1. Introduction
Protein-ligand interactions govern many biological processes. The specific ligand binding site (LBS) in a protein is essential for understanding its biological function and for structure-based drug design [
1]. LBSs can be directly obtained from known protein-ligand complex structures. The Protein Data Bank (PDB) [
2] provides the primary source of protein three-dimensional (3D) structures experimentally resolved. However, among the more than 190,000 experimentally-determined structures by 2021, only a small number were solved with bound ligand. In order to fill the gap between structures and binding sites, computational methods predicting LBSs from protein 3D structures have been developed [
3,
4]. Based on the known macromolecule structures in the PDB and diverse LBS detection tools, pocket databases have been constructed, which provide functional clues for protein domains of unknown functions [
5,
6,
7,
8,
9,
10,
11,
12,
13,
14,
15,
16,
17,
18,
19,
20,
21] (see
Table S1 for more details about typical pocket databases). The pocketome analysis bridges the gap between the protein structures and functions. For example, Bhagavat et al. proposed an augmented pocketome (PocketDB) which consists of 249,096 pockets detected from all the proteins with known 3D structures in the PDB. Combining the pocketome database and an effective pocket-comparison algorithm, functional relationships among protein folds, binding site types and ligand types could be deduced [
11]. Hedderich et al. focused on the pocketome of G-protein-coupled receptors and discovered previously untargeted allosteric sites [
22].
However, currently available pocket databases are limited to known protein structures. Only about 37% of human proteins have the corresponding PDB entries [
23]. Fortunately, protein structure prediction approaches have made great progress in the past several years [
24,
25,
26]. In 2021, AlphaFold, a deep neural network-based method developed by DeepMind, has made a major breakthrough and produced protein structures with atomic accuracy even where no similar structure is known [
27]. AlphaFold was then applied to build protein structure models for human proteome [
28,
29], which dramatically expanded the structural coverage of human proteins.
In this work, we analyzed potential ligand binding sites in the human protein structures predicted by AlphaFold and constructed a comprehensive human pocketome database, CavitySpace. CavitySpace expands the ligand binding site space from known human protein structures to predicted structures and provides a resource for human protein function study and drug design.
2. Materials and Methods
2.1. Data Collection
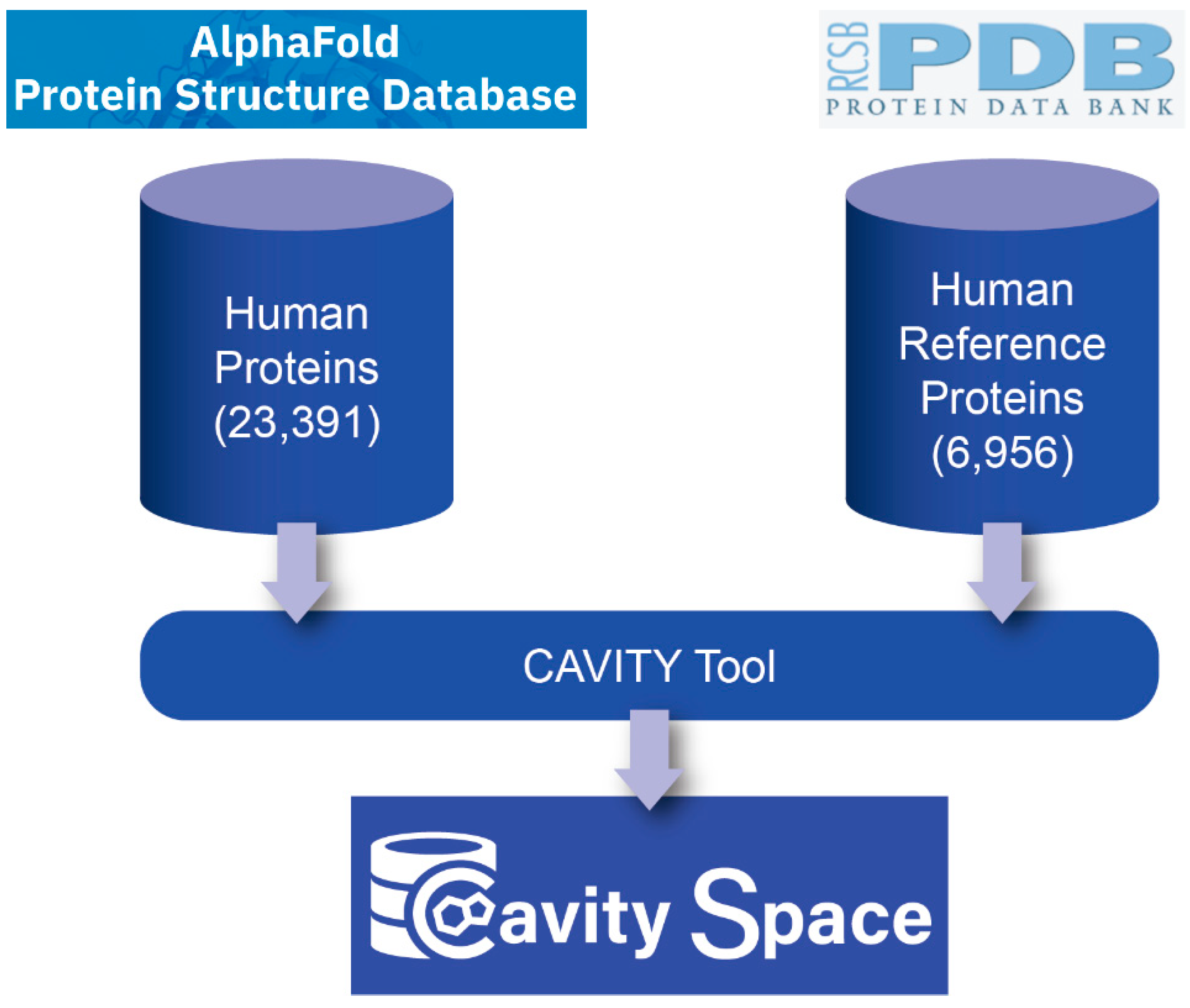
Figure 1 shows the flowchart for building the CavitySpace database. All data used to construct CavitySpace were obtained from public databases. The human protein structures predicted by AlphaFold were downloaded from the AlphaFold Protein Structure Database (
https://alphafold.ebi.ac.uk/, accessed on 1 September 2021). Only structures for Homo sapiens were downloaded, which contain 23,391 predicted structures of 20,504 sequence entries. The inequality is due to the fact that 208 sequence entries were divided into several fragments before structure prediction because of the extremely long sequence length.
About 37% of human proteins have corresponding PDB entries. It is very useful to compare the known and predicted structures. Therefore, we queried the UniProtKB database (
https://www.uniprot.org/, accessed on 1 October 2021) with the UniProt ID of each sequence to retrieve UniProt entries with known PDB structures and obtained a total of 7245 UniProt records. For each UniProt entry, the structure with the best resolution was selected as a representative structure. Sometimes, several PDB structures for one protein cover different domains of the same protein. In these cases, we selected representative structures for each domain of the sequence to cover the whole protein sequence as long as possible. In addition, all PDB structures not resolved by X-ray crystallography or with resolution larger than 3.5 Å were excluded. Finally, we screened 6956 PDB structures of 5725 UniProt entries, forming the known human PDB structure dataset (hrefPDB). All the structure files were downloaded from RCSB PDB (
https://www.rcsb.org/, accessed on 8 November 2021). Because each AlphaFold structure has only one single chain, we extracted one chain from each of the known structures to maintain consistency.
2.2. Cavity Detection
We applied the CAVITY tool developed by our lab to detect all the potential cavities on protein surfaces [
30]. CAVITY has been shown to be effective in detecting cavities and assessing pockets’ druggability on the NRDLD data set [
31]. For all the 23,391 AlphaFold structures, CAVITY successfully processed 18,820 (80.5%) structures. The remaining 4571 structures, for which CAVITY could not finish the job within a reasonable time, were mainly complicated structures with relatively long protein sequences and many irregular loops. For the hrefPDB dataset containing resolved PDB structures, all the 6956 structures can be processed by CAVITY and 86.9% (6043) of them have at least one cavity.
3. Results
3.1. Cavity Library for AlphaFold Structures
The cavity detection procedure found 237,872 cavities for the 18,672 AlphaFold structures. The druggability of each cavity was labelled as strong, medium or weak by CAVITY. Among the AlphaFold cavities, 16.3% (38,709) were predicted as strong druggable cavities (
Figure 2). However, for AlphaFold cavities, we need to further consider the structure accuracy, because inaccurate structures may produce fake cavities. In AlphaFold structures, the predicted Local Distance Difference Test (pLDDT) was given for each residue to measure the local accuracy [
27]. This confidence measure is a scale from 0 to 100. Structure regions with pLDDT > 90 are expected to be highly confident and can be used to characterize binding sites. Based on the pLDDT scores, we defined the ratio of the number of high confident residues (pLDDT > 90) to the total number of residues in the cavity as an
Index to evaluate the reliability of the cavity structure. Among all the AlphaFold cavities, about 40% are located in protein structure regions with relatively high confidence (
Index > 0.6), while only about 26% of strong cavities are constructed by reliable structures (
Figure 3).
As previously mentioned, about 37% of human proteins have corresponding PDB entries, so we split all the AlphaFold cavities into two subsets: one subset contains all cavities from proteins that have known 3D structures in our hrefPDB dataset (Subset_1), and the other subset contains the remaining cavities (Subset_2).
Figure 3 shows the
Index distributions of these two subsets. The median
Index of Subset_1 is higher than that of Subset_2, implying that AlphaFold gives more accurate structure predictions for proteins with known 3D structures. However, for cavities with strong druggability, the
Index values significantly decreased, indicating that the AlphaFold predicted protein structures may contain false-positive druggable pockets. Therefore, we include the
Index scores for each cavity in our library to help users pick reliable druggable pockets.
3.2. The Quality of Cavity Detection over AlphaFold Structures
One important question is how different are the hrefPDB structures from the AlphaFold predicted structures for cavity detection. We further performed cavity detection for all the hrefPDB structures and obtained 50,417 PDB cavities. To make a fair comparison, we extracted the subset of AlphaFold structures sharing the same UniProt IDs to the hrefPDB structures and then collected 64,897 corresponding AlphaFold cavities. Theoretically, the cavities detected from hrefPDB structures should also be detected from AlphaFold predicted structures if the AlphaFold predicted structures are as reliable as the experimentally resolved structures. Therefore, we defined a score of Tc = A∩B/A∪B to measure the overlap of two cavities, where A and B are the residue sets of two cavities. We defined two cavities as the same pocket if their Tc score is larger than a given threshold.
Figure 4a shows the recall rate of hrefPDB cavities in AlphaFold cavities at different thresholds. With a threshold of 0.5, only 22.8% of hrefPDB cavities can be detected from AlphaFold predicted structures, indicating non-negligible differences between the predicted structures and the real structures. This can be attributed to the following reasons. First, AlphaFold structures are always full-sequence structures, while part of the hrefPDB structures are not full-sequence structures and have missing residues, which affect the detection of cavities. Second, cavity detection results are influenced by protein conformation, which might be diverse under different ensemble states, binding states and experimental conditions. However, each AlphaFold structure from AlphaFold database gives only one single conformation, which may be different from the conformation of hrefPDB structures. For example, the AlphaFold structure of alpha-actinin-2 (UniProt ID: P35609) is different from its hrefPDB structure (PDB entry: 4D1E) [
32] (see
Figure S1 and Supplementary Results for details). Third, the AlphaFold program can produce relatively accurate secondary structures, but the relative position of different secondary structures and different domains might not be very accurate, not to mention the inaccurate position of flexible loops. These inaccurate predictions can significantly influence the cavity detection process.
In the above analysis, we checked if hrefPDB cavities can be detected from AlphaFold predicted structures. However, the hrefPDB cavities may change shape upon ligand binding. In order to obtain more accurate results, we further checked if experimentally confirmed ligand sites in hrefPDB structures can be correctly identified by CAVITY from AlphaFold structures. From the hrefPDB dataset, we only kept the structures with bound ligands and defined true ligand sites as residues within 4 Å around bound ligands. This gave 2439 true ligand binding sites as a test set. If we define that a true ligand binding site is successfully detected when 50% of true binding site residues are included in one of the CAVITY detected binding sites, 81% and 80% of true binding sites can be recovered from hrefPDB and AlphaFold structures, respectively (
Figure 4b). Such results demonstrate that our CAVITY program can successfully discover most of the true binding sites from protein structures and for the true binding site detection task, there is no significant difference between hrefPDB structures and AlphaFold predicted structures.
3.3. Applications of the Cavity Library
For the 63.6% of the AlphaFold predicted human protein structures with no experimental structure information, CAVITY detected 145,444 cavities and 17.4% of them are strong druggable cavities. As similar binding sites may bind the same or similar ligands and have similar functions, we used PocketMatch [
33] to compare binding sites and the PMSmax score to evaluate the overall pocket similarity (see the
Supplementary Methods for details). To obtain meaningful results, we only analyzed the 60,913 high-quality cavities, each of which contains at least 80% residues with pLDDT > 90. These high-quality cavities, together with 50,417 hrefPDB cavities, were used to perform an all-to-all pocket comparison. We then clustered the 111,330 cavities with the Butina algorithm [
34] (see
Supplementary Methods). With the threshold of PMSmax 0.6, 11,213 cavities did not have any similar cavities, which may be novel ligand binding sites. The other 100,117 cavities were grouped into 8015 clusters and 538 of them contain more than 10 members. The clusters that contain known ligand binding sites can be used to study the function of proteins that contain similar cavities or to find new targets for a known ligand. For example, the crystal structure of human cysteinyl leukotriene receptor one binding with its antagonist zafirlukast, an FDA approved drug for asthma treatment, has been solved [
35]. We selected seven top-scoring AlphaFold cavities of proteins without known PDB structures from the corresponding cavity cluster to which the zafirlukast binding site belongs (see Supplementary Results for details). Docking study with Autodock Vina 1.2 [
36] showed that zafirlukast can potentially bind to these cavities with high binding affinity (
Table S2).
3.4. The Webserver
We developed the CavitySpace webserver for public usage. Users can conveniently query the database with protein name, UniProt ID or gene name and obtain the cavity details for each structure visually. All data in the cavity library can be downloaded from the CavitySpace webserver, including the strong druggable cavities, the cavity clustering results, etc. Each cavity is represented by four files. The “cavity” file contains all the protein atoms that form the cavity. The “surface” file contains all the grid points on protein surface which form the cavity. The “vacant” file contains all the grid points in the cavity. Besides, the bounds, the surface area and the volume of each cavity can be found in the “surface” file and the “vacant” file, respectively. The “box” file contains the coordinates of the cavity center and the size of the docking box, which can be directly used for docking calculation by users. The webserver is freely available at
http://www.pkumdl.cn:8000/cavityspace/ (accessed on 8 November 2021).
4. Discussion
The shape and physicochemical properties of ligand binding sites provide useful information for protein function deduction. The large number of protein structures predicted by AlphaFold provides an unprecedented chance to analyze binding sites in previously unexplored structure space. The CavitySpace database can be used for various purposes, including identifying new druggable protein targets for drug design, predicting protein function based on pocket analysis, searching for new binding sites for known drugs for drug repurpose study, etc. For example, the cavities in CavitySpace provide a structural basis to screen potential active ligands. Molecular docking is commonly used to predict potential binding pose and affinity of a ligand in a binding site. For a given cavity, we can dock all the molecules in a compound library to the cavity to screen potential active ligands. In reverse, for a known drug or a compound of interest, we can also dock the compound to all the cavities in CavitySpace to find novel potential binding sites. Based on the hypothesis that similar binding sites may bind similar ligands, we can also calculate the cavity similarity between cavities and screen the similar cavities of a specific cavity with a known bound ligand. This ligand is likely to bind with these similar cavities.
At the same time, care should be taken when using these data. This can be attributed to several possible reasons. First, the AlphaFold method is currently restricted to single-chain structure prediction. In real cases, many proteins need to form oligomers to be functional. New methods, such as the AlphaFold-Multimer [
37] have been developed to address this problem. CavitySpace will be updated once the structure data of the protein oligomers is released. Second, AlphaFold provides the state-of-the-art structure predictions in general, but the accuracy varies between different domains and different structures. Pockets located on low confident domains are obviously unreliable to be treated as a real pocket. We recommend that based on the CavitySpace results, users further analyze the potential binding sites with more accurate structures after carefully considering inter-domain orientations and oligomeric states using our CavityPlus webserver [
38] or other cavity detection tools.
The development of AlphaFold and related methods have markedly advanced the protein 3D structure prediction field. However, for most drug discovery tasks, in addition to the overall correct protein structure, reliable binding site sub-structure is necessary. Based on the CavitySpace database, we found that the single-chain structures, the less reliable relative position between different secondary structures or domains, and the flexible loops of the currently available AlphaFold models are the major obstacles for highly accurate prediction of ligand binding sites. Care should be taken to check the reliability of the structures that form the binding sites before further analysis. More efforts are needed to solve the above problems in protein structure prediction. Learning the binding site structures in addition to the overall protein structures may provide a useful strategy to increase the accuracy of predicted protein structures.
5. Conclusions
We have developed CavitySpace, a comprehensive human protein pocketome based on the AlphaFold predicted human protein structures. CavitySpace not only captures known ligand binding sites, but also identifies novel ligand binding sites that can be used to predict protein functions or to virtually screen new binding compounds in drug discovery. CavitySpace can also be used to identify new targets for known drugs in drug repurposing or side effect studies. The cavity library can be easily applied to docking-based virtual screening or reverse virtual screening to screen active ligands or identify new targets for a given drug or natural product. Furthermore, the cavity library increases the diversity of ligand binding sites, which can be applied to the pre-training stage of AI-based drug discovery models. Though the current version of CavitySpace only contains human proteins, it can be easily extended to include more protein structures from other species in the future.
Supplementary Materials
The following supporting information can be downloaded at:
https://www.mdpi.com/article/10.3390/biom12070967/s1, Figure S1: An example of conformation difference between AlphaFold structures and hrefPDB structures; Table S1: Databases of protein pockets since 2004; Table S2: Seven representative AlphaFold cavities that are similar to the zafirlukast binding site in cysteinyl leukotriene receptor 1.
Author Contributions
Conceptualization, S.W., X.D., Y.X., J.P. and L.L.; methodology, S.W., Z.H. and Y.H.; webserver, Z.H. and H.L.; writing-original draft preparation, S.W., Z.H. and H.L.; writing—review and editing, J.P. and L.L.; funding acquisition, L.L. and J.P. All authors have read and agreed to the published version of the manuscript.
Funding
This work has been supported in part by the National Natural Science Foundation of China (22033001) and the Chinese Academy of Medical Sciences (2021-I2M-5-014).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Acknowledgments
We thank all members in Luhua Lai’s group for their helpful suggestions and testing of the CavitySpace database. We thank the high-performance computing platform of the Peking-Tsinghua Center for Life Sciences, Peking University for providing the computational resources.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pérot, S.; Sperandio, O.; Miteva, M.A.; Camproux, A.-C.; Villoutreix, B.O. Druggable pockets and binding site centric chemical space: A paradigm shift in drug discovery. Drug Discov. Today 2010, 15, 656–667. [Google Scholar] [CrossRef]
- Consortium, W. Protein Data Bank: The single global archive for 3D macromolecular structure data. Nucleic Acids Res. 2019, 47, D520–D528. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.-R.; Hwang, M.-J. Methods for predicting protein–ligand binding sites. Mol. Modeling Proteins 2015, 1215, 383–398. [Google Scholar]
- Dhakal, A.; McKay, C.; Tanner, J.J.; Cheng, J. Artificial intelligence in the prediction of protein–ligand interactions: Recent advances and future directions. Brief. Bioinform. 2021, 23, bbab476. [Google Scholar] [CrossRef]
- Ito, J.-I.; Tabei, Y.; Shimizu, K.; Tsuda, K.; Tomii, K. PoSSuM: A database of similar protein–ligand binding and putative pockets. Nucleic Acids Res. 2011, 40, D541–D548. [Google Scholar] [CrossRef] [Green Version]
- Kufareva, I.; Ilatovskiy, A.V.; Abagyan, R. Pocketome: An encyclopedia of small-molecule binding sites in 4D. Nucleic Acids Res. 2011, 40, D535–D540. [Google Scholar] [CrossRef] [Green Version]
- Maietta, P.; Lopez, G.; Carro, A.; Pingilley, B.J.; Leon, L.G.; Valencia, A.; Tress, M.L. FireDB: A compendium of biological and pharmacologically relevant ligands. Nucleic Acids Res. 2013, 42, D267–D272. [Google Scholar] [CrossRef] [Green Version]
- Meyer, T.; Knapp, E.-W. Database of protein complexes with multivalent binding ability: Bival-bind. Proteins 2014, 82, 744–751. [Google Scholar] [CrossRef]
- van Linden, O.P.J.; Kooistra, A.J.; Leurs, R.; de Esch, I.J.P.; de Graaf, C. KLIFS: A knowledge-based structural database to navigate Kinase–ligand interaction space. J. Med. Chem. 2014, 57, 249–277. [Google Scholar] [CrossRef]
- Desaphy, J.; Bret, G.; Rognan, D.; Kellenberger, E. sc-PDB: A 3D-database of ligandable binding sites—10 years on. Nucleic Acids Res. 2015, 43, D399–D404. [Google Scholar] [CrossRef]
- Bhagavat, R.; Sankar, S.; Srinivasan, N.; Chandra, N. An Augmented Pocketome: Detection and Analysis of Small-Molecule Binding Pockets in Proteins of Known 3D Structure. Structure 2018, 26, 499–512.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Qiu, J.; Liu, H.; Xu, Y.; Jia, Y.; Zhao, Y. HKPocket: Human kinase pocket database for drug design. BMC Bioinform. 2019, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Konc, J.; Lešnik, S.; Škrlj, B.; Janežič, D. ProBiS-Dock database: A web server and interactive web repository of small ligand–protein binding sites for drug design. J. Chem. Inf. Model. 2021, 61, 4097–4107. [Google Scholar] [CrossRef]
- Radusky, L.; Defelipe, L.A.; Lanzarotti, E. TuberQ: A Mycobacterium tuberculosis protein druggability database. Database 2014, 2014, bau035. [Google Scholar] [CrossRef] [Green Version]
- Tseng, Y.Y.; Chen, Z.J. and Li, W-H. fPOP: Footprinting functional pockets of proteins by comparative spatial patterns. Nucleic Acids Res. 2009, 38, D288–D295. [Google Scholar] [CrossRef] [Green Version]
- Schreyer, A.; Blundell, T. CREDO: A protein–ligand interaction database for drug discovery. Chem. Biol. Drug. Des. 2009, 73, 157–167. [Google Scholar] [CrossRef]
- Bauer, R.A.; Günther, S.; Jansen, D.; Heeger, C.; Thaben, P.F.; Preissner, R. SuperSite: Dictionary of metabolite and drug binding sites in proteins. Nucleic Acids Res. 2008, 37, D195–D200. [Google Scholar] [CrossRef] [Green Version]
- Dessailly, B.H.; Lensink, M.F.; Orengo, C.A.; Wodak, S.J. LigASite—a database of biologically relevant binding sites in proteins with known apo -structures. Nucleic Acids Res. 2007, 36, D667–D673. [Google Scholar] [CrossRef]
- Gold, N.D.; Jackson, R.M. SitesBase: A database for structure-based protein–ligand binding site comparisons. Nucleic Acids Res. 2006, 34, D231–D234. [Google Scholar] [CrossRef] [Green Version]
- Ivanisenko, V.A.; Pintus, S.S.; Grigorovich, D.A.; Kolchanov, N.A. PDBSite: A database of the 3D structure of protein functional sites. Nucleic Acids Res. 2005, 33 (Suppl. 1), D183–D187. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, A.; Iida, K.; Matsui, N.; Tomoda, S.; Yura, K.; Go, M. Het-PDB Navi.: A database for protein–small molecule interactions. J. Biochem. 2004, 135, 79–84. [Google Scholar] [CrossRef]
- Hedderich, J.B.; Persechino, M.; Becker, K.; Heydenreich, F.M.; Gutermuth, T.; Bouvier, M.; Bünemann, M.; Kolb, P. The pocketome of G-protein-coupled receptors reveals previously untargeted allosteric sites. Nat. Commun. 2022, 13, 2567. [Google Scholar] [CrossRef]
- Consortium, U. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef] [PubMed]
- Bonneau, R.; Baker, D. Ab initio protein structure prediction: Progress and prospects. Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 173–189. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y. Progress and challenges in protein structure prediction. Curr. Opin. Struct. Biol. 2008, 18, 342–348. [Google Scholar] [CrossRef] [Green Version]
- Kuhlman, B.; Bradley, P. Advances in protein structure prediction and design. Nat. Rev. Mol. Cell Biol. 2019, 20, 681–697. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.X.; Pei, J.F.; Lai, L.H. Binding site detection and druggability prediction of protein targets for structure-based drug design. Curr. Pharm. Des. 2013, 19, 2326–2333. [Google Scholar] [CrossRef]
- Krasowski, A.; Muthas, D.; Sarkar, A.; Schmitt, S.; Brenk, R. DrugPred: A structure-based approach to predict protein druggability developed using an extensive nonredundant data set. J. Chem. Inf. Model. 2011, 51, 2829–2842. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, J.E.A.; Pinotsis, N.; Ghisleni, A.; Salmazo, A.; Konarev, P.V.; Kostan, J.; Sjöblom, B.; Schreiner, C.; Polyansky, A.A.; Gkougkoulia, E.A.; et al. The structure and regulation of human muscle α-actinin. Cell 2014, 159, 1447–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeturu, K.; Chandra, N. PocketMatch: A new algorithm to compare binding sites in protein structures. BMC Bioinform. 2008, 9, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butina, D. Unsupervised data base clustering based on Daylight’s fingerprint and Tanimoto similarity: A fast and automated way To cluster small and large data sets. J. Chem. Inf. Comput. Sci. 1999, 39, 747–750. [Google Scholar] [CrossRef]
- Luginina, A.; Gusach, A.; Marin, E.; Mishin, A.; Brouillette, R.; Popov, P.; Shiriaeva, A.; Borshchevskiy, V.; Longpré, J.-M.; Lyapina, E.; et al. Structure-based mechanism of cysteinyl leukotriene receptor inhibition by antiasthmatic drugs. Sci. Adv. 2019, 5, eaax2518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F. AutoDock Vina 1.2.0: New docking methods, expanded force field, and Python bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv 2021. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, S.; Hu, Q.; Gao, S.; Ma, X.; Zhang, W.; Shen, Y.; Chen, F.; Lai, L.; Pei, J. CavityPlus: A web server for protein cavity detection with pharmacophore modelling, allosteric site identification and covalent ligand binding ability prediction. Nucleic Acids Res. 2018, 46, W374–W379. [Google Scholar] [CrossRef]
| Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}