wieczorek RM, Górna MW. The Repeating, Modular Architecture of the HtrA Proteases. Biomolecules. 2022; 12(6):793. https://doi.org/10.3390/biom12060793
Chicago/Turabian StyleMerski, Matthew, Sandra Macedo-Ribeiro, Rafal M. Wieczorek, and Maria W. Górna. 2022. "The Repeating, Modular Architecture of the HtrA Proteases" Biomolecules 12, no. 6: 793. https://doi.org/10.3390/biom12060793
APA StyleMerski, M., Macedo-Ribeiro, S., Wieczorek, R. M., & Górna, M. W. (2022). The Repeating, Modular Architecture of the HtrA Proteases. Biomolecules, 12(6), 793. https://doi.org/10.3390/biom12060793