
Comparative Study of Transcriptome in the Hearts Isolated from Mice, Rats, and Humans

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. RNA Extractions from Experimental Animals
2.2. RNA Extractions from Heart Donations
2.3. Mouse Gene Expression Microarrays (Illumina)
2.4. Rat Gene Expression Microarrays (Agilent)
2.5. RNA-Seq on the Donated Hearts
2.6. The Evaluation of Similarities of Expression Intensity among Species or Regions
2.7. Identification of Heart Region-Specific Expressed Genes
2.8. Comparison of Region-Specific Expressed Genes among Three Species
2.9. Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
3. Results
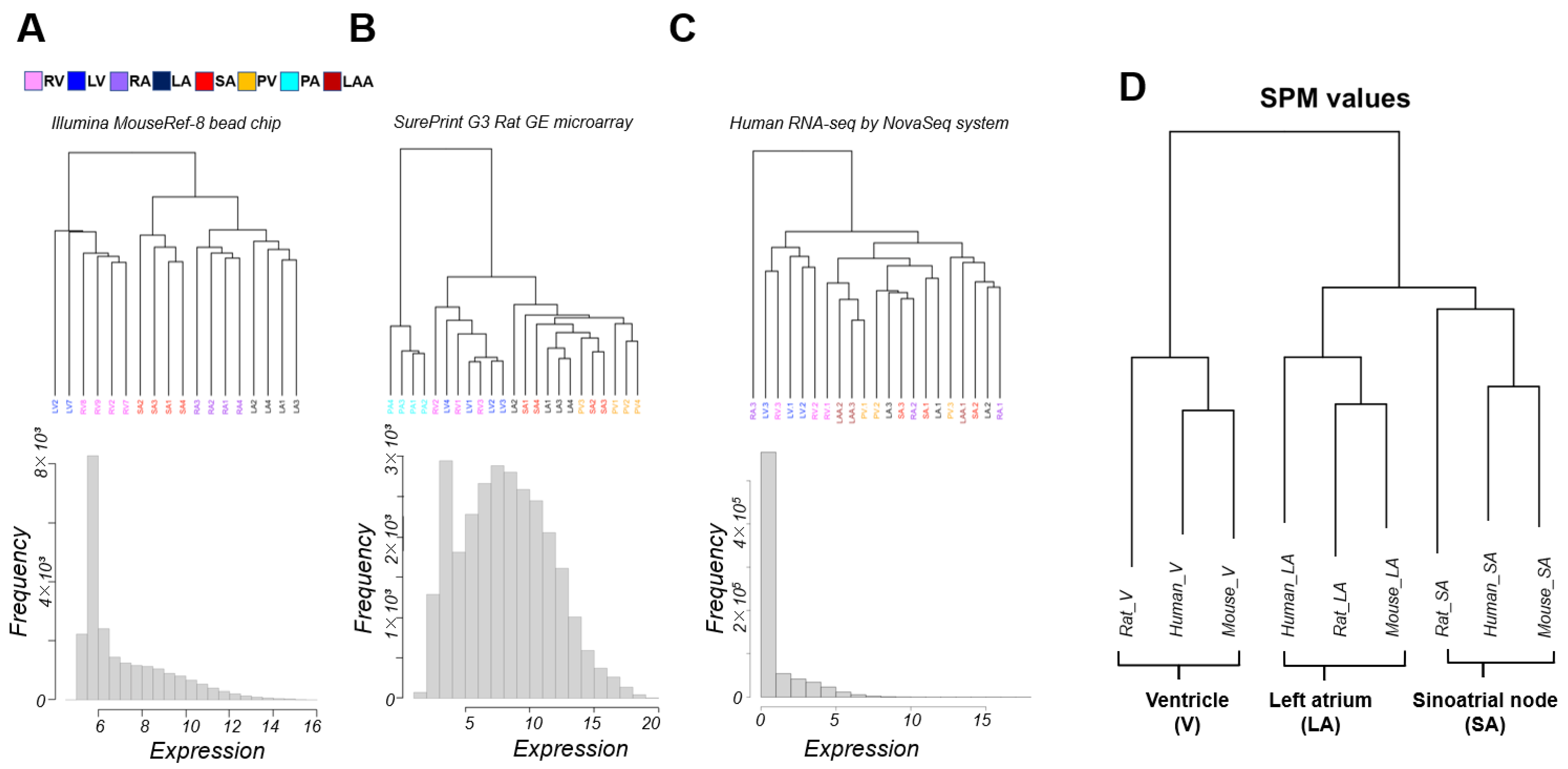
3.1. Classification of Cardiac Regions Based on Gene Expression Patterns
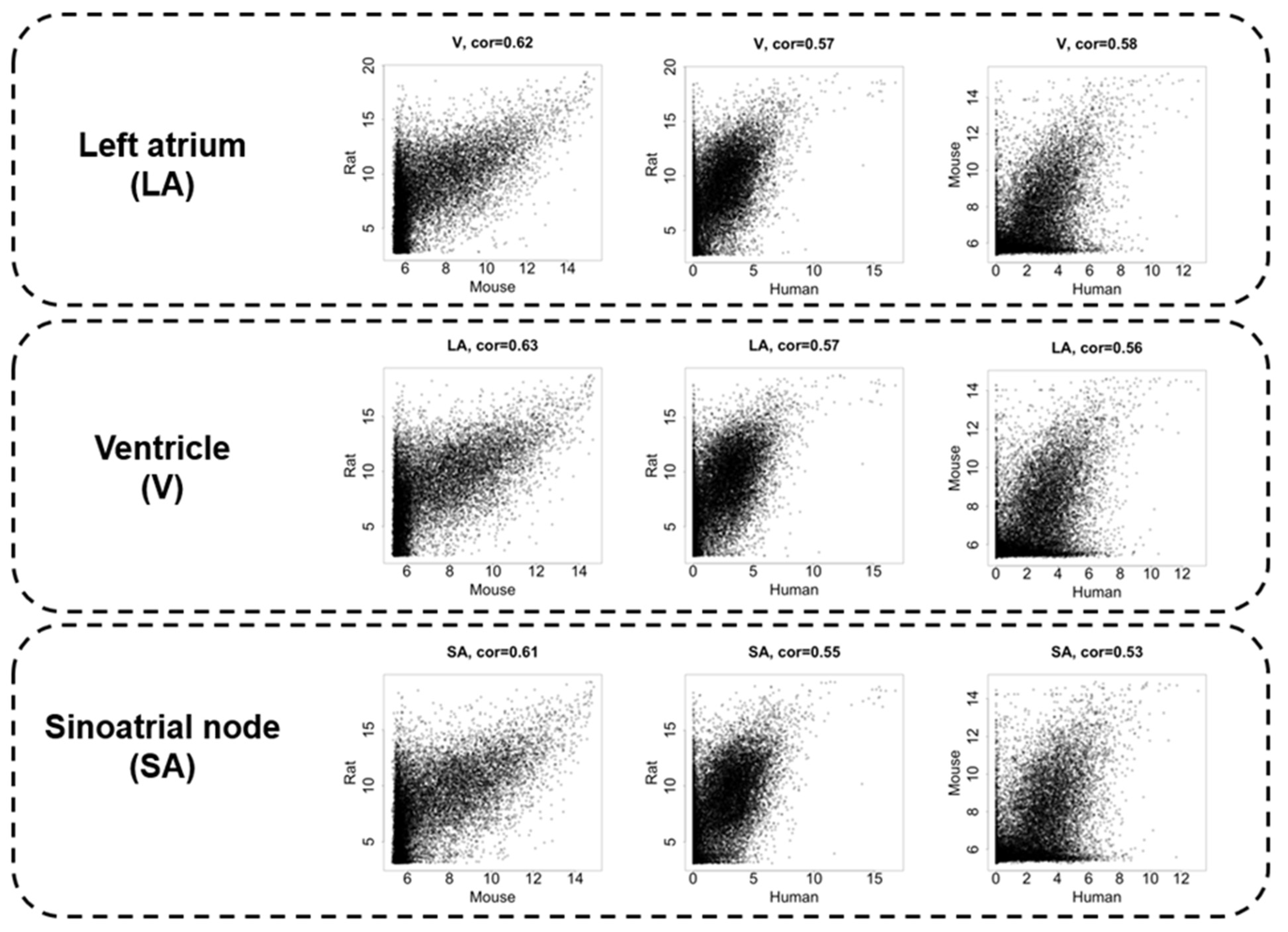
3.2. Interspecies Correlation of the Transcriptome
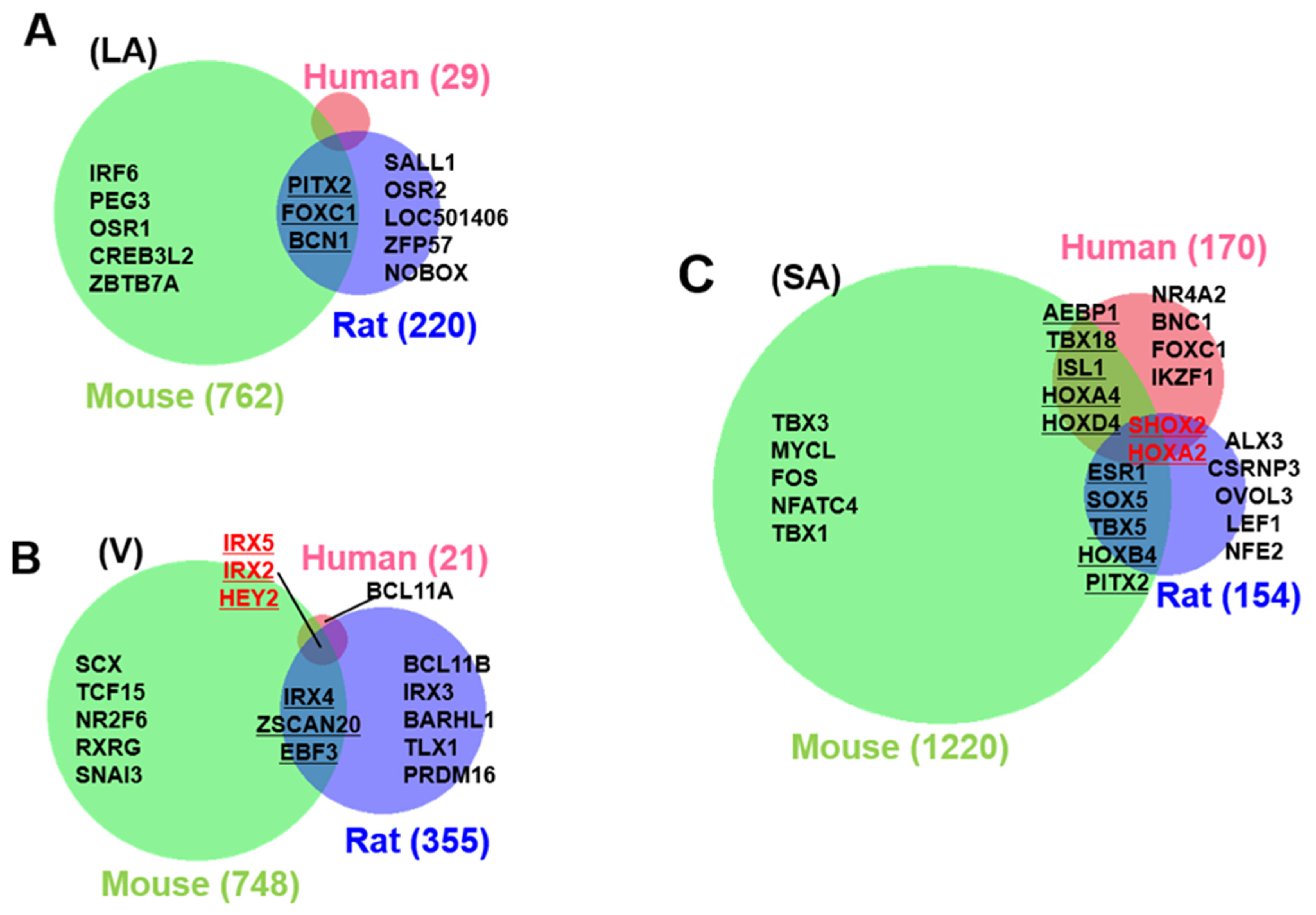
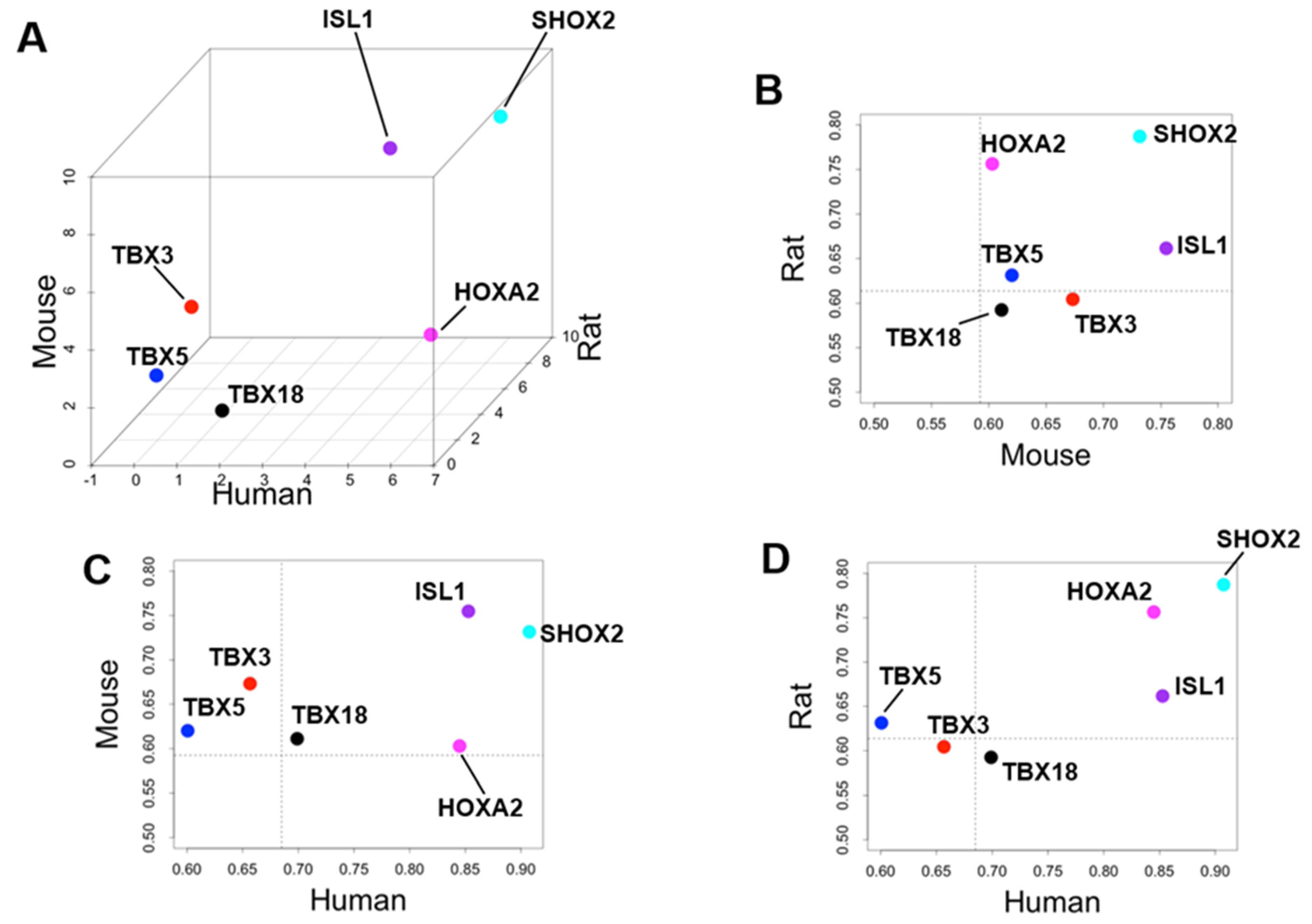
3.3. Identification of Cardiac Region-Specific Transcription Factors
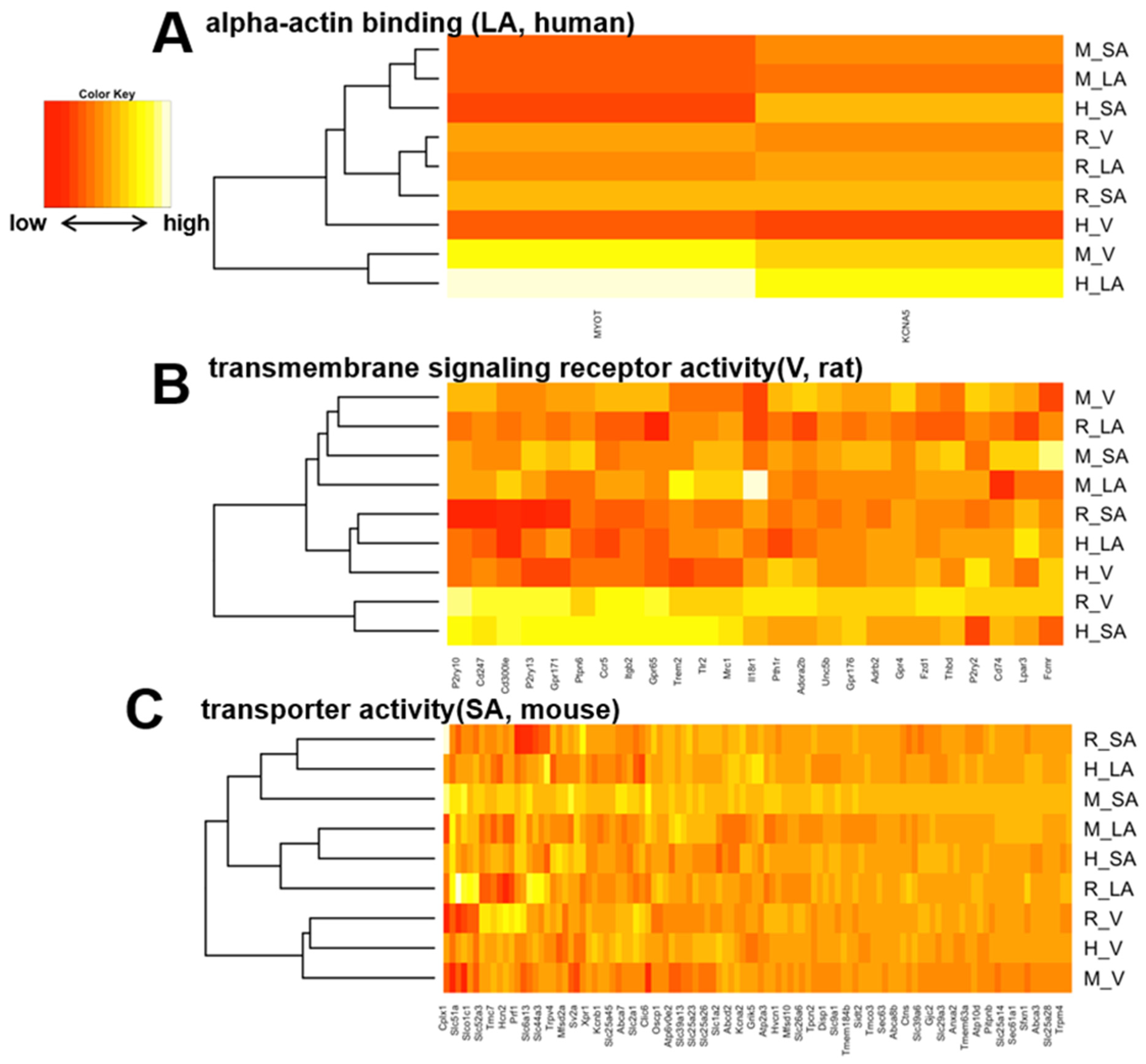
3.4. Heart Region-Specific Genes across Species, and Ontology Analysis of Heart Region and Species-Specific Genes
4. Discussions
4.1. Major Findings
4.2. Priority of SHOX2 over HOXA2 and ISL1
4.3. Validation of Our Analysis from Other Transcriptome Analysis
4.4. Limitations
4.5. Future Perspectives
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huxley, A.F.; Niedergerke, R. Structural changes in muscle during contraction: Interference microscopy of living muscle fibres. Nature 1954, 173, 971–973. [Google Scholar] [CrossRef] [PubMed]
- Huxley, H.; Hanson, J. Changes in the Cross-Striations of Muscle during Contraction and Stretch and their Structural Interpretation. AAPG Mem. 2011, 42, 61–102. [Google Scholar] [CrossRef] [PubMed]
- Ebashi, S.; Endo, M.; Ohtsuki, I. Control of muscle contraction. Q. Rev. Biophys. 1969, 2, 351–384. [Google Scholar] [CrossRef] [PubMed]
- Noda, M.; Shimizu, S.; Tanabe, T.; Takai, T.; Kayano, T.; Ikeda, T.; Takahashi, H.; Nakayama, H.; Kanaoka, Y.; Minamino, N.; et al. Primary structure of Electrophorus electricus sodium channel deduced from cDNA sequence. Nature 1984, 312, 121–127. [Google Scholar] [CrossRef]
- Tanabe, T.; Takeshima, H.; Hikami, A.; Flockerzi, V.; Takahashi, H.; Kangawa, K.; Kojima, M.; Matsuo, H.; Hirose, T.; Numa, S. Primary structure of the receptor for calcium channel blockers from skeletal muscle. Jpn. J. Pharmacol. 1988, 46, 33. [Google Scholar] [CrossRef]
- Login, S.; Takeshima, H.; Nishimura, S.; Matsumoto, T.; Ishida, H.; Kangawa, K.; Minamino, N.; Matsuo, H.; Hanaoka, M.; Hirose, T.; et al. Primary structure and expression from complementary DNA of skeletal muscle ryanodine receptor Hiroshi. Nature 1989, 339, 439–445. [Google Scholar]
- Wang, Q.; Curran, M.E.; Splawski, I.; Burn, T.C.; Millholland, J.M.; VanRaay, T.J.; Shen, J.; Timothy, K.W.; Vincent, G.M.; de Jager, T.; et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat. Genet. 1996, 12, 17–23. [Google Scholar] [CrossRef]
- Noma, A.; Irisawa, H. Membrane currents in the rabbit sinoatrial node cell as studied by the double microelectrode method. Pflügers Arch. Eur. J. Physiol. 1976, 364, 45–52. [Google Scholar] [CrossRef]
- Brown, B.Y.H.; Difrancesco, D. Voltege-clamp investigation of membrane current underlying pace-maker activity in rabbit sino-atrial node. J. Physiol. 1980, 308, 331–351. [Google Scholar] [CrossRef]
- Stieber, J.; Herrmann, S.; Feil, S.; Löster, J.; Feil, R.; Biel, M.; Hofmann, F.; Ludwig, A. The hyperpolarization-activated channel HCN4 is required for the generation of pacemaker action potentials in the embryonic heart. Proc. Natl. Acad. Sci. USA 2003, 100, 15235–15240. [Google Scholar] [CrossRef] [Green Version]
- Vinogradova, T.M.; Lyashkov, A.E.; Zhu, W.; Ruknudin, A.M.; Sirenko, S.; Yang, D.; Deo, S.; Barlow, M.; Johnson, S.; Caffrey, J.L.; et al. High basal protein kinase A-dependent phosphorylation drives rhythmic internal Ca2+ store oscillations and spontaneous beating of cardiac pacemaker cells. Circ. Res. 2006, 98, 505–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordan, R.; Gwathmey, J.K.; Xie, L.-H. Autonomic and endocrine control of cardiovascular function. World J. Cardiol. 2015, 7, 204. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.E.; Al-Khalidi, A.H.; Bonser, R.S.; Clutton-Brock, T.; Morton, D.; Paterson, D.; Townend, J.N.; Coote, J.H. Vagus nerve stimulation decreases left ventricular contractility in vivo in the human and pig heart. J. Physiol. 2001, 534, 547–552. [Google Scholar] [CrossRef]
- Himeno, Y.; Toyoda, F.; Satoh, H.; Amano, A.; Cha, C.Y.; Matsuura, H.; Noma, A. Minor contribution of cytosolic Ca2+ transients to the pacemaker rhythm in guinea pig sinoatrial node cells. Am. J. Physiol.—Heart Circ. Physiol. 2011, 300, 251–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namekata, I.; Jitsukata, K.; Fukuda, A.; Odaka, R.; Hamaguchi, S.; Tanaka, H. Intracellular Ca2+-Mediated Mechanisms for the Pacemaker Depolarization of the Mouse and Guinea Pig Sinus Node Tissue. Biomolecules 2022, 12, 377. [Google Scholar] [CrossRef]
- Ono, K.; Trautweint, W. Potentiation by cyclic GMP of beta-adrenergic effect on Ca2+ current in guinea-pig ventricular cells. J. Physiol. 1991, 443, 387–404. [Google Scholar] [CrossRef]
- Mary, P.O.I.S.; Lohmannt, S.M.; Waltert, U.; Fischmeister, R. Ca2+ current is regulated by cyclic GMP-dependent protein kinase in mammalian cardiac myocytes. Proc. Natl. Acad. Sci. USA 1991, 88, 1197–1201. [Google Scholar] [CrossRef] [Green Version]
- London, B.; Wang, D.W.; Hill, J.A.; Bennett, P.B. The transient outward current in mice lacking the potassium channel gene Kv1.4. J. Physiol. 1998, 509, 171–182. [Google Scholar] [CrossRef]
- Wettwer, E.; Amos, G.; Gath, J.; Zerkowski, H.R.; Reidemeister, J.C.; Ravens, U. Transient outward current in human and rat ventricular myocytes. Cardiovasc. Res. 1993, 27, 1662–1669. [Google Scholar] [CrossRef]
- Li, G.R.; Yang, B.; Sun, H.; Baumgarten, C.M. Existence of a transient outward K+ current in guinea pig cardiac myocytes. Am. J. Physiol.—Heart Circ. Physiol. 2000, 279, 130–138. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, S.; Ehara, T.; Noma, A. Chloride-sensitive nature of the adrenaline-induced current in guinea-pig cardiac myocytes. J. Physiol. 1990, 425, 579–598. [Google Scholar] [CrossRef] [PubMed]
- Takano, M.; Noma, A. Distribution of the isoprenaline-induced chloride current in rabbit heart. Pflügers Arch. Eur. J. Physiol. 1992, 420, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Kawamura, K.; Nakamura, Y.; Ono, K. Pathological impact of hyperpolarization-activated chloride current peculiar to rat pulmonary vein cardiomyocytes. J. Mol. Cell. Cardiol. 2014, 66, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Takagi, D.; Okamoto, Y.; Ohba, T.; Yamamoto, H.; Ono, K. Comparative study of hyperpolarization-activated currents in pulmonary vein cardiomyocytes isolated from rat, guinea pig, and rabbit. J. Physiol. Sci. 2020, 70, 6. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, J.R.; Cha, T.-J.; Zhang, L.; Chartier, D.; Villeneuve, L.; Hébert, T.E.; Nattel, S. Characterization of a hyperpolarization-activated time-dependent potassium current in canine cardiomyocytes from pulmonary vein myocardial sleeves and left atrium. J. Physiol. 2004, 557, 583–597. [Google Scholar] [CrossRef]
- Okamoto, Y.; Nagasawa, Y.; Obara, Y.; Ishii, K.; Takagi, D.; Ono, K. Molecular identification of HSPA8 as an accessory protein of a hyperpolarization-activated chloride channel from rat pulmonary vein cardiomyocytes. J. Biol. Chem. 2019, 294, 16049–16061. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Ruotti, V.; Stewart, R.M.; Thomson, J.A.; Dewey, C.N. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 2009, 26, 493–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.K.; Matkovich, S.J.; Nerbonne, J.M. Regional Differences in mRNA and lncRNA Expression Profiles in Non-Failing Human Atria and Ventricles. Sci. Rep. 2018, 8, 13919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Nishiyama, T.; Shimizu, K.; Kadota, K. TCC: An R package for comparing tag count data with robust normalization strategies. BMC Bioinform. 2013, 14, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galili, T. dendextend: An R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics 2015, 31, 3718–3720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, J.B.; Hu, S.C.; Wang, H.; Zou, Q.; Ji, Z.L. PaGeFinder: Quantitative identification of spatiotemporal pattern genes. Bioinformatics 2012, 28, 1544–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Costantini, D.L.; Arruda, E.P.; Agarwal, P.; Kim, K.H.; Zhu, Y.; Zhu, W.; Lebel, M.; Cheng, C.W.; Park, C.Y.; Pierce, S.A.; et al. The homeodomain transcription factor Irx5 establishes the mouse cardiac ventricular repolarization gradient. Cell 2005, 123, 347–358. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Rosen, A.; Bruneau, B.G.; Hui, C.C.; Backx, P.H. Iroquois homeodomain transcription factors in heart development and function. Circ. Res. 2012, 110, 1513–1524. [Google Scholar] [CrossRef] [Green Version]
- Gibb, N.; Lazic, S.; Yuan, X.; Deshwar, A.R.; Leslie, M.; Wilson, M.D.; Scott, I.C. Hey2 regulates the size of the cardiac progenitor pool during vertebrate heart development. Development 2018, 145, dev167510. [Google Scholar] [CrossRef] [Green Version]
- Blaschke, R.J.; Hahurij, N.D.; Kuijper, S.; Just, S.; Wisse, L.J.; Deissler, K.; Maxelon, T.; Anastassiadis, K.; Spitzer, J.; Hardt, S.E.; et al. Targeted Mutation Reveals Essential Functions of the Homeodomain Transcription Factor Shox2 in Sinoatrial and Pacemaking Development. Circulation 2007, 115, 1830–1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinoza-Lewis, R.A.; Yu, L.; He, F.; Liu, H.; Tang, R.; Shi, J.; Sun, X.; Martin, J.F.; Wang, D.; Yang, J.; et al. Shox2 is essential for the differentiation of cardiac pacemaker cells by repressing Nkx2-5. Dev. Biol. 2009, 327, 376–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Eif, V.W.W.; Stefanovic, S.; van Duijvenboden, K.; Bakker, M.; Wakker, V.; de Gier-de Vries, C.; Zaffran, S.; Verkerk, A.O.; Boukens, B.J.; Christoffels, V.M. Transcriptome analysis of mouse and human sinoatrial node cells reveals a conserved genetic program. Development 2019, 146, dev173161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Li, N.; Liu, L.; Zhang, H.; Xue, X.; Shao, X.; Zhang, Y.; Lang, X. Genetically Modified Porcine Mesenchymal Stem Cells by Lentiviral Tbx18 Create a Biological Pacemaker. Stem Cells Int. 2019, 2019, 3621314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grijalva, S.I.; Gu, J.M.; Li, J.; Fernandez, N.; Fan, J.; Sung, J.H.; Lee, S.Y.; Herndon, C.; Buckley, E.M.; Park, S.J.; et al. Engineered Cardiac Pacemaker Nodes Created by TBX18 Gene Transfer Overcome Source–Sink Mismatch. Adv. Sci. 2019, 6, 1901099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiese, C.; Grieskamp, T.; Airik, R.; Mommersteeg, M.T.M.; Gardiwal, A.; de Gier-de Vries, C.; Schuster-Gossler, K.; Moorman, A.F.M.; Kispert, A.; Christoffels, V.M. Formation of the Sinus Node Head and Differentiation of Sinus Node Myocardium Are Independently Regulated by Tbx18 and Tbx3. Circ. Res. 2009, 104, 388–397. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, N.; Liang, W.; Marbán, E.; Cho, H.C. Direct conversion of quiescent cardiomyocytes to pacemaker cells by expression of Tbx18. Nat. Biotechnol. 2013, 31, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Zhang, Q.; Cattaneo, P.; Zhuang, S.; Gong, X.; Spann, N.J.; Jiang, C.; Cao, X.; Zhao, X.; Zhang, X.; et al. Transcription factor ISL1 is essential for pacemaker development and function. J. Clin. Investig. 2015, 125, 3256–3268. [Google Scholar] [CrossRef] [Green Version]
- Vedantham, V.; Galang, G.; Evangelista, M.; Deo, R.C.; Srivastava, D. RNA sequencing of mouse sinoatrial node reveals an upstream regulatory role for Islet-1 in cardiac pacemaker cells. Circ. Res. 2015, 116, 797–803. [Google Scholar] [CrossRef]
- Zhao, H.; Wang, F.; Zhang, W.; Yang, M.; Tang, Y.; Wang, X.; Zhao, Q.; Huang, C. Overexpression of TBX3 in human induced pluripotent stem cells (hiPSCs) increases their differentiation into cardiac pacemaker-like cells. Biomed. Pharmacother. 2020, 130, 110612. [Google Scholar] [CrossRef]
- Hoogaars, W.M.H.; Engel, A.; Brons, J.F.; Verkerk, A.O.; de Lange, F.J.; Wong, L.Y.E.; Bakker, M.L.; Clout, D.E.; Wakker, V.; Barnett, P.; et al. Tbx3 controls the sinoatrial node gene program and imposes pacemaker function on the atria. Genes Dev. 2007, 21, 1098–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnolds, D.E.; Liu, F.; Fahrenbach, J.P.; Kim, G.H.; Schillinger, K.J.; Smemo, S.; McNally, E.M.; Nobrega, M.A.; Patel, V.V.; Moskowitz, I.P. TBX5 drives Scn5a expression to regulate cardiac conduction system function. J. Clin. Investig. 2012, 122, 2509–2518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barefield, D.Y.; Puckelwartz, M.J.; Kim, E.Y.; Wilsbacher, L.D.; Vo, A.H.; Waters, E.A.; Earley, J.U.; Hadhazy, M.; Dellefave-Castillo, L.; Pesce, L.L.; et al. Experimental Modeling Supports a Role for MyBP-HL as a Novel Myofilament Component in Arrhythmia and Dilated Cardiomyopathy. Circulation 2017, 136, 1477–1491. [Google Scholar] [CrossRef]
- Harris, S.P.; Lyons, R.G.; Bezold, K.L. In the thick of it: HCM-causing mutations in myosin binding proteins of the thick filament. Circ. Res. 2011, 108, 751–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Yuan, Y.; Wong, J.; Fishbein, M.C.; Chen, P.S.; Everett, T.H. Recording Intrinsic Nerve Activity at the Sinoatrial Node in Normal Dogs with High-Density Mapping. Circ. Arrhythm. Electrophysiol. 2021, 14, e008610. [Google Scholar] [CrossRef] [PubMed]
- Brahmajothi, M.V.; Morales, M.J.; Campbell, D.L.; Steenbergen, C.; Strauss, H.C. Expression and distribution of voltage-gated ion channels in ferret sinoatrial node. Physiol. Genom. 2010, 42 A, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Bruneau, B.G.; Nemer, G.; Schmitt, J.P.; Charron, F.; Robitaille, L.; Caron, S.; Conner, D.A.; Gessler, M.; Nemer, M.; Seidman, C.E.; et al. A murine model of Holt-Oram syndrome defines roles of the T-Box transcription factor Tbx5 in cardiogenesis and disease. Cell 2001, 106, 709–721. [Google Scholar] [CrossRef] [Green Version]
- Moskowitz, I.P.G.; Pizard, A.; Patel, V.V.; Bruneau, B.G.; Kim, J.B.; Kupershmidt, S.; Roden, D.; Berul, C.I.; Seidman, C.E.; Seidman, J.G. The T-Box transcription factor Tbx5 is required for the patterning and maturation of the murine cardiac conduction system. Development 2004, 131, 4107–4116. [Google Scholar] [CrossRef] [Green Version]
- Olson, T.M.; Alekseev, A.E.; Liu, X.K.; Park, S.; Zingman, L.V.; Bienengraeber, M.; Sattiraju, S.; Ballew, J.D.; Jahangir, A.; Terzic, A. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum. Mol. Genet. 2006, 15, 2185–2191. [Google Scholar] [CrossRef]
- Li, N.; Wang, Z.S.; Wang, X.H.; Xu, Y.J.; Qiao, Q.; Li, X.M.; Di, R.M.; Guo, X.J.; Li, R.G.; Zhang, M.; et al. A SHOX2 loss-of-function mutation underlying familial atrial fibrillation. Int. J. Med. Sci. 2018, 15, 1564–1572. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, S.; Berger, I.M.; Glaser, A.; Bacon, C.; Li, L.; Gretz, N.; Steinbeisser, H.; Rottbauer, W.; Just, S.; Rappold, G. Islet1 is a direct transcriptional target of the homeodomain transcription factor Shox2 and rescues the Shox2-mediated bradycardia. Basic Res. Cardiol. 2013, 108, 339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herriges, J.C.; Yi, L.; Hines, E.A.; Harvey, J.F.; Xu, G.; Gray, P.A.; Ma, Q.; Sun, X. Genome-scale study of transcription factor expression in the branching mouse lung. Dev. Dyn. 2012, 241, 1432–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, R.; Liang, X.; Cheedipudi, S.; Cordero, J.; Jiang, X.; Zhang, Q.; Caputo, L.; Günther, S.; Kuenne, C.; Ren, Y.; et al. Pioneering function of Isl1 in the epigenetic control of cardiomyocyte cell fate. Cell Res. 2019, 29, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Doisne, N.; Maupoil, V.; Cosnay, P.; Findlay, I. Catecholaminergic automatic activity in the rat pulmonary vein: Electrophysiological differences between cardiac muscle in the left atrium and pulmonary vein. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H102–H108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, Y.; Takano, M.; Ohba, T.; Ono, K. Arrhythmogenic coupling between the Na+–Ca2+ exchanger and inositol 1,4,5-triphosphate receptor in rat pulmonary vein cardiomyocytes. J. Mol. Cell. Cardiol. 2012, 52, 988–997. [Google Scholar] [CrossRef]
- Okada, D.; Zheng, C.; Cheng, J.H.; Yamada, R. Cell population-based framework of genetic epidemiology in the single-cell omics era. BioEssays 2022, 44, 2100118. [Google Scholar] [CrossRef]
- Litviňuková, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Worth, C.L.; Lindberg, E.L.; Kanda, M.; Polanski, K.; Heinig, M.; Lee, M.; et al. Cells of the adult human heart. Nature 2020, 588, 466–472. [Google Scholar] [CrossRef]
- Swindell, W.R.; List, E.O.; Berryman, D.E.; Kopchick, J.J. Transcriptional profiling identifies strain-specific effects of caloric restriction and opposite responses in human and mouse white adipose tissue. Aging 2018, 10, 701–746. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Nellåker, C.; McIntyre, R.E.; Buendia-Buendia, J.E.; Bumpstead, S.; Ponting, C.P.; Flint, J.; Durbin, R.; Keane, T.M.; Adams, D.J. High levels of RNA-editing site conservation amongst 15 laboratory mouse strains. Genome Biol. 2012, 13, r26. [Google Scholar] [CrossRef] [Green Version]
- Barnabei, M.S.; Palpant, N.J.; Metzger, J.M. Influence of genetic background on ex vivo and in vivo cardiac function in several commonly used inbred mouse strains. Physiol. Genom. 2010, 42 A, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Tanase, H.; Yamori, Y.; Hansen, C.T.; Lovenberg, W. Heart Size in Inbred Strains of Rats. Hypertension 1982, 4, 864–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aziz, Q.; Nobles, M.; Tinker, A. Acute Isolation of Cells from Murine Sino-atrial Node. Bio Protoc. 2020, 10, e3477. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Okada, D.; Okamoto, Y.; Io, T.; Oka, M.; Kobayashi, D.; Ito, S.; Yamada, R.; Ishii, K.; Ono, K. Comparative Study of Transcriptome in the Hearts Isolated from Mice, Rats, and Humans. Biomolecules 2022, 12, 859. https://doi.org/10.3390/biom12060859
Okada D, Okamoto Y, Io T, Oka M, Kobayashi D, Ito S, Yamada R, Ishii K, Ono K. Comparative Study of Transcriptome in the Hearts Isolated from Mice, Rats, and Humans. Biomolecules. 2022; 12(6):859. https://doi.org/10.3390/biom12060859
Chicago/Turabian StyleOkada, Daigo, Yosuke Okamoto, Toshiro Io, Miho Oka, Daiki Kobayashi, Suzuka Ito, Ryo Yamada, Kuniaki Ishii, and Kyoichi Ono. 2022. "Comparative Study of Transcriptome in the Hearts Isolated from Mice, Rats, and Humans" Biomolecules 12, no. 6: 859. https://doi.org/10.3390/biom12060859
APA StyleOkada, D., Okamoto, Y., Io, T., Oka, M., Kobayashi, D., Ito, S., Yamada, R., Ishii, K., & Ono, K. (2022). Comparative Study of Transcriptome in the Hearts Isolated from Mice, Rats, and Humans. Biomolecules, 12(6), 859. https://doi.org/10.3390/biom12060859