
COVID-19 and Parkinsonism: A Critical Appraisal

 ,
,  , , ,
, , ,
Abstract
1. Introduction
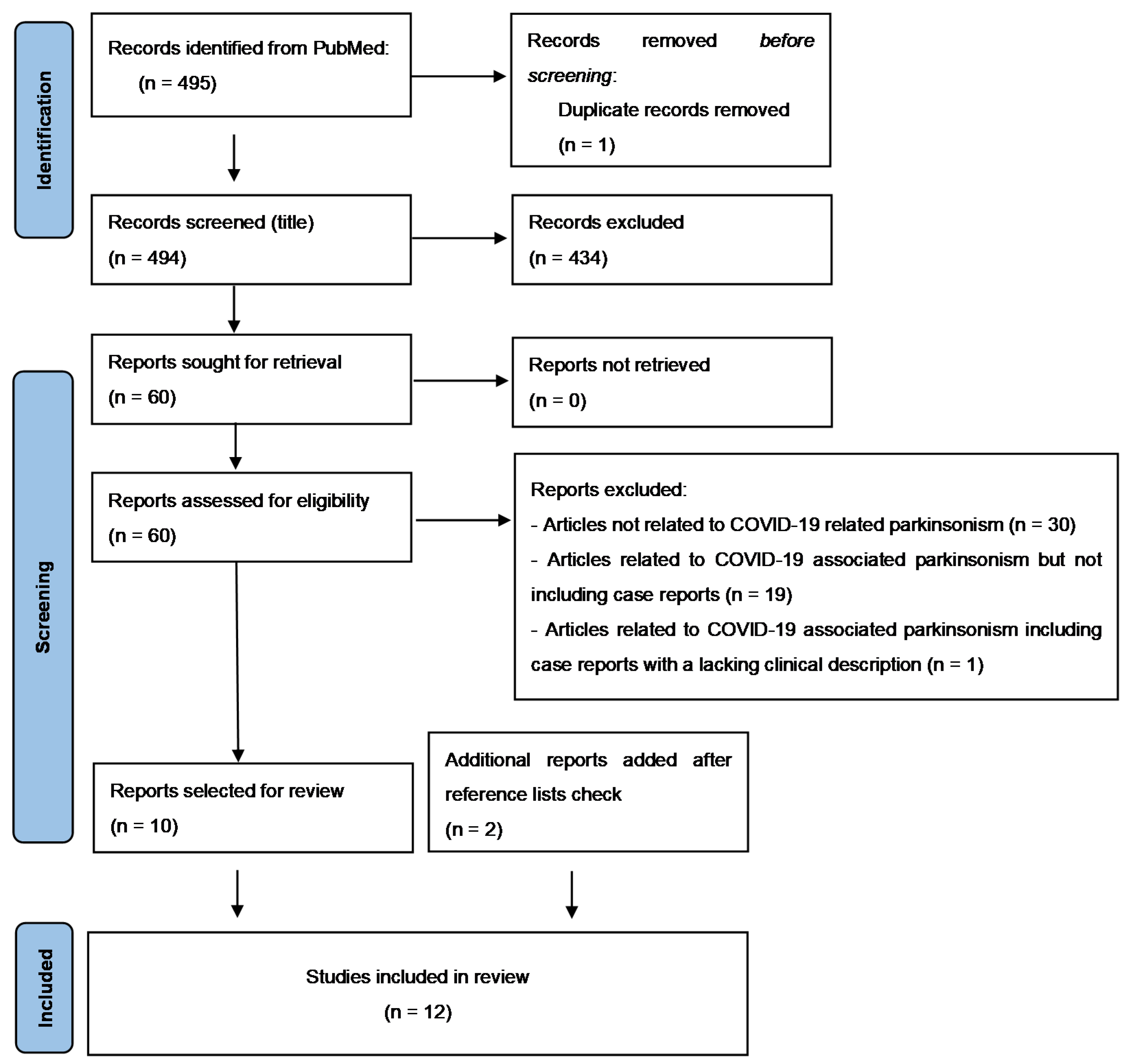
2. Materials and Methods
3. Results
3.1. Diagnostic Findings
3.1.1. Extensive Inflammation or Hypoxic Brain Injury within the Context of Encephalopathy (n = 5)
3.1.2. Unmasking of Underlying Still Non-Symptomatic PD (n = 5)
3.1.3. Structural and Functional Basal Ganglia Damage (n = 3)
4. Discussion
4.1. Data from Systematic Review
4.2. Possible Pathophysiological Mechanisms of COVID-19 Associated Parkinsonism
4.2.1. Vascular Damage
4.2.2. Neuroinflammation
4.2.3. SARS-CoV-2 Neuroinvasivity
4.2.4. SARS-CoV-2 and α-Synuclein
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chou, S.H.-Y.; Beghi, E.; Helbok, R.; Moro, E.; Sampson, J.; Altamirano, V.; Mainali, S.; Bassetti, C.; Suarez, J.I.; McNett, M.; et al. Global Incidence of Neurological Manifestations among Patients Hospitalized with COVID-19-A Report for the GCS-NeuroCOVID Consortium and the ENERGY Consortium. JAMA Netw. Open. 2021, 4, e2112131. [Google Scholar] [CrossRef] [PubMed]
- Fraiman, P.; Junior, C.G.; Moro, E.; Cavallieri, F.; Zedde, M. COVID-19 and Cerebrovascular Diseases: A Systematic Review and Perspectives for Stroke Management. Front. Neurol. 2020, 11, 574694. [Google Scholar] [CrossRef] [PubMed]
- Fasano, A.; Cavallieri, F.; Canali, E.; Valzania, F. First motor seizure as presenting symptom of SARS-CoV-2 infection. Neurol. Sci. 2020, 41, 1651–1653. [Google Scholar] [CrossRef] [PubMed]
- Cavallieri, F.; Marti, A.; Fasano, A.; Salda, A.D.; Ghirarduzzi, A.; Moratti, C.; Bonacini, L.; Ghadirpour, R.; Pascarella, R.; Valzania, F.; et al. Prothrombotic state induced by COVID-19 infection as trigger for stroke in young patients: A dangerous association. ENeurologicalsci 2020, 20, 100247. [Google Scholar] [CrossRef] [PubMed]
- Moro, E.; Priori, A.; Beghi, E.; Helbok, R.; Campiglio, L.; Bassetti, C.; Bianchi, E.; Maia, L.; Ozturk, S.; Cavallieri, F.; et al. The international European Academy of Neurology survey on neurological symptoms in patients with COVID-19 infection. Eur. J. Neurol. 2020, 27, 1727–1737. [Google Scholar] [CrossRef]
- Brandão, P.R.P.; Grippe, T.C.; Pereira, D.A.; Munhoz, R.P.; Cardoso, F. New-Onset Movement Disorders Associated with COVID-19. Tremor Other Hyperkinet. Mov. 2021, 11, 26. [Google Scholar] [CrossRef]
- Romero-Sánchez, C.M.; Díaz-Maroto, I.; Fernández-Díaz, E.; Sánchez-Larsen, Á.; Layos-Romero, A.; García-García, J.; González, E.; Redondo-Peñas, I.; Perona-Moratalla, A.B.; Del Valle-Pérez, J.A.; et al. Neurologic manifestations in hospitalized patients with COVID-19: The ALBACOVID registry. Neurology 2020, 95, e1060–e1070. [Google Scholar] [CrossRef]
- Cavallieri, F.; Fioravanti, V.; Toschi, G.; Grisanti, S.; Napoli, M.; Moratti, C.; Pascarella, R.; Versari, A.; Fraternali, A.; Casali, M.; et al. COVID-19 and Parkinson’s disease: A casual association or a possible second hit in neurodegeneration? J. Neurol. 2022, 269, 59–61. [Google Scholar] [CrossRef]
- Makhoul, K.; Jankovic, J. Parkinson’s disease after COVID-19. J. Neurol. Sci. 2021, 422, 117331. [Google Scholar] [CrossRef]
- Cohen, M.E.; Eichel, R.; Steiner-Birmanns, B.; Janah, A.; Ioshpa, M.; Bar-Shalom, R.; Paul, J.J.; Gaber, H.; Skrahina, V.; Bornstein, N.M.; et al. A case of probable Parkinson’s disease after SARS-CoV-2 infection. Lancet Neurol. 2020, 19, 804–805. [Google Scholar] [CrossRef]
- Sulzer, D.; Antonini, A.; Leta, V.; Nordvig, A.; Smeyne, R.J.; Goldman, J.E.; Al-Dalahmah, O.; Zecca, L.; Sette, A.; Bubacco, L.; et al. COVID-19 and possible links with Parkinson’s disease and parkinsonism: From bench to bedside. NPJ Parkinsons Dis. 2020, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- Merello, M.; Bhatia, K.P.; Obeso, J.A. SARS-CoV-2 and the risk of Parkinson’s disease: Facts and fantasy. Lancet Neurol. 2021, 20, 94–95. [Google Scholar] [CrossRef]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Clinical Management of COVID-19: Interim Guidance, 27 May 2020 and Last Updated as “COVID-19 Clinical Management: Living Guidance, 25 January 2021”. Available online: https://www.who.int/publications/i/item/WHO-2019-nCoV-clinical-2021-2 (accessed on 1 February 2022).
- Singh, B.; Lant, S.; Cividini, S.; Cattrall, J.W.S.; Goodwin, L.C.; Benjamin, L.; Michael, B.D.; Khawaja, A.; Matos, A.D.M.B.; Alkeridy, W.; et al. Prognostic indicators and outcomes of hospitalised COVID-19 patients with neurological disease: An individual patient data meta-analysis. PLoS ONE 2022, 17, e0263595. [Google Scholar] [CrossRef]
- Ayele, B.A.; Demissie, H.; Awraris, M.; Amogne, W.; Shalash, A.; Ali, K.; Zenebe, Y.; Tafesse, A.; Rao, C.P.V. SARS-COV-2 induced Parkinsonism: The first case from the sub-Saharan Africa. Clin. Park Relat. Disord. 2021, 5, 100116. [Google Scholar] [CrossRef]
- Ong, T.L.; Nor, K.M.; Yusoff, Y.; Sapuan, S. COVID-19 Associated Acute Necrotizing Encephalopathy Presenting as Parkinsonism and Myorhythmia. J. Mov. Disord. 2022, 15, 89–92. [Google Scholar] [CrossRef]
- Fearon, C.; Mikulis, D.J.; Lang, A.E. Parkinsonism as a Sequela of SARS-CoV-2 Infection: Pure Hypoxic Injury or Additional COVID-19-Related Response? Mov. Disord. 2021, 36, 1483–1484. [Google Scholar] [CrossRef]
- Tiraboschi, P.; Xhani, R.; Zerbi, S.M.; Corso, A.; Martinelli, I.; Fusi, L.; Grampa, G.; Lombardo, A.; Cavalcante, P.; Cappelletti, C.; et al. Postinfectious Neurologic Complications in COVID-19: A Complex Case Report. J. Nucl. Med. 2021, 62, 1171–1176. [Google Scholar] [CrossRef]
- Morassi, M.; Palmerini, F.; Nici, S.; Magni, E.; Savelli, G.; Guerra, U.P.; Chieregato, M.; Morbelli, S.; Vogrig, A. SARS-CoV-2-related encephalitis with prominent parkinsonism: Clinical and FDG-PET correlates in two patients. J. Neurol. 2021, 268, 3980–3987. [Google Scholar] [CrossRef]
- Faber, I.; Brandão, P.R.P.; Menegatti, F.; Bispo, D.D.D.C.; Maluf, F.B.; Cardoso, F. Coronavirus Disease 2019 and Parkinsonism: A Non-post-encephalitic Case. Mov. Disord. 2020, 35, 1721–1722. [Google Scholar] [CrossRef]
- Méndez-Guerrero, A.; Laespada-García, M.I.; Gómez-Grande, A.; Ruiz-Ortiz, M.; Blanco-Palmero, V.A.; Azcarate-Diaz, F.J.; Rábano-Suárez, P.; Álvarez-Torres, E.; Hoz, C.P.D.F.-F.D.L.; Pérez, D.V.; et al. Acute hypokinetic-rigid syndrome following SARS-CoV-2 infection. Neurology 2020, 95, e2109–e2118. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.; Song, J.; Awad, N.; Zamudio, P. Treatment of unexplained coma and hypokinetic-rigid syndrome in a patient with COVID-19. BMJ Case Rep. 2021, 14, e239781. [Google Scholar] [CrossRef] [PubMed]
- von Economo, K. Encepahlitis lethargica. Wien. Klin. Wochenschrift. 1917, 30, 581–585. [Google Scholar]
- Jang, H.; Boltz, D.A.; Webster, R.G.; Smeyne, R.J. Viral parkinsonism. Biochim. Biophys. Acta 2009, 1792, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Wang, P.; Jankovic, J. The genetics of Parkinson disease. Ageing Res. Rev. 2018, 42, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Gegg, M.E.; Menozzi, E.; Schapira, A.H.V. Glucocerebrosidase-associated Parkinson disease: Pathogenic mechanisms and potential drug treatments. Neurobiol. Dis. 2022, 166, 105663. [Google Scholar] [CrossRef] [PubMed]
- von Linstow, C.U.; Gan-Or, Z.; Brundin, P. Precision medicine in Parkinson’s disease patients with LRRK2 and GBA risk variants-Let’s get even more personal. Transl. Neurodegener. 2020, 9, 39. [Google Scholar] [CrossRef]
- Mascalchi, M.; Vella, A.; Ceravolo, R. Movement disorders: Role of imaging in diagnosis. J. Magn. Reason. Imaging 2012, 35, 239–256. [Google Scholar] [CrossRef]
- Meyer, P.T.; Frings, L.; Rücker, G.; Hellwig, S. 18F-FDG PET in Parkinsonism: Differential Diagnosis and Evaluation of Cognitive Impairment. J. Nucl. Med. 2017, 58, 1888–1898. [Google Scholar] [CrossRef]
- Eckert, T.; Barnes, A.; Dhawan, V.; Frucht, S.; Gordon, M.F.; Feigin, A.S.; Eidelberg, D. FDG PET in the differential diagnosis of parkinsonian disorders. Neuroimage 2005, 26, 912–921. [Google Scholar] [CrossRef]
- Peralta, C.; Strafella, A.P.; van Eimeren, T. International Parkinson Movement Disorders Society-Neuroimaging Study Group. Pragmatic Approach on Neuroimaging Techniques for the Differential Diagnosis of Parkinsonisms. Mov. Disord. Clin. Pract. 2021, 9, 6–19. [Google Scholar] [CrossRef]
- Goldman, S.; Amrom, D.; Szliwowski, H.B.; Detemmerman, D.; Goldman, S.; Bidaut, L.M.; Stanus, E. Reversible striatal hypermetabolism in a case of Sydenham’s chorea. Mov. Disord. 1993, 8, 355–358. [Google Scholar] [CrossRef]
- Stancu, P.; Uginet, M.; Assal, F.; Allali, G.; Lovblad, K.O. COVID-19 associated stroke and cerebral endotheliitis. J. Neuroradiol. 2021, 48, 291–292. [Google Scholar] [CrossRef] [PubMed]
- Kirschenbaum, D.; Imbach, L.L.; Rushing, E.J.; Frauenknecht, K.B.M.; Gascho, D.; Ineichen, B.V.; Keller, E.; Kohler, S.; Lichtblau, M.; Reimann, R.R.; et al. Intracerebral endotheliitis and microbleeds are neuropathological features of COVID-19. Neuropathol. Appl. Neurobiol. 2021, 47, 454–459. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Maccio, U.; Zinkernagel, A.S.; Shambat, S.M.; Zeng, X.; Cathomas, G.; Ruschitzka, F.; Schuepbach, R.A.; Moch, H.; Varga, Z. SARS-CoV-2 leads to a small vessel endotheliitis in the heart. EBioMedicine 2021, 63, 103182. [Google Scholar] [CrossRef] [PubMed]
- Rossouw, T.M.; Anderson, R.; Manga, P.; Feldman, C. Emerging Role of Platelet-Endothelium Interactions in the Pathogenesis of Severe SARS-CoV-2 Infection-Associated Myocardial Injury. Front. Immunol. 2022, 13, 776861. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, R.H.; Love, G.L.; Da Silva Lameira, F.; Sadough, M.S.; Fox, S.E.; Vander Heide, R.S. Acute Endotheliitis (Type 3 Hypersensitivity Vasculitis) in Ten COVID-19 Autopsy Brains. medRxiv 2021. Available online: https://www.medrxiv.org/content/10.1101/2021.01.16.21249632v1 (accessed on 27 May 2022).
- Sashindranath, M.; Nandurkar, H.H. Endothelial Dysfunction in the Brain: Setting the Stage for Stroke and Other Cerebrovascular Complications of COVID-19. Stroke 2021, 52, 1895–1904. [Google Scholar] [CrossRef]
- Kadi, R.; Rumy, A.; Stadnik, T.; Cannie, M.; Mabiglia, C.; Divano, L. Bilateral lesions of the globus pallidus in a young woman. JBR-BTR 2014, 97, 118–120. [Google Scholar] [CrossRef][Green Version]
- Kelley, M.; Samson, K. Bipallidal lesions in COVID 19 patients: Case series and review of literature. Neurology 2021, 96, 1583. [Google Scholar]
- Ashok, S.; Shastri, K.; Beryl Guterman, L.; Guterman, L.R. Bipallidal Lesions in a COVID-19 Patient: A Case Report and Brief Review of Literature. Res. Sq. Prepr. 2020. [Google Scholar] [CrossRef]
- Thomas, A.; Bonanni, L.; Luciano, A. Bipallidal lesions induce delayed and progressive parkinsonism. J. Neurol. 2006, 253, 1. [Google Scholar]
- Mizuguchi, M.; Hayashi, M.; Nakano, I.; Kuwashima, M.; Yoshida, K.; Nakai, Y.; Itoh, M.; Takashima, S. Concentric structure of thalamic lesions in acute necrotizing encephalopathy. Neuroradiology 2002, 44, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Araújo, R.; Gouveia, P.; Fineza, I. Bilateral Thalamic Lesions in Acute Necrotizing Encephalopathy Due to H1N1 Infection. Pediatr. Neurol. 2016, 65, 96–97. [Google Scholar] [CrossRef]
- Maiese, A.; Manetti, A.C.; Bosetti, C.; Del Duca, F.; La Russa, R.; Frati, P.; Di Paolo, M.; Turillazzi, E.; Fineschi, V. SARS-CoV-2 and the brain: A review of the current knowledge on neuropathology in COVID-19. Brain Pathol. 2021, 31, e13013. [Google Scholar] [CrossRef]
- Matschke, J.; Lütgehetmann, M.; Hagel, C.; Sperhake, J.P.; Schröder, A.S.; Edler, C.; Mushumba, H.; Fitzek, A.; Allweiss, L.; Dandri, M.; et al. Neuropathology of patients with COVID-19 in Germany: A post-mortem case series. Lancet Neurol. 2020, 19, 919–929. [Google Scholar] [CrossRef]
- Awogbindin, I.O.; Ben-Azu, B.; Olusola, B.A.; Akinluyi, E.T.; Adeniyi, P.A.; Di Paolo, T.; Tremblay, M.E. Microglial Implications in SARS-CoV-2 Infection and COVID-19: Lessons from Viral RNA Neurotropism and Possible Relevance to Parkinson’s Disease. Front. Cell Neurosci. 2021, 15, 670298. [Google Scholar] [CrossRef]
- Deigendesch, N.; Sironi, L.; Kutza, M.; Wischnewski, S.; Fuchs, V.; Hench, J.; Frank, A.; Nienhold, R.; Mertz, K.D.; Cathomas, G.; et al. Correlates of critical illness-related encephalopathy predominate postmortem COVID-19 neuropathology. Acta Neuropathol. 2020, 140, 583–586. [Google Scholar] [CrossRef]
- Lou, J.J.; Movassaghi, M.; Gordy, D.; Olson, M.G.; Zhang, T.; Khurana, M.S.; Chen, Z.; Perez-Rosendahl, M.; Thammachantha, S.; Singer, E.J.; et al. Neuropathology of COVID-19 (neuro-COVID): Clinicopathological update. Free Neuropathol. 2021, 2, 2. [Google Scholar]
- Hirsch, E.C.; Standaert, D.G. Ten Unsolved Questions About Neuroinflammation in Parkinson’s Disease. Mov. Disord. 2021, 36, 16–24. [Google Scholar] [CrossRef]
- Olanow, C.W.; Savolainen, M.; Chu, Y.; Halliday, G.M.; Kordower, J.H. Temporal evolution of microglia and alpha-synuclein accumulation following foetal grafting in Parkinson’s disease. Brain 2019, 142, 1690–1700. [Google Scholar] [CrossRef]
- Arlehamn, C.S.L.; Dhanwani, R.; Pham, J.; Kuan, R.; Frazier, A.; Dutra, J.R.; Phillips, E.; Mallal, S.; Roederer, M.; Marder, K.S.; et al. α-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease. Nat. Commun. 2020, 11, 1875. [Google Scholar] [CrossRef] [PubMed]
- Hunter, R.L.; Dragicevic, N.; Seifert, K.; Choi, D.Y.; Liu, M.; Kim, H.-C.; Cass, W.A.; Sullivan, P.G.; Bing, G. Inflammation induces mitochondrial dysfunction and dopaminergic neurodegeneration in the nigrostriatal system. J. Neurochem. 2007, 100, 1375–1386. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; O’Reilly, E.J.; Schwarzschild, M.A.; Ascherio, A. Peripheral inflammatory biomarkers and risk of Parkinson’s disease. Am. J. Epidemiol. 2008, 167, 90–95. [Google Scholar] [CrossRef]
- Ferrari, C.C.; Tarelli, R. Parkinson’s disease and systemic inflammation. Parkinsons Dis. 2011, 2011, 436813. [Google Scholar] [CrossRef]
- Beauchamp, L.C.; Finkelstein, D.I.; Bush, A.I.; Evans, A.H.; Barnham, K.J. Parkinsonism as a Third Wave of the COVID-19 Pandemic? J. Parkinsons Dis. 2020, 10, 1343–1353. [Google Scholar] [CrossRef]
- Chaná-Cuevas, P.; Salles-Gándara, P.; Rojas-Fernandez, A.; Salinas-Rebolledo, C.; Milán-Solé, A. The Potential Role of SARS-COV-2 in the Pathogenesis of Parkinson’s Disease. Front. Neurol. 2020, 11, 1044. [Google Scholar] [CrossRef]
- Krey, L.; Huber, M.K.; Hoglinger, G.U.; Wegner, F. Can SARS-CoV-2 Infection Lead to Neurodegeneration and Parkinson’s Disease? Brain Sci. 2021, 11, 1654. [Google Scholar] [CrossRef]
- Li, Z.; Liu, T.; Yang, N.; Han, D.; Mi, X.; Li, Y.; Liu, K.; Vuylsteke, A.; Xiang, H.; Guo, X. Neurological manifestations of patients with COVID-19: Potential routes of SARS-CoV-2 neuroinvasion from the periphery to the brain. Front. Med. 2020, 14, 533–541. [Google Scholar] [CrossRef]
- Dubé, M.; Le Coupanec, A.; Wong, A.H.M.; Rini, J.M.; Desforges, M.; Talbot, P.J. Axonal transport enables neuron-to-neuron propagation of human coronavirus OC43. J. Virol. 2018, 92, e00404-18. [Google Scholar] [CrossRef] [PubMed]
- Song, E.; Zhang, C.; Israelow, B.; Lu-Culligan, A.; Prado, A.V.; Skriabine, S.; Lu, P.; Weizman, O.-E.; Liu, F.; Dai, Y.; et al. Neuroinvasion of SARS-CoV-2 in human and mouse brain. J. Exp. Med. 2021, 218, e20202135. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A multibasic cleavage site in the spike protein of SARS-CoV-2 is essential for infection of human lung cells. Mol. Cell 2020, 78, 779–784.e5. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.L.C.; Lely, A.T.; Navis, G.J.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Khan, S.; Gomes, J. Neuropathogenesis of SARS-CoV-2 infection. Elife 2020, 9, e59136. [Google Scholar] [CrossRef]
- Chen, R.; Wang, K.; Yu, J.; Howard, D.; French, L.; Chen, Z.; Wen, C.; Xu, Z. The spatial and cell-type distribution of SARS-CoV-2 receptor ACE2 in human and mouse brain. Front. Neurol. 2021, 11, 573095. [Google Scholar] [CrossRef]
- Doobay, M.F.; Talman, L.S.; Obr, T.D.; Tian, X.; Davisson, R.L.; Lazartigues, E. Differential expression of neuronal ACE2 in transgenic mice with overexpression of the brain renin-angiotensin system. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R373–R381. [Google Scholar] [CrossRef]
- Rodriguez-Perez, A.I.; Garrido-Gil, P.; Pedrosa, M.A.; Garcia-Garrote, M.; Valenzuela, R.; Navarro, G.; Franco, R.; Labandeira-Garcia, J.L. Angiotensin type 2 receptors: Role in aging and neuroinflammation in the substantia nigra. Brain Behav. Immun. 2020, 87, 256–271. [Google Scholar] [CrossRef]
- Nataf, S. An alteration of the dopamine synthetic pathway is possibly involved in the pathophysiology of COVID-19. J. Med. Virol. 2020, 92, 1743–1744. [Google Scholar] [CrossRef]
- Puelles, V.G.; Lütgehetmann, M.; Lindenmeyer, M.T.; Sperhake, J.P.; Wong, M.N.; Allweiss, L.; Chilla, S.; Heinemann, A.; Wanner, N.; Liu, S.; et al. Multiorgan and renal tropism of SARS-CoV-2. N. Engl. J. Med. 2020, 383, 590–592. [Google Scholar] [CrossRef] [PubMed]
- Solomon, I.H.; Normandin, E.; Bhattacharyya, S.; Mukerji, S.S.; Keller, K.; Ali, A.S.; Adams, G.; Hornick, J.L.; Padera, R.F., Jr.; Sabeti, P. Neuropathological features of Covid-19. N. Engl. J. Med. 2020, 383, 989–992. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, T.; Harii, N.; Goto, J.; Harada, D.; Sugawara, H.; Takamino, J.; Ueno, M.; Sakata, H.; Kondo, K.; Myose, N.; et al. A first case of meningitis/encephalitis associated with SARS-coronavirus-2. Int. J. Infect. Dis. 2020, 94, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Domingues, R.B.; Mendes-Correa, M.C.; Leite, F.B.V.D.M.; Sabino, E.C.; Salarini, D.Z.; Claro, I.; Santos, D.W.; De Jesus, J.G.; Ferreira, N.E.; Romano, C.M.; et al. First case of SARS-COV-2 sequencing in cerebrospinal fluid of a patient with suspected demyelinating disease. J. Neurol. 2020, 267, 3154–3156. [Google Scholar] [CrossRef]
- Paniz-Mondolfi, A.; Bryce, C.; Grimes, Z.; Gordon, R.E.; Reidy, J.; Lednicky, J.; Sordillo, E.M.; Fowkes, M. Central nervous system involvement by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2). J. Med. Virol. 2020, 92, 699–702. [Google Scholar] [CrossRef]
- Al Saiegh, F.; Mouchtouris, N.; Khanna, O.; Baldassari, M.; Theofanis, T.; Ghosh, R.; Tjoumakaris, S.; Gooch, M.R.; Herial, N.; Zarzour, H.; et al. Battle-tested guidelines and operational protocols for neurosurgical practice in times of a pandemic: Lessons learned from COVID-19. World Neurosurg. 2021, 146, 20–25. [Google Scholar] [CrossRef]
- Yin, R.; Feng, W.; Wang, T.; Chen, G.; Wu, T.; Chen, D.; Lv, T.; Xiang, D. Concomitant neurological symptoms observed in a patient diagnosed with coronavirus disease 2019. J. Med. Virol. 2020, 92, 1782–1784. [Google Scholar] [CrossRef]
- Cosentino, G.; Todisco, M.; Hota, N.; Della Porta, G.; Morbini, P.; Tassorelli, C.; Pisani, A. Neuropathological findings from COVID-19 patients with neurological symptoms argue against a direct brain invasion of SARS-CoV-2: A critical systematic review. Eur. J. Neurol. 2021, 28, 3856–3865. [Google Scholar] [CrossRef]
- Jiang, R.-D.; Liu, M.-Q.; Chen, Y.; Shan, C.; Zhou, Y.-W.; Shen, X.-R.; Li, Q.; Zhang, L.; Zhu, Y.; Si, H.-R.; et al. Pathogenesis of SARS-CoV-2 in transgenic mice expressing human angiotensin-converting enzyme 2. Cell 2020, 182, 50–58.e8. [Google Scholar] [CrossRef]
- Sun, S.-H.; Chen, Q.; Gu, H.-J.; Yang, G.; Wang, Y.-X.; Huang, X.-Y.; Liu, S.-S.; Zhang, N.-N.; Li, X.-F.; Xiong, R.; et al. A mouse model of SARS-CoV-2 infection and pathogenesis. Cell Host Microbe 2020, 28, 124–133. [Google Scholar] [CrossRef]
- Meinhardt, J.; Radke, J.; Dittmayer, C.; Franz, J.; Thomas, C.; Mothes, R.; Laue, M.; Schneider, J.; Brünink, S.; Greuel, S.; et al. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat. Neurosci. 2021, 24, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Leda, A.; Bertrand, L.; Andras, I.E.; El-Hage, N.; Nair, M.; Toborek, M. Selective disruption of the blood–brain barrier by Zika virus. Front. Microbiol. 2019, 10, 2158. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.-F.; Chu, L.-W.; Liao, I.-C.; Simanjuntak, Y.; Lin, Y.-L.; Juan, C.-C.; Ping, Y.-H. The mechanism of the Zika virus crossing the placental barrier and the blood-brain barrier. Front. Microbiol. 2020, 11, 214. [Google Scholar] [CrossRef] [PubMed]
- Fleischer, M.; Köhrmann, M.; Dolff, S.; Szepanowski, F.; Schmidt, K.; Herbstreit, F.; Güngör, C.; Stolte, B.; Steiner, K.M.; Stadtler, C.; et al. Observational cohort study of neurological involvement among patients with SARSCoV-2 infection. Ther. Adv. Neurol. Disord. 2021, 14, 1756286421993701. [Google Scholar] [CrossRef] [PubMed]
- Bellon, M.; Schweblin, C.; Lambeng, N.; Cherpillod, P.; Vazquez, J.; Lalive, P.H.; Schibler, M.; Deffert, C. Cerebrospinal fluid features in SARS-CoV-2 RT-PCR positive patients. Clin. Infect. Dis. 2021, 73, e3102–e3105. [Google Scholar] [CrossRef]
- Xu, J.; Zhong, S.; Liu, J.; Li, L.; Li, Y.; Wu, X.; Li, Z.; Deng, P.; Zhang, J.; Zhong, N.; et al. Detection of severe acute respiratory syndrome coronavirus in the brain: Potential role of the chemokine mig in pathogenesis. Clin. Infect. Dis. 2005, 41, 1089–1096. [Google Scholar] [CrossRef]
- Sinha, S.; Mittal, S.; Roy, R. Parkinson’s Disease and the COVID-19 Pandemic: A Review Article on the Association between SARS-CoV-2 and α-Synucleinopathy. J. Mov. Disord. 2021, 14, 184–192. [Google Scholar] [CrossRef]
- Hawkes, C.H.; Del Tredici, K.; Braak, H. Parkinson’s disease: A dual-hit hypothesis. Neuropathol. Appl. Neurobiol. 2007, 33, 599–614. [Google Scholar] [CrossRef]
- Blanco-Palmero, V.A.; Azcárate-Díaz, F.J.; Ruiz-Ortiz, M.; Laespada-García, M.I.; Rábano-Suárez, P.; Méndez-Guerrero, A.; Aramendi-Ramos, M.; Eguiburu, J.L.; Pérez-Rivilla, A.; Marchán-López, A.; et al. Serum and CSF alpha-synuclein levels do not change in COVID-19 patients with neurological symptoms. J. Neurol. 2021, 268, 3116–3124. [Google Scholar] [CrossRef]
- Chen, W.W.; Zhang, X.; Huang, W.J. Role of neuroinflammation in neurodegenerative diseases (Review). Mol. Med. Rep. 2016, 13, 3391–3396. [Google Scholar] [CrossRef]
- Que, Y.; Hu, C.; Wan, K.; Hu, P.; Wang, R.; Luo, J.; Li, T.; Ping, R.; Hu, Q.; Sun, Y.; et al. Cytokine release syndrome in COVID-19: A major mechanism of morbidity and mortality. Int. Rev. Immunol. 2022, 41, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Picard, K.; St-Pierre, M.-K.; Vecchiarelli, H.A.; Bordeleau, M.; Tremblay, M. Neuroendocrine, neuroinflammatory and pathological outcomes of chronic stress: A story of microglial remodeling. Neurochem. Int. 2021, 145, 104987. [Google Scholar] [CrossRef] [PubMed]
- Sharaf, A.; Krieglstein, K.; Spittau, B. Distribution of microglia in the postnatal murine nigrostriatal system. Cell Tissue Res. 2013, 351, 373–382. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Reference | Age | Sex | Comorbidities | COVID-19 Symptoms | COVID-19 Severity | COVID-19 Treatment | Days to Parkinsonian Features Onset after First COVID Symptoms | Genetic Analysis |
|---|---|---|---|---|---|---|---|---|
| Ayele et al., 2021 [16] | 35 | Female | None | Fluctuating mentation, abnormal behavior, fever, visual hallucination | Mild | 13 | N/A | |
| Ong et al., 2021 [17] | 31 | Male | None | Fever, cough, shortness of breath | Severe | Oxygen, dexamethasone, favipiravir, subcutaneous low-molecular-weight heparin. | 15 | N/A |
| Cavallieri et al., 2021 [8] | 67 | Male | None | Dyspnea, fever, anosmia, ageusia | Severe | Tocilizumab | 120 | Heterozygous variant in the GBA gene (NM_000157.3:c.1223C > T-p.(Thr408Met); [T369M]). |
| Cavallieri et al., 2021 [8] | 45 | Male | None | Fever, anosmia and ageusia | Mild | 90 | Heterozygous variant in the PRKN gene (chr6:162683546–1 62683807NM_004562; exons:3) | |
| Fearon et al., 2021 [18] | 46 | Male | None | Fever, dyspnea, cough, ARDS, acute renal failure, DIC | Critical | ICU admission, Intubation and ventilation, dialysis | N/A | N/A |
| Tiraboschi et al., 2021 [19] | 40 | Female | Overweight | Fever, anosmia, fatigue, dyspnea and one syncope | Critical | ICU admission, intubation, and ventilation | 82 | N/A |
| Morassi et al., 2021 [20] | 70 | Female | Hypertension, anxiety–depressive disorder. | Fever, cough, dysgeusia, bilateral pneumonia | Severe | Darunavir, ritonavir, hydroxychloroquine | 47 | N/A |
| Morassi et al., 2021 [20] | 73 | Female | Hypertension, mixed anxiety–depressive disorder. | Fever, unilateral pneumonia | Moderate | 0 | N/A | |
| Makhoul et al., 2021 [9] | 64 | Female | N/A | Fever, fatigue, loss of smell | Mild | 5 | N/A | |
| Cohen et al., 2020 [10] | 45 | Male | Hypertension, asthma | Dry cough, muscle pain, loss of smell, fatigue, shortness of breath. | Moderate | 17 | Negative | |
| Faber et al., 2020 [21] | 35 | Female | N/A | Fever, cough, diarrhea, myalgia, anosmia, hypogeusia | Mild | 10 | N/A | |
| Mendez-Guerrero et al., 2020 [22] | 58 | Male | Hypertension, asthma | Cough, fever, nausea, and shortness of breath, ARDS | Critical | Hydroxychloroquine; Lopinavir/ritonavir, tocilizumab; INF-beta | 32 | N/A |
| Roy et al., 2020 [23] | 60 | Male | Hypertension, diabetes, and hypercholesterolemia | Hypoxic respiratory failure, septic shock, ventricular tachycardia, acute renal failure | Critical | Intubation and mechanic ventilation, convalescent plasma, hemodialysis | 41 | N/A |
| Reference | Parkinsonian Features | Side Involved | Functional Imaging | Brain-MRI | CSF Analysis | Dopaminergic Treatment | Other Treatments | Outcome | Possible Mechanisms of Post-COVID-19 Parkinsonism |
|---|---|---|---|---|---|---|---|---|---|
| Ayele et al., 2021 [16] | Right hand resting tremor, bradykinesia, oromandibular dystonia, rigidity, hypomimia, hypophonia | Bilateral | N/A | Symmetrical T2 and FLAIR hyperintense lesions in both pallidal regions | Unremarkable | Levodopa/carbidopa 250/25 mg half tablet 3/day | IV acyclovir, dexamethasone | Improvement | BG damage |
| Ong et al., 2021 [17] | Reduced eye blinking, mild bilateral upper limb rigidity, slow finger tapping and absence of arm swing | Bilateral | N/A | Symmetrical T2/FLAIR thalamic hyperintensities with hemosiderin deposition and patchy contrast enhancement. | Mildly elevated protein level. | N/A | IV methylprednisolone, trihexyphenidyl | Significant improvement | Inflammation or hypoxic brain injury in encephalopathy |
| Cavallieri et al., 2021 [8] | Right hand resting tremor, bilateral bradykinesia rigidity, (MDS-UPDRS-III: 12/132). | bilateral | Mild bilateral reduction in presynaptic dopaminergic uptake | Bilateral mild white matter hyperintensities in the centrum semiovale and external capsule | N/A | Levodopa 300 mg/day | N/A | Good outcome | Unmasking non symptomatic PD |
| Cavallieri et al., 2021 [8] | Mild resting tremor in left leg and left hand bradykinesia (MDS-UPDRS- III: 4/132) | Left | Decreased dopamine transporter density in both putamens | Unremarkable | N/A | Pramipexole 1.05 mg extended release 1/day | N/A | Good outcome | Unmasking non symptomatic PD |
| Fearon et al., 2021 [18] | Hypophonia, hypomimia, asymmetric rigidity and bradykinesia, freezing of gait, postural instability. | Bilateral | N/A | CT scan/Brain MRI: bilateral edema in the globus pallidus and deep cerebellar nuclei with hemorrhagic foci. | N/A | Levodopa 450 mg/day | N/A | Lack of improvement one year after COVID-19 infection | BG damage |
| Tiraboschi et al., 2021 [19] | Parkinsonism | Bilateral | N/A | Unremarkable | Positive for anti-SARS-CoV-2 IgG antibodies and elevated pro-inflammatory cytokines. | N/A | Two IVIg cycles | Complete resolution of symptoms | Inflammation or hypoxic brain injury in encephalopathy |
| Morassi et al., 2021 [20] | Generalized hypertonia, cogwheel rigidity, bradykinesia hypomimia, hypophonia | Bilateral | Bilateral decrease in presynaptic dopamine involving both putamina, more severe on the left side | Slight enlargement of the ventricular system; fronto-parietal and occipital cortical thinning; fronto-temporal increased cortical thickness. | Decreased amyloid β42, increased total Tau protein | Carbidopa/levodopa (100/25 mg qid) | Corticosteroids followed by five days IVIGs 0.4 g/Kg/die | Modest effect of levodopa. 9 months after presentation: mRS = 4. | Inflammation or hypoxic brain injury in encephalopathy |
| Morassi et al., 2021 [20] | Bilateral hypokinetic-rigid syndrome. | Bilateral | N/A | Unremarkable | Increased protein content and four oligoclonal bands. | Levodopa/carbidopa up to 100/25 mg qid | Corticosteroids, IVIGs | The patient died of medical complications | Inflammation or hypoxic brain injury in encephalopathy |
| Makhoul et al., 2021 [9] | Rest tremor in her left arm, minimal hypomimia and mild left-sided bradykinesia and rigidity | bilateral | decreased uptake in the right putamen | N/A | N/A | N/A | N/A | N/A | Unmasking non symptomatic PD |
| Cohen et al., 2020 [10] | Right more than left tremor, bradykinesia, rigidity | Bilateral | Decreased uptake in bilateral putamen more apparent on the left | Normal | Unremarkable | 0.375 mg pramipexole extended release, once daily, biperiden 4 mg daily. | N/A | Tremor improvement after biperiden introduction | Unmasking non symptomatic PD |
| Faber et al., 2020 [21] | Right rigidity, bradykinesia hypophonia, hypomimia, MDS-UPDRS part III: 49. | Bilateral | Decreased left putamen uptake | Unremarkable | Levodopa/benserazide 200/50 mg three times a day | N/A | Improvement after levodopa introduction | Unmasking non symptomatic PD | |
| Mendez-Guerrero et al., 2020 [22] | Right side–dominant hypokinetic-rigid syndrome, with mixed postural and resting tremor. | Bilateral | Decreased uptake in bilateral putamen more apparent on the left | Unremarkable | Unremarkable | Apomorphine test (3 mg) | N/A | Improvement without any specific treatment | Inflammation or hypoxic brain injury in encephalopathy |
| Roy et al., 2020 [23] | Diffuse hypokinetic rigid syndrome | Bilateral | N/A | Basal ganglia and corona radiata stroke. | Carbidopa–levodopa 100/25 mg three times a day | N/A | The patient was able to discharge to home after 30 days at the acute rehabilitation center. | BG damage |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cavallieri, F.; Fioravanti, V.; Bove, F.; Del Prete, E.; Meoni, S.; Grisanti, S.; Zedde, M.; Pascarella, R.; Moro, E.; Valzania, F. COVID-19 and Parkinsonism: A Critical Appraisal. Biomolecules 2022, 12, 970. https://doi.org/10.3390/biom12070970
Cavallieri F, Fioravanti V, Bove F, Del Prete E, Meoni S, Grisanti S, Zedde M, Pascarella R, Moro E, Valzania F. COVID-19 and Parkinsonism: A Critical Appraisal. Biomolecules. 2022; 12(7):970. https://doi.org/10.3390/biom12070970
Chicago/Turabian StyleCavallieri, Francesco, Valentina Fioravanti, Francesco Bove, Eleonora Del Prete, Sara Meoni, Sara Grisanti, Marialuisa Zedde, Rosario Pascarella, Elena Moro, and Franco Valzania. 2022. "COVID-19 and Parkinsonism: A Critical Appraisal" Biomolecules 12, no. 7: 970. https://doi.org/10.3390/biom12070970
APA StyleCavallieri, F., Fioravanti, V., Bove, F., Del Prete, E., Meoni, S., Grisanti, S., Zedde, M., Pascarella, R., Moro, E., & Valzania, F. (2022). COVID-19 and Parkinsonism: A Critical Appraisal. Biomolecules, 12(7), 970. https://doi.org/10.3390/biom12070970