
Potential to Eradicate Cancer Stemness by Targeting Cell Surface GRP78


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
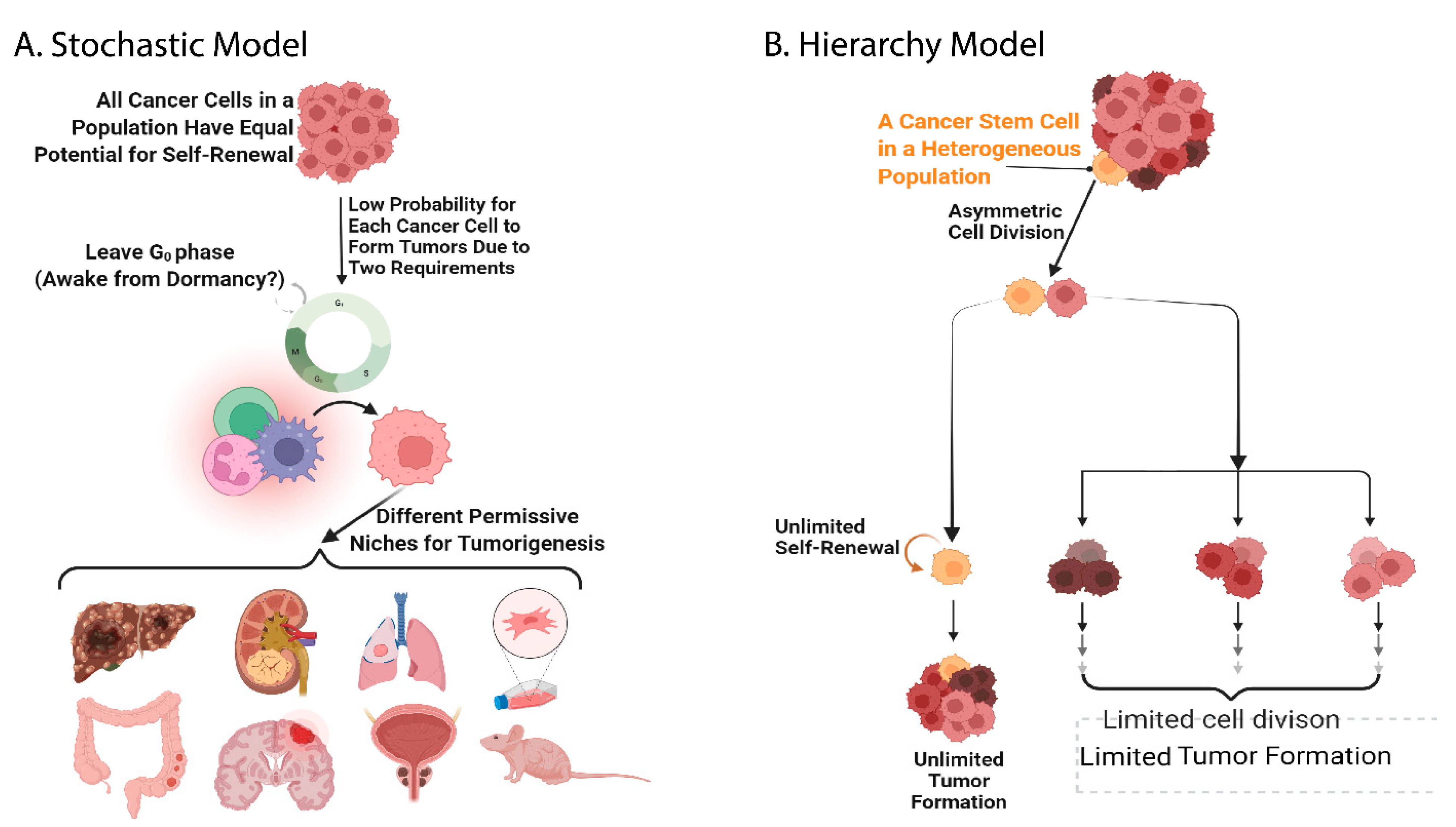
2. The History of Stem Cell Investigation and Evidence of the Existence of Cancer Stem Cells
3. Identification of Cancer Stem Cell Surface Markers in Different Cancer Models
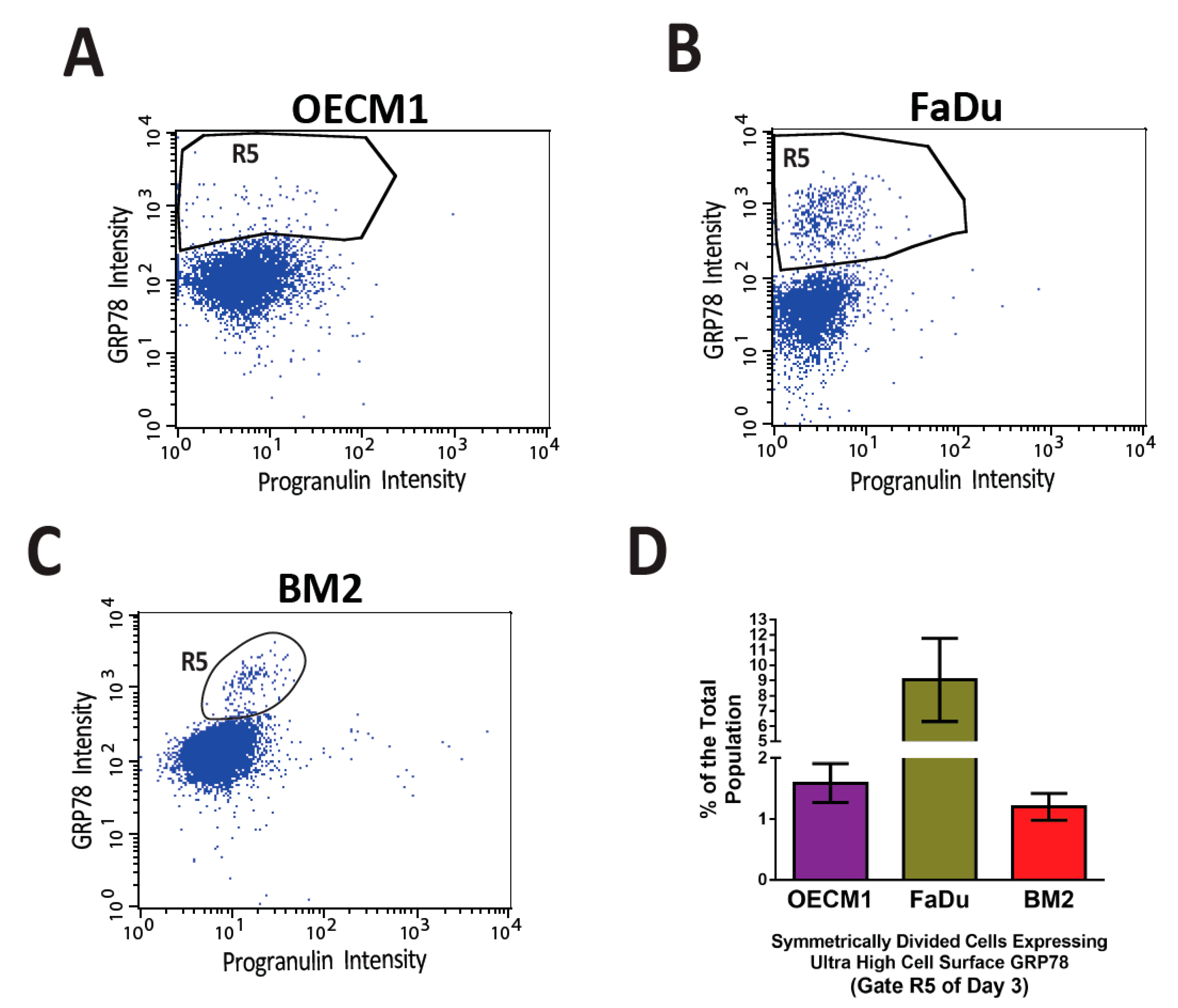
4. The Conventional Functions of GRP78, and Following Its Emerging Role in Cancer Stemness
5. GRP78-Based Therapy for Multiple Cancer Models, and Specificity Concerns of GRP78 Targeting
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Cancer. 2022. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 3 February 2022).
- Verhoef, M.J. Declining conventional cancer treatment and using Complementary and Alternative Medicine: A problem or a challenge? Curr. Oncol. 2008, 15, S31–S36-6. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, E.; Long, B.; Sullivan, P.; McClellan, S.; Finan, M.A.; Reed, E.; Shevde, L.; Rocconi, R.P. CD44+/CD24− ovarian cancer cells demonstrate cancer stem cell properties and correlate to survival. Clin. Exp. Metastasis 2012, 29, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Bruce, W.R.; Van Der Gaag, H. A Quantitative Assay for the Number of Murine Lymphoma Cells capable of Proliferation in vivo. Nature 1963, 199, 79–80. [Google Scholar] [CrossRef]
- Chen, H.-Y.; Chang, J.T.-C.; Chien, K.-Y.; Lee, Y.-S.; You, G.-R.; Cheng, A.-J. The Endogenous GRP78 Interactome in Human Head and Neck Cancers: A Deterministic Role of Cell Surface GRP78 in Cancer Stemness. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Haeckel, E. Natü Rliche Schö Pfungsgeschichte; Georg Reimer: Berlin, Germany, 1868. [Google Scholar]
- Ramalho-Santos, M.; Willenbring, H. On the Origin of the Term “Stem Cell”. Cell Stem Cell 2007, 1, 35–38. [Google Scholar] [CrossRef] [Green Version]
- Häcker, V. Die Kerntheilungsvorgänge bei der Mesodermund Entodermbildung von Cyclops. Archiv. F. Mikr. Anat. 1892, 39, 556–581. [Google Scholar] [CrossRef] [Green Version]
- Maximow, A. Der Lymphocyt als gemeinsame Stammzelle der verschiedenen Blutelemente in der embryonalen Entwicklung und im postfoetalen Leben der Säugetiere. Fol. Haematol. 1909, 8, 125–134. [Google Scholar]
- Neumann, E. Über die Bedeutung des Knochenmarks für die Blutbildung. Zentralbl. F.D. Mediz. Wiss. 1868, 44, 689. [Google Scholar]
- Pappenheim, A. Ueber Entwickelung und Ausbildung der Erythroblasten. Virchows Arch. 1896, 145, 587–643. [Google Scholar] [CrossRef] [Green Version]
- Till, J.E.; McCulloch, E.A. A Direct Measurement of the Radiation Sensitivity of Normal Mouse Bone Marrow Cells. Radiat. Res. 1961, 14, 213. [Google Scholar] [CrossRef] [PubMed]
- Briggs, R.; King, T.J. Transplantation of living nuclei from blastula cells into enucleated frogs’ eggs. Proc. Natl. Acad. Sci. USA 1952, 38, 455–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurdon, J. The Developmental Capacity of Nuclei taken from Intestinal Epithelium Cells of Feeding Tadpoles. Development 1962, 10, 622–640. [Google Scholar] [CrossRef]
- Gurdon, J.B.; Uehlinger, V. “Fertile” Intestine Nuclei. Nature 1966, 210, 1240–1241. [Google Scholar] [CrossRef]
- Wilmut, I.; Schnieke, A.; McWhir, J.; Kind, A.J.; Campbell, K.H.S. Viable offspring derived from fetal and adult mammalian cells. Nature 1997, 385, 810–813. [Google Scholar] [CrossRef]
- Shin, T.; Kraemer, D.; Pryor, J.; Liu, L.; Rugila, J.; Howe, L.; Buck, S.; Murphy, K.; Lyons, L.; Westhusin, M. A cat cloned by nuclear transplantation. Nature 2002, 415, 859. [Google Scholar] [CrossRef]
- Stevens, L.C.; Little, C.C. Spontaneous Testicular Teratomas in an Inbred Strain of Mice. Proc. Natl. Acad. Sci. USA 1954, 40, 1080–1087. [Google Scholar] [CrossRef] [Green Version]
- Brinster, R. A Method for in vitro cultivation of mouse ova from two-cell to blastocyst. Exp. Cell Res. 1963, 32, 205–208. [Google Scholar] [CrossRef] [Green Version]
- Brinster, R.L. Studies on the development of mouse embyrosin vitro. II. The effect of energy source. J. Exp. Zool. 1965, 158, 59–68. [Google Scholar] [CrossRef]
- Brinster, R.L. Studies on the development of mouse embyrosin vitro. III. The effect of fixed-nitrogen source. J. Exp. Zool. 1965, 158, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Brinster, R.L. The effect of cells transferred into the mouse blastocyst on subsequent development. J. Exp. Med. 1974, 140, 1049–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Niwa, H.; Burdon, T.; Chambers, I.; Smith, A. Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev. 1998, 12, 2048–2060. [Google Scholar] [CrossRef] [Green Version]
- Vastag, B. Suddenly, 64 Stem Cell Lines. JAMA 2001, 286, 1163–1164. [Google Scholar] [CrossRef]
- Powell, K. US labs bemoan lack of stem cells. Nature 2002, 418, 358. [Google Scholar] [CrossRef] [Green Version]
- Lovell-Badge, R. The regulation of human embryo and stem-cell research in the United Kingdom. Nat. Rev. Mol. Cell Biol. 2008, 9, 998–1003. [Google Scholar] [CrossRef]
- Ayer, A.C. Stem cell research: The laws of nations and a proposal for international guidelines. Connect. J. Int. Law 2002, 17, 393–428. [Google Scholar]
- Vogel, G. British Parliament Approves New Rules. Science 2001, 291, 23. [Google Scholar] [CrossRef]
- Gopalan, N.; Nor, S.N.M.; Mohamed, M.S. Regulation of Stem Cell Technology in Malaysia: Current Status and Recommendations. Sci. Eng. Ethics 2019, 26, 1–25. [Google Scholar] [CrossRef]
- Then, S.-N. Stem cell technologies: Regulation, patents and problems. J. Law Med. 2004, 12, 188–204. [Google Scholar] [PubMed]
- Curley, D.; Sharples, A. Ethical questions to ponder in the European stem cell patent debate. J Biolaw Bus. 2006, 9, 12–16. [Google Scholar] [PubMed]
- Charitos, I.A.; Ballini, A.; Cantore, S.; Boccellino, M.; Di Domenico, M.; Borsani, E.; Nocini, R.; Di Cosola, M.; Santacroce, L.; Bottalico, L. Stem Cells: A Historical Review about Biological, Religious, and Ethical Issues. Stem Cells Int. 2021, 2021, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Nandi, S.K.; Roychowdhury, T.; Chattopadhyay, S.; Basu, S.; Chatterjee, K.; Choudhury, P.; Banerjee, N.; Saha, P.; Mukhopadhyay, S.; Mukhopadhyay, A.; et al. Deregulation of the CD44-NANOG-MDR1 associated chemoresistance pathways of breast cancer stem cells potentiates the anti-cancer effect of Kaempferol in synergism with Verapamil. Toxicol. Appl. Pharmacol. 2022, 437, 115887. [Google Scholar] [CrossRef]
- Zhou, T.; Liu, J.; Xie, Y.; Yuan, S.; Guo, Y.; Bai, W.; Zhao, K.; Jiang, W.; Wang, H.; Wang, H.; et al. ESE3/EHF, a promising target of rosiglitazone, suppresses pancreatic cancer stemness by downregulating CXCR4. Gut 2021, 71, 357–371. [Google Scholar] [CrossRef]
- Yiyan, S.Y.; Yang, S.; Li, D.; Li, W. Vitamin D Affects the Warburg Effect and Stemness Maintenance of Non-Small-Cell Lung Cancer Cells by Regulating the PI3K/AKT/mTOR Signaling Pathway. Curr. Cancer Drug Targets 2022, 22, 86–95. [Google Scholar] [CrossRef]
- Yu, W.; Ma, Y.; Shrivastava, S.K.; Srivastava, R.K.; Shankar, S. Chronic alcohol exposure induces hepatocyte damage by inducing oxidative stress, SATB2 and stem cell-like characteristics, and activating lipogenesis. J. Cell. Mol. Med. 2022, 26, 2119–2131. [Google Scholar] [CrossRef]
- Pouraghajan, K.; Mahdiuni, H.; Ghobadi, S.; Khodarahmi, R. LRH-1 (liver receptor homolog-1) derived affinity peptide ligand to inhibit interactions between β-catenin and LRH-1 in pancreatic cancer cells: From computational design to experimental validation. J. Biomol. Struct. Dyn. 2022, 40, 1–16. [Google Scholar] [CrossRef]
- Kim, J.Y.; Cheng, X.; Alborzinia, H.; Wölfl, S. Modified STAP conditions facilitate bivalent fate decision between pluripotency and apoptosis in Jurkat T-lymphocytes. Biochem. Biophys. Res. Commun. 2016, 472, 585–591. [Google Scholar] [CrossRef]
- Huntly, B.J.P.; Gilliland, D.G. Leukaemia stem cells and the evolution of cancer-stem-cell research. Nat. Cancer 2005, 5, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solter, D. From teratocarcinomas to embryonic stem cells and beyond: A history of embryonic stem cell research. Nat. Rev. Genet. 2006, 7, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Virchow, R. Editorial archive für pathologische. Pathol. Anat. Physiol. Klin. Med. 1855, 8, 23–54. [Google Scholar]
- Salk Insitute Released. Back to the Future—Breast Cancer Reprises Pathways Found in Fetal Cells. Available online: https://medicalxpress.com/news/2018-08-treatment-breast-cancer.html (accessed on 7 August 2018).
- Cohnheim, J.F. Quergestreiftes Muskelsarkon der Nireren. Virchows Arch. 1875, 65, 64. [Google Scholar] [CrossRef] [Green Version]
- Chandler, J.M.; Lagasse, E. Cancerous stem cells: Deviant stem cells with cancer-causing misbehavior. Stem Cell Res. Ther. 2010, 1, 13. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Wang, Y.; Tong, C.; Xi, H.; Zhao, X.; Wang, Y.; Chen, L. A unified model of the hierarchical and stochastic theories of gastric cancer. Br. J. Cancer 2017, 116, 973–989. [Google Scholar] [CrossRef]
- Wu, M.-J.; Jan, C.-I.; Tsay, Y.-G.; Yu, Y.-H.; Huang, C.-Y.; Lin, S.-C.; Liu, C.-J.; Chen, Y.-S.; Lo, J.-F.; Yu, C.-C. Elimination of head and neck cancer initiating cells through targeting glucose regulated protein78 signaling. Mol. Cancer 2010, 9, 283. [Google Scholar] [CrossRef] [Green Version]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Kennedy, J.A.; Barabé, F.; Poeppl, A.G.; Wang, J.C.Y.; Dick, J.E. Comment on “Tumor Growth Need Not Be Driven by Rare Cancer Stem Cells”. Science 2007, 318, 1722. [Google Scholar] [CrossRef] [Green Version]
- Kelly, P.N.; Dakic, A.; Adams, J.M.; Nutt, S.L.; Strasser, A. Tumor Growth Need Not Be Driven by Rare Cancer Stem Cells. Science 2007, 317, 337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, J.M.; Strasser, A. Is Tumor Growth Sustained by Rare Cancer Stem Cells or Dominant Clones? Cancer Res. 2008, 68, 4018–4021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, M.H.; Hatfield, D.L. The cancer stem cell theory: Is it correct? Mol. Cells 2008, 26, 514–516. [Google Scholar] [PubMed]
- Pine, S.R.; Ryan, B.M.; Varticovski, L.; Robles, A.I.; Harris, C.C. Microenvironmental modulation of asymmetric cell division in human lung cancer cells. Proc. Natl. Acad. Sci. USA 2010, 107, 2195–2200. [Google Scholar] [CrossRef] [Green Version]
- Allman, D.; Lindsley, R.C.; DeMuth, W.; Rudd, K.; Shinton, S.A.; Hardy, R.R. Resolution of Three Nonproliferative Immature Splenic B Cell Subsets Reveals Multiple Selection Points During Peripheral B Cell Maturation. J. Immunol. 2001, 167, 6834–6840. [Google Scholar] [CrossRef]
- Borowski, C.; Martin, C.; Gounari, F.; Haughn, L.; Aifantis, I.; Grassi, F.; von Boehmer, H. On the brink of becoming a T cell. Curr. Opin. Immunol. 2002, 14, 200–206. [Google Scholar] [CrossRef]
- Hurt, E.M.; Kawasaki, B.T.; Klarmann, G.J.; Thomas, S.B.; Farrar, W.L. CD44+CD24− prostate cells are early cancer progenitor/stem cells that provide a model for patients with poor prognosis. Br. J. Cancer 2008, 98, 756–765. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Jeganathan, G.; Amiri, S.; Morgan, K.M.; Ryan, B.M.; Pine, S.R. Asymmetric segregation of template DNA strands in basal-like human breast cancer cell lines. Mol. Cancer 2013, 12, 139. [Google Scholar] [CrossRef] [Green Version]
- Chiu, C.-C.; Lee, L.-Y.; Li, Y.-C.; Chen, Y.-J.; Lu, Y.-C.; Wang, H.-M.; Chang, J.T.; Cheng, A.-J.; Cheng, A.-J. Grp78 as a therapeutic target for refractory head-neck cancer with CD24−CD44+ stemness phenotype. Cancer Gene Ther. 2013, 20, 606–615. [Google Scholar] [CrossRef] [Green Version]
- Roudi, R.; Madjd, Z.; Ebrahimi, M.; Samani, F.S.; Samadikuchaksaraei, A. CD44 and CD24 cannot act as cancer stem cell markers in human lung adenocarcinoma cell line A549. Cell. Mol. Biol. Lett. 2014, 19, 23–36. [Google Scholar] [CrossRef]
- Soeda, A.; Hara, A.; Kunisada, T.; Yoshimura, S.-I.; Iwama, T.; Park, D.M. The Evidence of Glioblastoma Heterogeneity. Sci. Rep. 2015, 5, srep07979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.-T.; Ryu, A.C.J. Cancer stem cell surface markers on normal stem cells. BMB Rep. 2017, 50, 285–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behrooz, A.B.; Syahir, A.; Ahmad, S. CD133: Beyond a cancer stem cell biomarker. J. Drug Target. 2018, 27, 257–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Hsu, H.-S.; Chen, Y.-W.; Tsai, T.-H.; How, C.-K.; Wang, C.-Y.; Hung, S.-C.; Chang, Y.-L.; Tsai, M.-L.; Lee, Y.-Y.; et al. Oct-4 Expression Maintained Cancer Stem-Like Properties in Lung Cancer-Derived CD133-Positive Cells. PLoS ONE 2008, 3, e2637. [Google Scholar] [CrossRef] [Green Version]
- Chang, W.-W.; Lee, C.H.; Lee, P.; Lin, J.; Hsu, C.-W.; Hung, J.-T.; Lin, J.-J.; Yu, J.-C.; Shao, L.-E.; Yu, J.; et al. Expression of Globo H and SSEA3 in breast cancer stem cells and the involvement of fucosyl transferases 1 and 2 in Globo H synthesis. Proc. Natl. Acad. Sci. USA 2008, 105, 11667–11672. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.-G.; Zhang, X.; Xue, X.-Y.; Guo, G.; Wang, P.; Zhang, W.; Fei, Z.; Zhen, H.-N.; You, S.-W.; Yang, H. Brain Tumor Stem-Like Cells Identified by Neural Stem Cell Marker CD15. Transl. Oncol. 2009, 2, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Stone, K.R.; Smith, R.E.; Joklik, W.K. Changes in membrane polypeptides that occur when chick embryo fibroblasts and NRK cells are transformed with avian sarcoma viruses. Virology 1974, 58, 86–100. [Google Scholar] [CrossRef]
- Shiu, R.P.; Pouyssegur, J.; Pastan, I. Glucose depletion accounts for the induction of two transformation-sensitive membrane proteinsin Rous sarcoma virus-transformed chick embryo fibroblasts. Proc. Natl. Acad. Sci. USA 1977, 74, 3840–3844. [Google Scholar] [CrossRef] [Green Version]
- Haas, I.G.; Wabl, M. Immunoglobulin heavy chain binding protein. Nature 1983, 306, 387–389. [Google Scholar] [CrossRef]
- Haas, I.G.; Meo, T. cDNA cloning of the immunoglobulin heavy chain binding protein. Proc. Natl. Acad. Sci. USA 1988, 85, 2250–2254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knittler, M.; Haas, I. Interaction of BiP with newly synthesized immunoglobulin light chain molecules: Cycles of sequential binding and release. EMBO J. 1992, 11, 1573–1581. [Google Scholar] [CrossRef] [PubMed]
- Knittler, M.R.; Dirks, S.; Haas, I.G. Molecular chaperones involved in protein degradation in the endoplasmic reticulum: Quantitative interaction of the heat shock cognate protein BiP with partially folded immunoglobulin light chains that are degraded in the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 1995, 92, 1764–1768. [Google Scholar] [CrossRef] [Green Version]
- Haas, I.G. BiP (GRP78), an essential hsp70 resident protein in the endoplasmic reticulum. Experientia 1994, 50, 1012–1020. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. Mammalian stress response: Induction of the glucose-regulated protein family. Curr. Opin. Cell Biol. 1992, 4, 267–273. [Google Scholar] [CrossRef]
- Little, E.; Ramakrishnan, M.; Roy, B.; Gazit, G.; Lee, A. The Glucose-Regulated Proteins (GRP78 and GRP94): Functions, Gene Regulation, and Applications. Crit. Rev. Eukaryot. Gene Expr. 1994, 4, 1–18. [Google Scholar] [CrossRef]
- Quinones, Q.J.; Gustaaf, R.G.; Pizzo, S.V. GRP78: A chaperone with diverse roles beyond the endoplasmic reticulum. Histol. Histopathol. 2008, 23, 1409–1416. [Google Scholar] [CrossRef]
- Chiu, C.-C.; Lin, C.-Y.; Lee, L.-Y.; Chen, Y.-J.; Kuo, T.-F.; Chang, J.T.-C.; Liao, C.-T.; Wang, H.-M.; Yen, T.-C.; Shen, C.-R.; et al. Glucose-regulated protein 78 regulates multiple malignant phenotypes in head and neck cancer and may serve as a molecular target of therapeutic intervention. Mol. Cancer Ther. 2008, 7, 2788–2797. [Google Scholar] [CrossRef] [Green Version]
- Daneshmand, S.; Quek, M.L.; Lin, E.; Lee, C.; Cote, R.J.; Hawes, D.; Cai, J.; Groshen, S.; Lieskovsky, G.; Skinner, D.G.; et al. Glucose-regulated protein GRP78 is up-regulated in prostate cancer and correlates with recurrence and survival. Hum. Pathol. 2007, 38, 1547–1552. [Google Scholar] [CrossRef]
- Uramoto, H.; Sugio, K.; Oyama, T.; Nakata, S.; Ono, K.; Yoshimastu, T.; Morita, M.; Yasumoto, K. Expression of endoplasmic reticulum molecular chaperone Grp78 in human lung cancer and its clinical significance. Lung Cancer 2005, 49, 55–62. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Chen, W.-H.; Liao, C.-T.; Chen, I.-H.; Chiu, C.-C.; Wang, H.-M.; Yen, T.-C.; Lee, L.-Y.; Chang, J.T.-C.; Cheng, A.-J.; et al. Positive association of glucose-regulated protein 78 during oral cancer progression and the prognostic value in oral precancerous lesions. Head Neck 2009, 32, 1028–1039. [Google Scholar] [CrossRef] [PubMed]
- Reddy, R.K.; Mao, C.; Baumeister, P.; Austin, R.C.; Kaufman, R.J.; Lee, A.S. Endoplasmic Reticulum Chaperone Protein GRP78 Protects Cells from Apoptosis Induced by Topoisomerase Inhibitors. J. Biol. Chem. 2003, 278, 20915–20924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Yang, J.; Lv, W.; Wang, S.; Du, T.; Zhang, K.; Wu, Y.; Feng, X. Down-regulating GRP78 reverses pirarubicin resistance of triple negative breast cancer by miR-495-3p mimics and involves the p-AKT/mTOR pathway. Biosci. Rep. 2022, 42, BSR20210245. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yin, Y.; Hua, H.; Li, M.; Luo, T.; Xu, L.; Wang, R.; Liu, D.; Zhang, Y.; Jiang, Y. Blockade of GRP78 sensitizes breast cancer cells to microtubules-interfering agents that induce the unfolded protein response. J. Cell. Mol. Med. 2009, 13, 3888–3897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.K.; Kang, K.A.; Piao, M.J.; Ryu, Y.S.; Han, X.; Fernando, P.M.D.J.; Oh, M.C.; Park, J.E.; Shilnikova, K.; Boo, S.J.; et al. Endoplasmic reticulum stress induces 5-fluorouracil resistance in human colon cancer cells. Environ. Toxicol. Pharmacol. 2016, 44, 128–133. [Google Scholar] [CrossRef]
- Chen, D.; Rauh, M.; Buchfelder, M.; Eyupoglu, I.Y.; Savaskan, N. The oxido-metabolic driver ATF4 enhances temozolamide chemo-resistance in human gliomas. Oncotarget 2017, 8, 51164–51176. [Google Scholar] [CrossRef] [Green Version]
- Visioli, F.; Wang, Y.; Alam, G.N.; Ning, Y.; Rados, P.; Nör, J.E.; Polverini, P.J. Glucose-Regulated Protein 78 (Grp78) Confers Chemoresistance to Tumor Endothelial Cells under Acidic Stress. PLoS ONE 2014, 9, e101053. [Google Scholar] [CrossRef] [Green Version]
- Cook, K.L.; Soto-Pantoja, D.R.; Clarke, P.A.; Cruz, M.I.; Zwart, A.; Wärri, A.; Hilakivi-Clarke, L.; Roberts, D.D.; Clarke, R. Endoplasmic Reticulum Stress Protein GRP78 Modulates Lipid Metabolism to Control Drug Sensitivity and Antitumor Immunity in Breast Cancer. Cancer Res. 2016, 76, 5657–5670. [Google Scholar] [CrossRef] [Green Version]
- Li, L. Correlation of the expression of neurotrophins family and its receptors with cell proliferation and invasion in ovarian cancer. J. Hainan Med. Univ. 2018, 24, 50–53. [Google Scholar]
- Zhang, G.; Wang, X.; Bi, X.; Li, C.; Deng, Y.; Al-Hashimi, A.A.; Luo, X.; Gillette, T.G.; Austin, R.C.; Wang, Y.; et al. GRP78 (Glucose-Regulated Protein of 78 kDa) Promotes Cardiomyocyte Growth Through Activation of GATA4 (GATA-Binding Protein 4). Hypertension 2019, 73, 390–398. [Google Scholar] [CrossRef]
- Ma, N.; Xu, N.; Yin, D.; Zheng, P.; Liu, W.; Wang, G.; Hui, Y.; Han, G.; Yang, C.; Cheng, X. Levels of circulating GRP78 and CHOP in endoplasmic reticulum stress pathways in Chinese type 2 diabetic kidney disease patients. Medicine 2021, 100, e26879. [Google Scholar] [CrossRef] [PubMed]
- Delpino, A.; Castelli, M. The 78 kDa Glucose-Regulated Protein (GRP78/BIP) Is Expressed on the Cell Membrane, Is Released into Cell Culture Medium and Is Also Present in Human Peripheral Circulation. Biosci. Rep. 2002, 22, 407–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Gronow, M.; Cuchacovich, M.; Llanos, C.; Urzua, C.; Gawdi, G.; Pizzo, S.V. Prostate Cancer Cell Proliferation In vitro Is Modulated by Antibodies against Glucose-Regulated Protein 78 Isolated from Patient Serum. Cancer Res. 2006, 66, 11424–11431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misra, U.K.; Deedwania, R.; Pizzo, S.V. Binding of Activated α2-Macroglobulin to Its Cell Surface Receptor GRP78 in 1-LN Prostate Cancer Cells Regulates PAK-2-dependent Activation of LIMK. J. Biol. Chem. 2005, 280, 26278–26286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shani, G.; Fischer, W.H.; Justice, N.J.; Kelber, J.A.; Vale, W.; Gray, P.C. GRP78 and Cripto Form a Complex at the Cell Surface and Collaborate to Inhibit Transforming Growth Factor β Signaling and Enhance Cell Growth. Mol. Cell. Biol. 2008, 28, 666–677. [Google Scholar] [CrossRef] [Green Version]
- Burikhanov, R.; Zhao, Y.; Goswami, A.; Qiu, S.; Schwarze, S.R.; Rangnekar, V.M.; Schwarze, S. The tumor suppressor par-4 activates an extrinsic pathway for apoptosis. Cell 2009, 138, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Davidson, D.J.; Haskell, C.; Majest, S.; Kherzai, A.; Egan, D.A.; Walter, K.A.; Schneider, A.; Gubbins, E.F.; Solomon, L.; Chen, Z.; et al. Kringle 5 of Human Plasminogen Induces Apoptosis of Endothelial and Tumor Cells through Surface-Expressed Glucose-Regulated Protein 78. Cancer Res. 2005, 65, 4663–4672. [Google Scholar] [CrossRef] [Green Version]
- Corrigall, V.M.; Bodman-Smith, M.D.; Fife, M.S.; Canas, B.; Myers, L.K.; Wooley, P.H.; Soh, C.; Staines, N.A.; Pappin, D.J.C.; Berlo, S.E.; et al. The human endoplasmic reticulum molecular chaperone BiP is an autoantigen for rheumatoid arthritis and prevents the induction of experimental arthritis. J. Immunol. 2001, 166, 1492–1498. [Google Scholar] [CrossRef] [Green Version]
- Bläss, S.; Union, A.; Raymackers, J.; Schumann, F.; Ungethüm, U.; Müller-Steinbach, S.; De Keyser, F.; Engel, J.M.; Burmester, G.R. The stress protein BiP is overexpressed and is a major B and T cell target in rheumatoid arthritis. Arthritis Rheum. 2001, 44, 761–771. [Google Scholar] [CrossRef]
- Triantafilou, K.; Fradelizi, D.; Wilson, K.; Triantafilou, M. GRP78, a Coreceptor for Coxsackievirus A9, Interacts with Major Histocompatibility Complex Class I Molecules Which Mediate Virus Internalization. J. Virol. 2002, 76, 633–643. [Google Scholar] [CrossRef] [Green Version]
- Honda, T.; Horie, M.; Daito, T.; Ikuta, K.; Tomonaga, K. Molecular Chaperone BiP Interacts with Borna Disease Virus Glycoprotein at the Cell Surface. J. Virol. 2009, 83, 12622–12625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.-Y.; Cheng, C.-W.; Kung, C.-C.; Liao, K.-S.; Jan, J.-T.; Ma, C.; Wong, C.-H. Glycosite-deleted mRNA of SARS-CoV-2 spike protein as a broad-spectrum vaccine. Proc. Natl. Acad. Sci. USA 2022, 119, e2119995119. [Google Scholar] [CrossRef] [PubMed]
- Kabakov, A.; Yakimova, A.; Matchuk, O. Molecular Chaperones in Cancer Stem Cells: Determinants of Stemness and Potential Targets for Antitumor Therapy. Cells 2020, 9, 892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzales, K.A.U.; Liang, H.; Lim, Y.-S.; Chan, Y.-S.; Yeo, J.-C.; Tan, C.-P.; Gao, B.; Le, B.; Tan, Z.-Y.; Low, K.-Y.; et al. Deterministic Restriction on Pluripotent State Dissolution by Cell-Cycle Pathways. Cell 2015, 162, 564–579. [Google Scholar] [CrossRef] [Green Version]
- Conner, C.; Lager, T.W.; Guldner, I.H.; Wu, M.-Z.; Hishida, Y.; Hishida, T.; Ruiz, S.; Yamasaki, A.E.; Gilson, R.C.; Belmonte, J.C.I.; et al. Cell surface GRP78 promotes stemness in normal and neoplastic cells. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Xiu, Z.; He, Y.; Huang, W.; Li, Y.; Sun, T. Bip inhibition in glioma stem cells promotes radiation-induced immunogenic cell death. Cell Death Dis. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Chen, Z.; Wang, H.; Zhang, Z.; Xu, J.; Qi, Y.; Xue, H.; Gao, Z.; Zhao, R.; Wang, S.; Zhang, S.; et al. Cell surface GRP78 regulates BACE2 via lysosome-dependent manner to maintain mesenchymal phenotype of glioma stem cells. J. Exp. Clin. Cancer Res. 2021, 40, 1–17. [Google Scholar] [CrossRef]
- Ei, Z.Z.; Choochuay, K.; Tubsuwan, A.; Pinkaew, D.; Suksomtip, M.; Vinayanuwattikun, C.; Chanvorachote, P.; Chunhacha, P. GRP78/BiP determines senescence evasion cell fate after cisplatin-based chemotherapy. Sci. Rep. 2021, 11, 1–15. [Google Scholar] [CrossRef]
- Borah, A.; Raveendran, S.; Rochani, A.; Maekawa, T.; Kumar, S. Targeting self-renewal pathways in cancer stem cells: Clinical implications for cancer therapy. Oncogenesis 2015, 4, e177. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, I.; Cohen, M. Linking cell-surface GRP78 to cancer: From basic research to clinical value of GRP78 antibodies. Cancer Lett. 2021, 524, 1–14. [Google Scholar] [CrossRef]
- Rai, R.; Kennedy, A.L.; Isingizwe, Z.R.; Javadian, P.; Benbrook, D.M. Similarities and Differences of Hsp70, hsc70, Grp78 and Mortalin as Cancer Biomarkers and Drug Targets. Cells 2021, 10, 2996. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Shi, D.; Shang, M.; Sun, X.; Guo, L.; Meng, D.; Liu, X.; Zhou, X.; Li, J. GRP78-targeted and doxorubicin-loaded nanodroplets combined with ultrasound: A potential novel theranostics for castration-resistant prostate cancer. Drug Deliv. 2022, 29, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Lu, L.; Zhou, J.; Ran, D.; Wang, S.; Xu, Q.; Xu, W.; Wang, J.; Liu, Y.; Xie, C.; et al. All-stage targeted therapy for glioblastoma based on lipid membrane coated cabazitaxel nanocrystals. J. Control. Release 2022, 345, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Farshbaf, M.; Mojarad-Jabali, S.; Hemmati, S.; Khosroushahi, A.Y.; Motasadizadeh, H.; Zarebkohan, A.; Valizadeh, H. Enhanced BBB and BBTB penetration and improved anti-glioma behavior of Bortezomib through dual-targeting nanostructured lipid carriers. J. Control. Release 2022, 345, 371–384. [Google Scholar] [CrossRef]
- Hebbar, N.; Epperly, R.; Vaidya, A.; Thanekar, U.; Moore, S.E.; Umeda, M.; Ma, J.; Patil, S.L.; Langfitt, D.; Huang, S.; et al. CAR T cells redirected to cell surface GRP78 display robust anti-acute myeloid leukemia activity and do not target hematopoietic progenitor cells. Nat. Commun. 2022, 13, 1–14. [Google Scholar] [CrossRef]
- Liu, S.; Li, Y.; Li, Z. Salidroside suppresses the activation of nasopharyngeal carcinoma cells via targeting miR-4262/GRP78 axis. Cell Cycle 2022, 21, 720–729. [Google Scholar] [CrossRef]
- Luo, S.; Mao, C.; Lee, B.; Lee, A.S. GRP78/BiP Is Required for Cell Proliferation and Protecting the Inner Cell Mass from Apoptosis during Early Mouse Embryonic Development. Mol. Cell. Biol. 2006, 26, 5688–5697. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.; Jin, Y.; Lan, X.; Yang, Y.; Chen, F.; Wang, N.; Li, X.; Sun, Y.; Wang, A. GRP78 expression and regulation in the mouse uterus during embryo implantation. Histochem. J. 2013, 45, 259–268. [Google Scholar] [CrossRef]
- Xiong, Z.; Jiang, R.; Li, X.; Liu, Y.; Guo, F. Different Roles of GRP78 on Cell Proliferation and Apoptosis in Cartilage Development. Int. J. Mol. Sci. 2015, 16, 21153–21176. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.-C.; Fu, H.-C.; Hsiao, B.-L.; Sobue, G.; Adachi, H.; Huang, F.-J.; Hsuuw, Y.-D.; Wei, K.-T.; Chang, C.; Huang, K.-E.; et al. Androgen receptor inclusions acquire GRP78/BiP to ameliorate androgen-induced protein misfolding stress in embryonic stem cells. Cell Death Dis. 2013, 4, e607. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.-Y.; Cheng, A.-J. Potential to Eradicate Cancer Stemness by Targeting Cell Surface GRP78. Biomolecules 2022, 12, 941. https://doi.org/10.3390/biom12070941
Chen H-Y, Cheng A-J. Potential to Eradicate Cancer Stemness by Targeting Cell Surface GRP78. Biomolecules. 2022; 12(7):941. https://doi.org/10.3390/biom12070941
Chicago/Turabian StyleChen, Hsin-Ying, and Ann-Joy Cheng. 2022. "Potential to Eradicate Cancer Stemness by Targeting Cell Surface GRP78" Biomolecules 12, no. 7: 941. https://doi.org/10.3390/biom12070941