
TRPV6 Regulation by Cis-22a and Cholesterol

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Constructs and Reagents
2.2. Cell Culture and Transfection
2.3. Calcium Imaging
2.4. Electrophysiology
3. Results
3.1. Structure-Guided Mutagenesis within LBS-2 of Human TRPV6
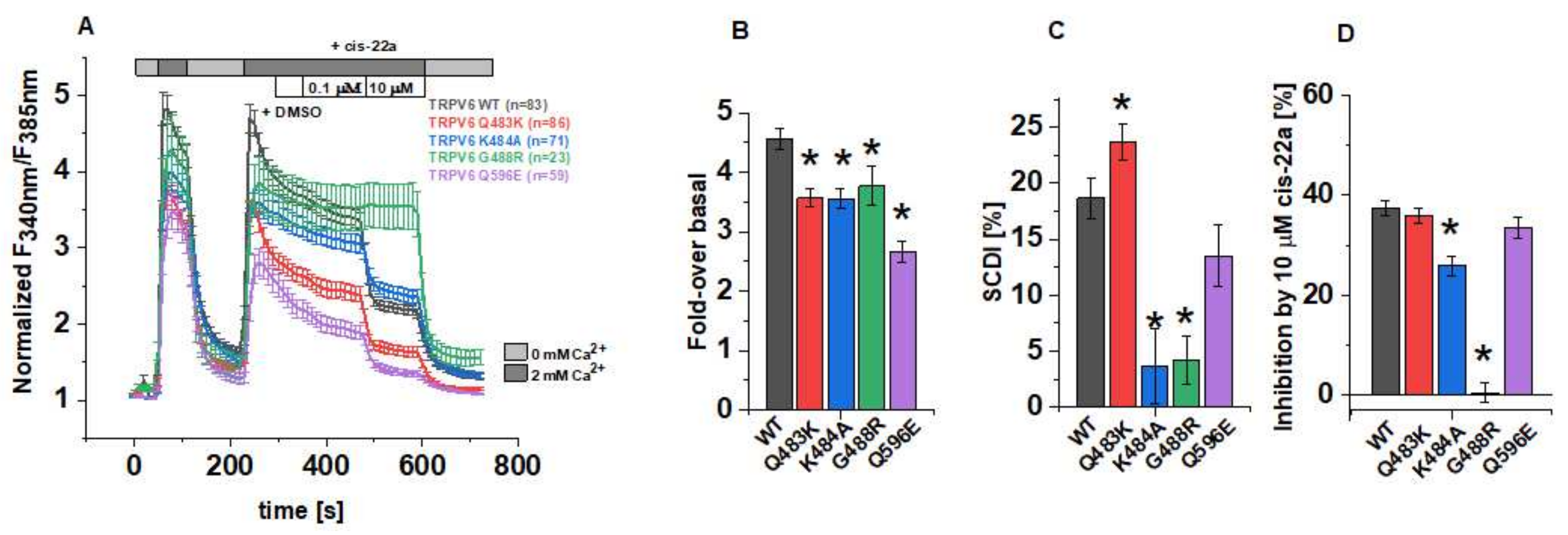
3.2. Point Mutations within LBS-2 Alter TRPV6 Activity and Cytosolic Ca2+ Levels in Fura-2 Measurements
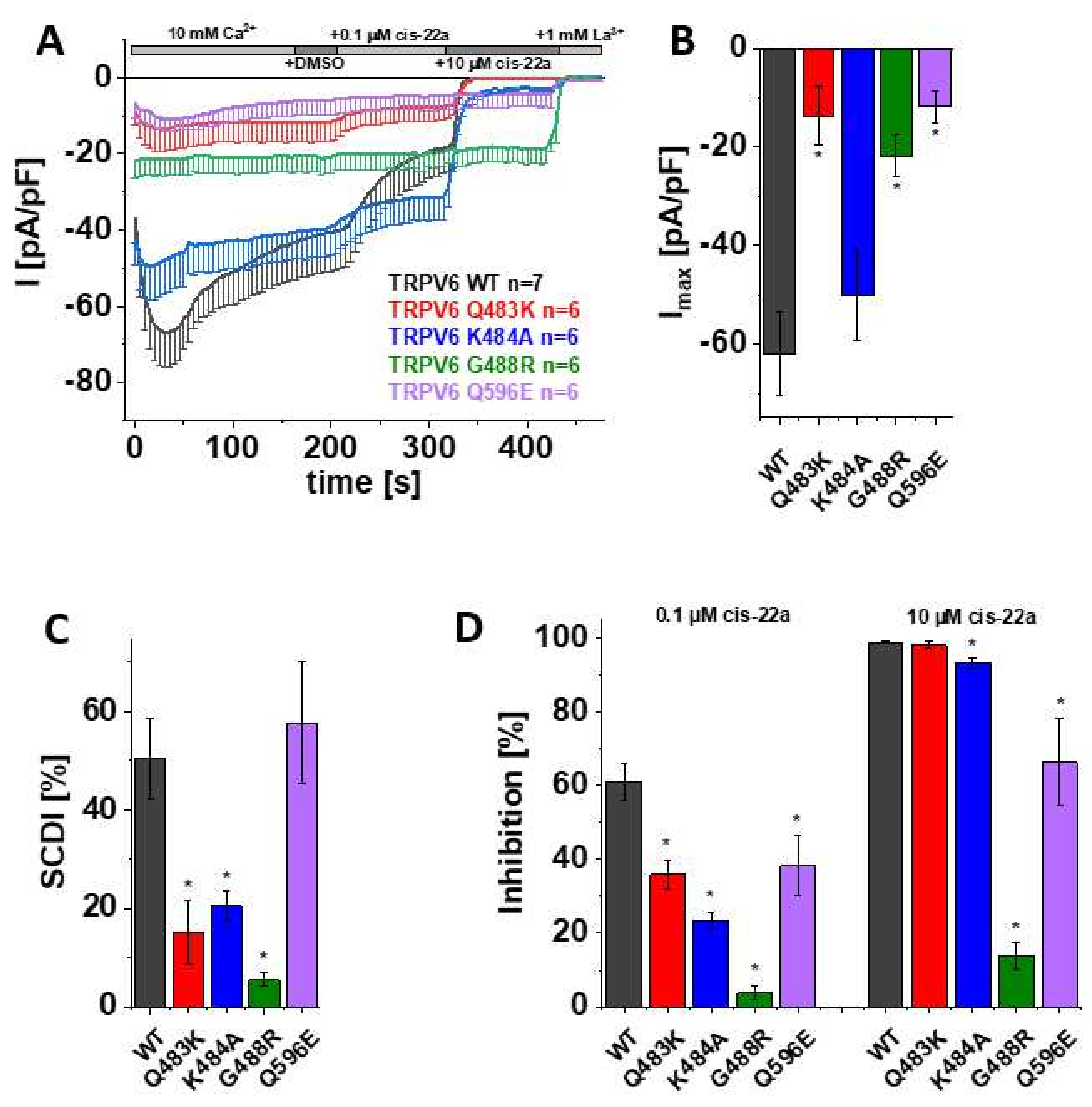
3.3. Mutations in LBS-2 Lead to Reduced Maximum Currents, SCDI and Inhibition by Cis-22a
3.4. Preincubation with Cholesterol Oxidase (CO) Has No Effect on Maximum Currents and SCDI of TRPV6 WT
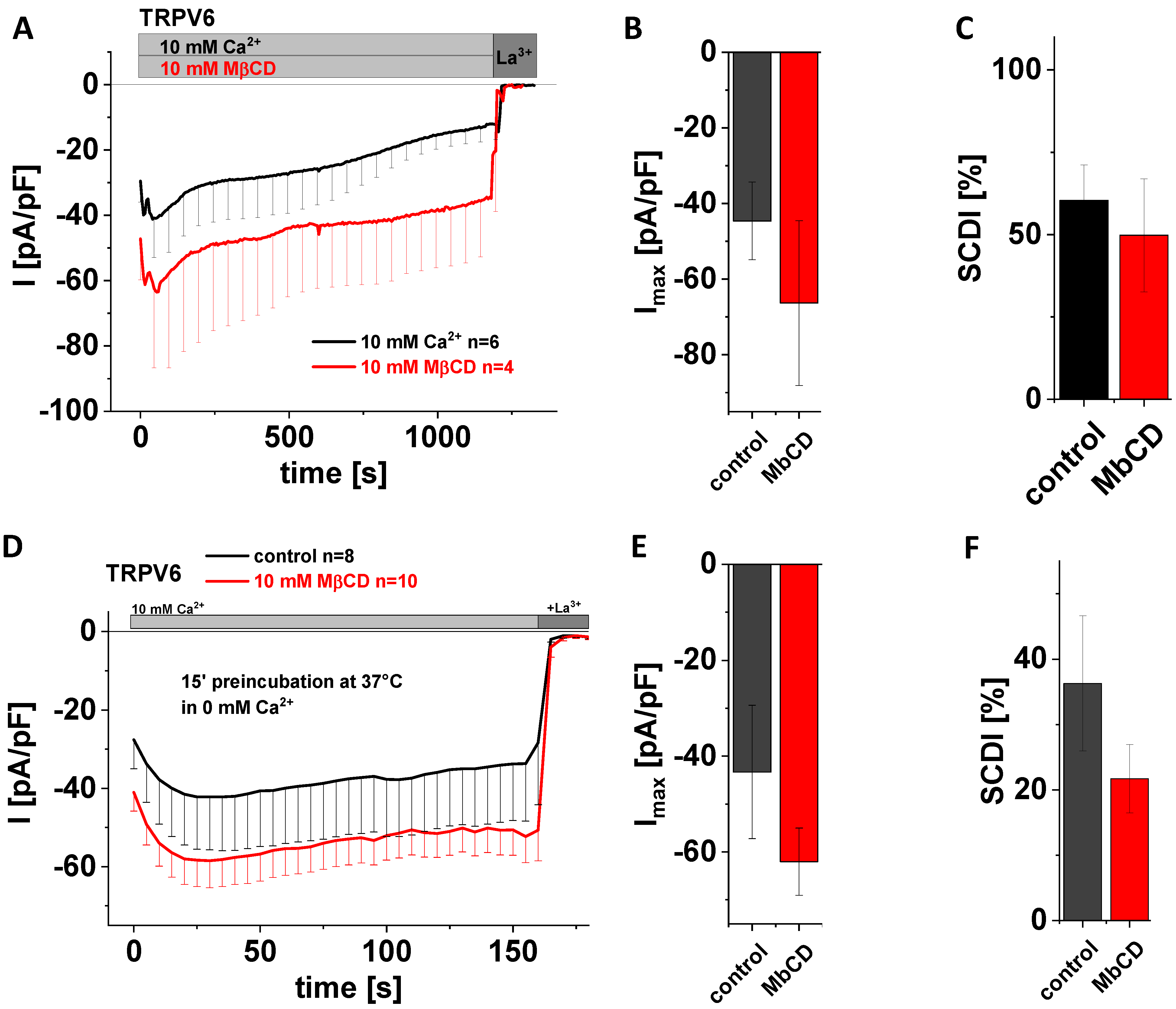
3.5. Application of MβCD Lacks a Significant Effect on Maximum Currents and SCDI of TRPV6 WT
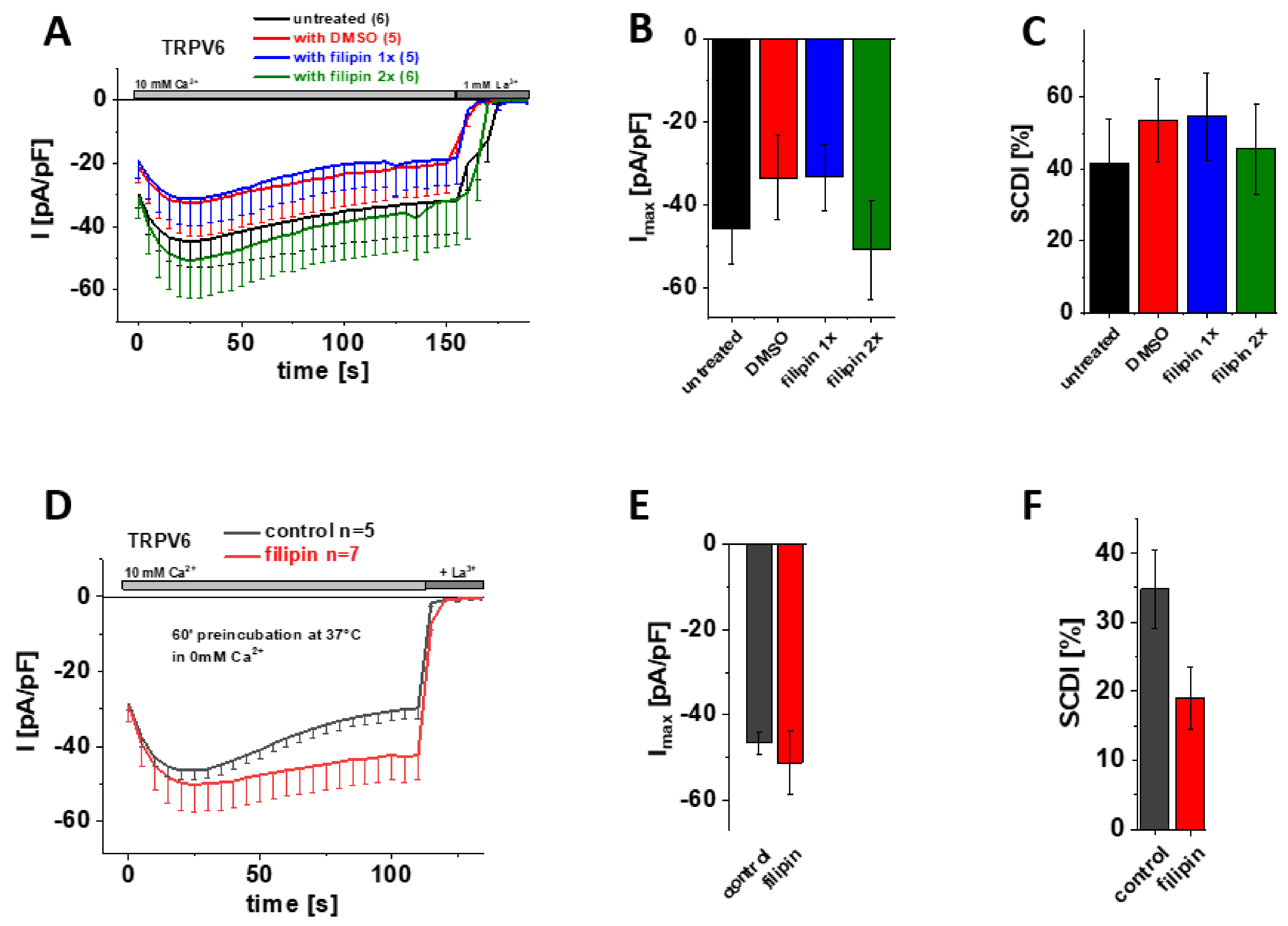
3.6. Cholesterol Sequestration at Different Stages of Protein Expression Using Filipin Does Not Significantly Alter Maximum Currents and SCDI of TRPV6 WT
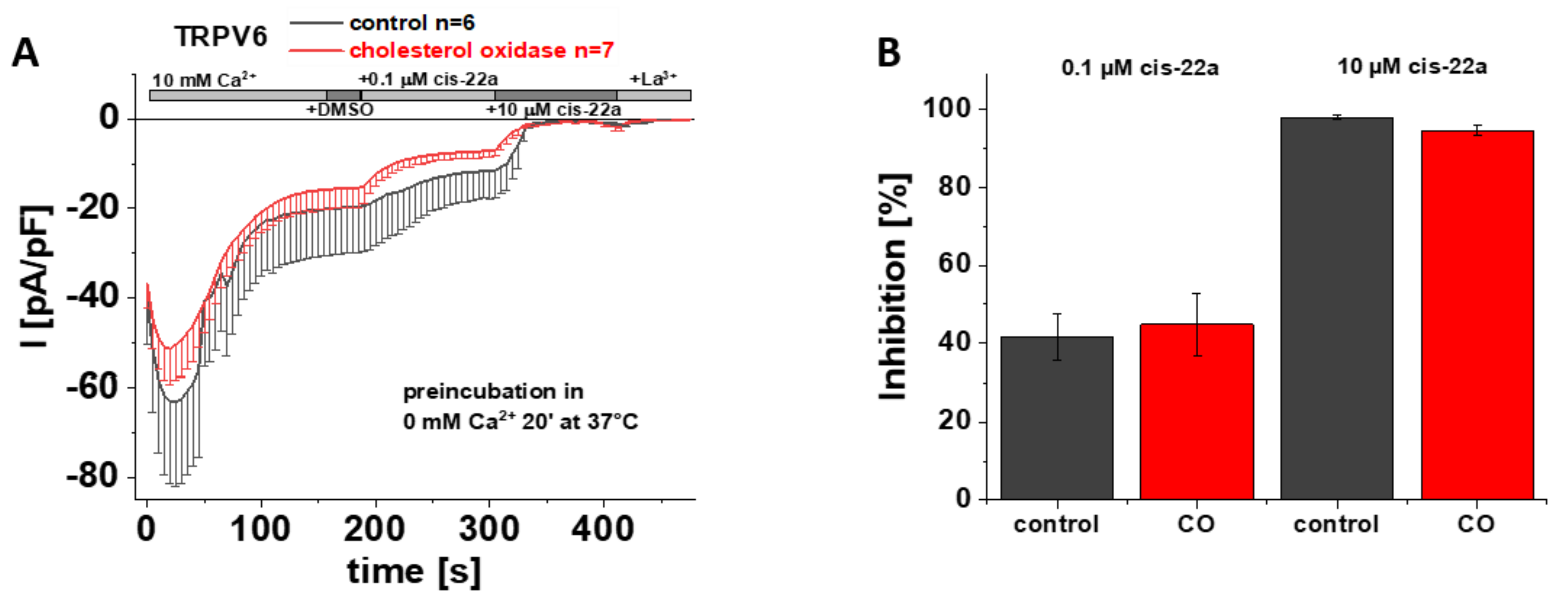
3.7. Preincubation with Cholesterol Oxidase Has No Effect on TRPV6 WT Inhibition by Cis-22a
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bootman, M.D. Calcium signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a011171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bootman, M.D.; Bultynck, G. Fundamentals of Cellular Calcium Signaling: A Primer. Cold Spring Harb. Perspect. Biol. 2020, 12, a038802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Vennekens, R.; Prenen, J.; Hoenderop, J.G.J.; Droogmans, G.; Bindels, R.J.M. The Single Pore Residue Asp542 Determines Ca2+ Permeation and Mg2+ Block of the Epithelial Ca2+ Channel*. J. Biol. Chem. 2001, 276, 1020–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vennekens, R.; Hoenderop, J.G.J.; Prenen, J.; Stuiver, M.; Willems, P.H.G.M.; Droogmans, G.; Nilius, B.; Bindels, R.J.M. Permeation and Gating Properties of the Novel Epithelial Ca2+ Channel*. J. Biol. Chem. 2000, 275, 3963–3969. [Google Scholar] [CrossRef] [Green Version]
- Nilius, B.; Vennekens, R.; Prenen, J.; Hoenderop, J.G.; Bindels, R.J.; Droogmans, G. Whole-cell and single channel monovalent cation currents through the novel rabbit epithelial Ca2+ channel ECaC. J. Physiol. 2000, 527 Part 2, 239–248. [Google Scholar] [CrossRef]
- Nilius, B.; Prenen, J.; Vennekens, R.; Hoenderop, J.G.J.; Bindels, R.J.M.; Droogmans, G. Modulation of the epithelial calcium channel, ECaC, by intracellular Ca2+. Cell Calcium 2001, 29, 417–428. [Google Scholar] [CrossRef]
- Voets, T.; Janssens, A.; Prenen, J.; Droogmans, G.; Nilius, B. Mg2+-dependent gating and strong inward rectification of the cation channel TRPV6. J. Gen. Physiol. 2003, 121, 245–260. [Google Scholar] [CrossRef] [Green Version]
- Nilius, B.; Prenen, J.; Hoenderop, J.G.J.; Vennekens, R.; Hoefs, S.; Weidema, A.F.; Droogmans, G.; Bindels, R.J.M. Fast and Slow Inactivation Kinetics of the Ca2+Channels ECaC1 and ECaC2 (TRPV5 and TRPV6): ROLE OF THE INTRACELLULAR LOOP LOCATED BETWEEN TRANSMEMBRANE SEGMENTS 2 AND 3*. J. Biol. Chem. 2002, 277, 30852–30858. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.-B.; Chen, X.-Z.; Berger, U.V.; Weremowicz, S.; Morton, C.C.; Vassilev, P.M.; Brown, E.M.; Hediger, M.A. Human Calcium Transport Protein CaT1. Biochem. Biophys. Res. Commun. 2000, 278, 326–332. [Google Scholar] [CrossRef]
- Yelshanskaya, M.V.; Nadezhdin, K.D.; Kurnikova, M.G.; Sobolevsky, A.I. Structure and function of the calcium-selective TRP channel TRPV6. J. Physiol. 2021, 599, 2673–2697. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Chitayat, D.; Sawada, H.; Deardorff, M.A.; McLaughlin, H.M.; Begtrup, A.; Millar, K.; Harrington, J.; Chong, K.; Roifman, M.; et al. TRPV6 Variants Interfere with Maternal-Fetal Calcium Transport through the Placenta and Cause Transient Neonatal Hyperparathyroidism. Am. J. Hum. Genet. 2018, 102, 1104–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, Y.; Sawada, H.; Tokumasu, T.; Suzuki, S.; Ninomiya, S.; Shirai, M.; Mukai, T.; Saito, C.T.; Nishimura, G.; Tominaga, M. Novel TRPV6 mutations in the spectrum of transient neonatal hyperparathyroidism. J. Physiol. Sci. 2020, 70, 33. [Google Scholar] [CrossRef] [PubMed]
- Oracz, G.; Zaród, M.; Ewers, M.; Laumen, H.; Gambin, T.; Kamiński, P.; Grabowska, I.; Drożak, A.; Kwiatkowski, S.; Wertheim-Tysarowska, K.; et al. Loss of function TRPV6 variants are associated with chronic pancreatitis in nonalcoholic early-onset Polish and German patients. Pancreatology 2021, 21, 1434–1442. [Google Scholar] [CrossRef]
- Hamada, S.; Masson, E.; Chen, J.A.-O.; Sakaguchi, R.; Rebours, V.; Buscail, L.; Matsumoto, R.; Tanaka, Y.; Kikuta, K.; Kataoka, F.; et al. Functionally deficient TRPV6 variants contribute to hereditary and familial chronic pancreatitis. Hum. Mutat. 2022, 43, 228–239. [Google Scholar] [CrossRef]
- Xu, X.; Li, N.; Wang, Y.; Yu, J.; Mi, J. Calcium channel TRPV6 promotes breast cancer metastasis by NFATC2IP. Cancer Lett. 2021, 519, 150–160. [Google Scholar] [CrossRef]
- Song, H.; Dong, M.; Zhou, J.; Sheng, W.; Li, X.; Gao, W. Expression and prognostic significance of TRPV6 in the development and progression of pancreatic cancer. Oncol. Rep. 2018, 39, 1432–1440. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.-S.; Xie, X.; Wen, J.; Luo, K.-J.; Liu, Q.-W.; Yang, H.; Hu, Y.; Fu, J.-H. TRPV6 plays a new role in predicting survival of patients with esophageal squamous cell carcinoma. Diagn. Pathol. 2016, 11, 14. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.Y.; Kim, S.H.; Yang, E.K. RNA interference mediated suppression of TRPV6 inhibits the progression of prostate cancer in vitro by modulating cathepsin B and MMP9 expression. Investig. Clin. Urol. 2021, 62, 447–454. [Google Scholar] [CrossRef]
- Wu, Y.; Miyamoto, T.; Li, K.; Nakagomi, H.; Sawada, N.; Kira, S.; Kobayashi, H.; Zakohji, H.; Tsuchida, T.; Fukazawa, M.; et al. Decreased expression of the epithelial Ca2+ channel TRPV5 and TRPV6 in human renal cell carcinoma associated with vitamin D receptor. J. Urol. 2011, 186, 2419–2425. [Google Scholar] [CrossRef]
- Hofer, A.; Kovacs, G.; Zappatini, A.; Leuenberger, M.; Hediger, M.A.; Lochner, M. Design, synthesis and pharmacological characterization of analogs of 2-aminoethyl diphenylborinate (2-APB), a known store-operated calcium channel blocker, for inhibition of TRPV6-mediated calcium transport. Bioorganic Med. Chem. 2013, 21, 3202–3213. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Saotome, K.; McGoldrick, L.L.; Sobolevsky, A.I. Structural bases of TRP channel TRPV6 allosteric modulation by 2-APB. Nat. Commun. 2018, 9, 2465-2465. [Google Scholar] [CrossRef] [PubMed]
- Neuberger, A.; Nadezhdin, K.D.; Sobolevsky, A.I. Structural mechanisms of TRPV6 inhibition by ruthenium red and econazole. Nat. Commun. 2021, 12, 6284-6284. [Google Scholar] [CrossRef] [PubMed]
- Bowen, C.V.; DeBay, D.; Ewart, H.S.; Gallant, P.; Gormley, S.; Ilenchuk, T.T.; Iqbal, U.; Lutes, T.; Martina, M.; Mealing, G.; et al. In vivo detection of human TRPV6-rich tumors with anti-cancer peptides derived from soricidin. PLoS ONE 2013, 8, e58866. [Google Scholar] [CrossRef]
- Xue, H.; Wang, Y.; MacCormack, T.J.; Lutes, T.; Rice, C.; Davey, M.; Dugourd, D.; Ilenchuk, T.T.; Stewart, J.M. Inhibition of Transient Receptor Potential Vanilloid 6 channel, elevated in human ovarian cancers, reduces tumour growth in a xenograft model. J. Cancer 2018, 9, 3196–3207. [Google Scholar] [CrossRef]
- Fu, S.; Hirte, H.; Welch, S.; Ilenchuk, T.T.; Lutes, T.; Rice, C.; Fields, N.; Nemet, A.; Dugourd, D.; Piha-Paul, S.; et al. First-in-human phase I study of SOR-C13, a TRPV6 calcium channel inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 324–333. [Google Scholar] [CrossRef] [Green Version]
- Simonin, C.; Awale, M.; Brand, M.; van Deursen, R.; Schwartz, J.; Fine, M.; Kovacs, G.; Häfliger, P.; Gyimesi, G.; Sithampari, A.; et al. Optimization of TRPV6 Calcium Channel Inhibitors Using a 3D Ligand-Based Virtual Screening Method. Angew. Chem. Int. Ed. 2015, 54, 14748–14752. [Google Scholar] [CrossRef]
- Cunha, M.R.; Bhardwaj, R.; Lindinger, S.; Butorac, C.; Romanin, C.; Hediger, M.A.; Reymond, J.-L. Photoswitchable Inhibitor of the Calcium Channel TRPV6. ACS Med. Chem. Lett. 2019, 10, 1341–1345. [Google Scholar] [CrossRef]
- Cunha, M.R.; Bhardwaj, R.; Carrel, A.L.; Lindinger, S.; Romanin, C.; Parise-Filho, R.; Hediger, M.A.; Reymond, J.-L. Natural product inspired optimization of a selective TRPV6 calcium channel inhibitor. RSC Med. Chem. 2020, 11, 1032–1040. [Google Scholar] [CrossRef]
- Bhardwaj, R.; Lindinger, S.; Neuberger, A.; Nadezhdin, K.D.; Singh, A.K.; Cunha, M.R.; Derler, I.; Gyimesi, G.; Reymond, J.-L.; Hediger, M.A.; et al. Inactivation-mimicking block of the epithelial calcium channel TRPV6. Sci. Adv. 2020, 6, eabe1508. [Google Scholar] [CrossRef]
- Saotome, K.; Singh, A.K.; Yelshanskaya, M.V.; Sobolevsky, A.I. Crystal structure of the epithelial calcium channel TRPV6. Nature 2016, 534, 506–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGoldrick, L.L.; Singh, A.K.; Saotome, K.; Yelshanskaya, M.V.; Twomey, E.C.; Grassucci, R.A.; Sobolevsky, A.I. Opening of the human epithelial calcium channel TRPV6. Nature 2018, 553, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Kever, L.; Cherezova, A.; Zenin, V.; Negulyaev, Y.; Komissarchik, Y.; Semenova, S. Downregulation of TRPV6 channel activity by cholesterol depletion in Jurkat T cell line. Cell Biol. Int. 2019, 43, 965–975. [Google Scholar] [CrossRef] [PubMed]
- Bobkov, D.; Yudintceva, N.; Lomert, E.; Shatrova, A.; Kever, L.; Semenova, S. Lipid raft integrity is required for human leukemia Jurkat T-cell migratory activity. Biochim. Et Biophys. Acta (BBA)—Mol. Cell Biol. Lipids 2021, 1866, 158917. [Google Scholar] [CrossRef]
- Thyagarajan, B.; Benn, B.S.; Christakos, S.; Rohacs, T. Phospholipase C-mediated regulation of transient receptor potential vanilloid 6 channels: Implications in active intestinal Ca2+ transport. Mol. Pharmacol. 2009, 75, 608–616. [Google Scholar] [CrossRef] [Green Version]
- Velisetty, P.; Borbiro, I.; Kasimova, M.A.; Liu, L.; Badheka, D.; Carnevale, V.; Rohacs, T. A molecular determinant of phosphoinositide affinity in mammalian TRPV channels. Sci. Rep. 2016, 6, 27652-27652. [Google Scholar] [CrossRef] [Green Version]
- Cai, R.; Liu, X.; Zhang, R.; Hofmann, L.; Zheng, W.; Amin, M.R.; Wang, L.; Hu, Q.; Peng, J.-B.; Michalak, M.; et al. Autoinhibition of TRPV6 Channel and Regulation by PIP2. Iscience 2020, 23, 101444-101444. [Google Scholar] [CrossRef]
- Vachel, L.; Norez, C.; Jayle, C.; Becq, F.; Vandebrouck, C. The low PLC-δ1 expression in cystic fibrosis bronchial epithelial cells induces upregulation of TRPV6 channel activity. Cell Calcium 2015, 57, 38–48. [Google Scholar] [CrossRef]
- Thyagarajan, B.; Lukacs, V.; Rohacs, T. Hydrolysis of phosphatidylinositol 4,5-bisphosphate mediates calcium-induced inactivation of TRPV6 channels. J. Biol. Chem. 2008, 283, 14980–14987. [Google Scholar] [CrossRef] [Green Version]
- Zakharian, E.; Cao, C.; Rohacs, T. Intracellular ATP supports TRPV6 activity via lipid kinases and the generation of PtdIns(4,5) P2. FASEB J. 2011, 25, 3915–3928. [Google Scholar] [CrossRef] [Green Version]
- Cao, C.; Zakharian, E.; Borbiro, I.; Rohacs, T. Interplay between calmodulin and phosphatidylinositol 4,5-bisphosphate in Ca2+-induced inactivation of transient receptor potential vanilloid 6 channels. J. Biol. Chem. 2013, 288, 5278–5290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W.; Cai, R.; Hofmann, L.; Nesin, V.; Hu, Q.; Long, W.; Fatehi, M.; Liu, X.; Hussein, S.; Kong, T.; et al. Direct Binding between Pre-S1 and TRP-like Domains in TRPP Channels Mediates Gating and Functional Regulation by PIP2. Cell Rep. 2018, 22, 1560–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bousova, K.; Barvik, I.; Herman, P.; Hofbauerová, K.; Monincova, L.; Majer, P.; Zouharova, M.; Vetyskova, V.; Postulkova, K.; Vondrasek, J. Mapping of CaM, S100A1 and PIP2-Binding Epitopes in the Intracellular N- and C-Termini of TRPM4. Int. J. Mol. Sci. 2020, 21, 4323. [Google Scholar] [CrossRef]
- Ufret-Vincenty, C.A.; Klein, R.M.; Hua, L.; Angueyra, J.; Gordon, S.E. Localization of the PIP2 sensor of TRPV1 ion channels. J. Biol. Chem. 2011, 286, 9688–9698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohacs, T. Phosphoinositide regulation of TRPV1 revisited. Pflugers Arch. 2015, 467, 1851–1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fathizadeh, A.; Senning, E.; Elber, R. Impact of the Protonation State of Phosphatidylinositol 4,5-Bisphosphate (PIP2) on the Binding Kinetics and Thermodynamics to Transient Receptor Potential Vanilloid (TRPV5): A Milestoning Study. J. Phys. Chem. B 2021, 125, 9547–9556. [Google Scholar] [CrossRef]
- Hughes, T.E.T.; Pumroy, R.A.; Yazici, A.T.; Kasimova, M.A.; Fluck, E.C.; Huynh, K.W.; Samanta, A.; Molugu, S.K.; Zhou, Z.H.; Carnevale, V.; et al. Structural insights on TRPV5 gating by endogenous modulators. Nat. Commun. 2018, 9, 4198-4198. [Google Scholar] [CrossRef] [Green Version]
- Castro, B.M.; Torreno-Pina, J.A.; van Zanten, T.S.; Garcia-Parajo, M.F. Chapter 6—Biochemical and Imaging Methods to Study Receptor Membrane Organization and Association with Lipid Rafts. In Methods in Cell Biology; Conn, P.M., Ed.; Academic Press: New York, NY, USA, 2013; Volume 117, pp. 105–122. [Google Scholar]
- Lenne, P.-F.; Wawrezinieck, L.; Conchonaud, F.; Wurtz, O.; Boned, A.; Guo, X.-J.; Rigneault, H.; He, H.-T.; Marguet, D. Dynamic molecular confinement in the plasma membrane by microdomains and the cytoskeleton meshwork. EMBO J. 2006, 25, 3245–3256. [Google Scholar] [CrossRef] [Green Version]
- Neuvonen, M.; Manna, M.; Mokkila, S.; Javanainen, M.; Rog, T.; Liu, Z.; Bittman, R.; Vattulainen, I.; Ikonen, E. Enzymatic oxidation of cholesterol: Properties and functional effects of cholestenone in cell membranes. PLoS ONE 2014, 9, e103743. [Google Scholar] [CrossRef] [Green Version]
- Derler, I.; Jardin, I.; Stathopulos, P.B.; Muik, M.; Fahrner, M.; Zayats, V.; Pandey, S.K.; Poteser, M.; Lackner, B.; Absolonova, M.; et al. Cholesterol modulates Orai1 channel function. Sci. Signal. 2016, 9, ra10. [Google Scholar] [CrossRef] [Green Version]
- Nishijo, J.; Moriyama, S.; Shiota, S. Interactions of Cholesterol with Cyclodextrins in Aqueous Solution. Chem. Pharm. Bull. 2003, 51, 1253–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahammad, S.; Parmryd, I. Cholesterol depletion using methyl-β-cyclodextrin. Methods Mol. Biol. 2015, 1232, 91–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behnke, O.; Tranum-Jensen, J.; van Deurs, B. Filipin as a cholesterol probe. II. Filipin-cholesterol interaction in red blood cell membranes. Eur. J. Cell Biol. 1984, 35, 200–215. [Google Scholar] [PubMed]
- Gimpl, G.; Gehrig-Burger, K. Cholesterol reporter molecules. Biosci. Rep. 2007, 27, 335–358. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Barrantes, F.J. How cholesterol interacts with membrane proteins: An exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains. Front. Physiol. 2013, 4, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fantini, J.; Di Scala, C.; Evans, L.S.; Williamson, P.T.F.; Barrantes, F.J. A mirror code for protein-cholesterol interactions in the two leaflets of biological membranes. Sci. Rep. 2016, 6, 21907-21907. [Google Scholar] [CrossRef] [Green Version]
- Levitan, I.; Singh, D.K.; Rosenhouse-Dantsker, A. Cholesterol binding to ion channels. Front. Physiol. 2014, 5, 65. [Google Scholar] [CrossRef] [Green Version]
- Kumar, G.A.; Chattopadhyay, A. Membrane cholesterol regulates endocytosis and trafficking of the serotonin(1A) receptor: Insights from acute cholesterol depletion. Biochim. Et Biophys. Acta. Mol. Cell Biol. Lipids 2021, 1866, 158882. [Google Scholar] [CrossRef]
- Oddi, S.; Stepniewski, T.M.; Totaro, A.; Selent, J.; Scipioni, L.; Dufrusine, B.; Fezza, F.; Dainese, E.; Maccarrone, M. Palmitoylation of cysteine 415 of CB(1) receptor affects ligand-stimulated internalization and selective interaction with membrane cholesterol and caveolin 1. Biochim. Et Biophys. Acta. Mol. Cell Biol. Lipids 2017, 1862, 523–532. [Google Scholar] [CrossRef]
- Banach-Orłowska, M.; Wyszyńska, R.; Pyrzyńska, B.; Maksymowicz, M.; Gołąb, J.; Miączyńska, M. Cholesterol restricts lymphotoxin β receptor-triggered NF-κB signaling. Cell Commun. Signal. 2019, 17, 171. [Google Scholar] [CrossRef] [Green Version]
- Gwozdz, T.; Dutko-Gwozdz, J.; Schafer, C.; Bolotina, V.M. Overexpression of Orai1 and STIM1 proteins alters regulation of store-operated Ca2+ entry by endogenous mediators. J. Biol. Chem. 2012, 287, 22865–22872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fecher-Trost, C.; Wissenbach, U.; Beck, A.; Schalkowsky, P.; Stoerger, C.; Doerr, J.; Dembek, A.; Simon-Thomas, M.; Weber, A.; Wollenberg, P.; et al. The in vivo TRPV6 protein starts at a non-AUG triplet, decoded as methionine, upstream of canonical initiation at AUG. J. Biol. Chem. 2013, 288, 16629–16644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohórquez-Hernández, A.; Gratton, E.; Pacheco, J.; Asanov, A.; Vaca, L. Cholesterol modulates the cellular localization of Orai1 channels and its disposition among membrane domains. Biochim. Et Biophys. Acta. Mol. Cell Biol. Lipids 2017, 1862, 1481–1490. [Google Scholar] [CrossRef] [PubMed]
- Pani, B.; Singh, B.B. Lipid rafts/caveolae as microdomains of calcium signaling. Cell Calcium 2009, 45, 625–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dionisio, N.; Galán, C.; Jardín, I.; Salido, G.M.; Rosado, J.A. Lipid rafts are essential for the regulation of SOCE by plasma membrane resident STIM1 in human platelets. Biochim. Et Biophys. Acta 2011, 1813, 431–437. [Google Scholar] [CrossRef] [Green Version]
- Pacheco, J.; Dominguez, L.; Bohórquez-Hernández, A.; Asanov, A.; Vaca, L. A cholesterol-binding domain in STIM1 modulates STIM1-Orai1 physical and functional interactions. Sci. Rep. 2016, 6, 29634-29634. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Lin, W.Y.; Leibow, S.; Morateck, A.; Ahuja, M.; Muallem, S. TRPC3 channel gating by lipids requires localization at the ER/PM junctions defined by STIM1. J. Cell Biol. 2022, 221, e202107120. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Humer, C.; Lindinger, S.; Carrel, A.L.; Romanin, C.; Höglinger, C. TRPV6 Regulation by Cis-22a and Cholesterol. Biomolecules 2022, 12, 804. https://doi.org/10.3390/biom12060804
Humer C, Lindinger S, Carrel AL, Romanin C, Höglinger C. TRPV6 Regulation by Cis-22a and Cholesterol. Biomolecules. 2022; 12(6):804. https://doi.org/10.3390/biom12060804
Chicago/Turabian StyleHumer, Christina, Sonja Lindinger, Aline L. Carrel, Christoph Romanin, and Carmen Höglinger. 2022. "TRPV6 Regulation by Cis-22a and Cholesterol" Biomolecules 12, no. 6: 804. https://doi.org/10.3390/biom12060804
APA StyleHumer, C., Lindinger, S., Carrel, A. L., Romanin, C., & Höglinger, C. (2022). TRPV6 Regulation by Cis-22a and Cholesterol. Biomolecules, 12(6), 804. https://doi.org/10.3390/biom12060804