Abstract
Mature neurotrophic factors and their propeptides play key roles ranging from the regulation of neuronal growth and differentiation to prominent participation in neuronal survival and recovery after injury. Their signaling pathways sculpture neuronal circuits during brain development and regulate adaptive neuroplasticity. In addition, neurotrophic factors provide trophic support for damaged neurons, giving them a greater capacity to survive and maintain their potential to regenerate their axons. Therefore, the modulation of these factors can be a valuable target for treating or preventing neurologic disorders and age-dependent cognitive decline. Neuroregenerative medicine can take great advantage by the deepening of our knowledge on the molecular mechanisms underlying the properties of neurotrophic factors. It is indeed an intriguing topic that a significant interplay between neurotrophic factors and various metals can modulate the outcome of neuronal recovery. This review is particularly focused on the roles of GDNF, BDNF and NGF in motoneuron survival and recovery from injuries and evaluates the therapeutic potential of various neurotrophic factors in neuronal regeneration. The key role of metal homeostasis/dyshomeostasis and metal interaction with neurotrophic factors on neuronal pathophysiology is also highlighted as a novel mechanism and potential target for neuronal recovery. The progress in mechanistic studies in the field of neurotrophic factor-mediated neuroprotection and neural regeneration, aiming at a complete understanding of integrated pathways, offers possibilities for the development of novel neuroregenerative therapeutic approaches.
1. Introduction
Neurotrophin (NT) modulation can be a valuable target for treating or preventing neurologic disorders and age-dependent cognitive decline. Neuroregenerative medicine can take particular advantage through the deepening of knowledge of the molecular mechanisms underlying the NT properties. It is indeed intriguing that a significant interplay between NTs and essential metals can modulate the neuronal recovery outcome.
Motoneuron recovery represents a well-known example in the studies of neuroregeneration. Indeed, motoneuron degeneration and regenerative outcome received large attention, owing to their implications for muscular control and skilled activities. Remarkably, in contrast to CNS, motor neurons are autonomously able, under certain conditions, to support robust regenerative responses after peripheral nerve injuries [1].
This feature makes motoneuron recovery from insults a compelling model to study the molecular events that govern neuroregenerative pathways. On the contrary, regeneration after severe acute trauma or chronic neurodegeneration from underlying neurodegenerative disease is often irreversible with a profound impact on neuromuscular functions. Thus, amyotrophic lateral sclerosis (ALS), a disease that shows selective motor neuron degeneration with typical signs including axonal damage, mitochondrial dysfunction, cytoskeletal deterioration, and muscle denervation, gave a further model system for studies on motoneurons pathophysiology [2].
This review aims to emphasize the relevant role of NTs, particularly GDNF, BDNF, and NGF, in the process of neural recovery and motoneuron regeneration, being able to protect motoneurons from various insults and support restoration of lost neuromuscular interplay. NTs have been widely explored as therapeutic targets in different experimental models of motor neuron damage and ALS, and also in age-related neurodegenerative diseases. This review will also evaluate the key role of metal homeostasis/dyshomeostasis and metal interactivity with NTs on neuronal pathophysiology. This is a new scheme and potential target for neurorecovery that suggests further strategies and tools to deepen the knowledge on multiple NT activities.
2. Motoneuron Injury and Degeneration
Injury close to the cell body of motoneurons results in the death of the vast majority of injured motoneurons [3,4,5]. The morphological changes of the damaged motoneurons are initiated within a few days following ventral root avulsion injury. The cell body of the motoneurons becomes swollen and the nucleus migrates toward the periphery of cells. The Nissl bodies (tigroid) break up into several small units and appear to dissolve, which is a process known as chromatolysis [6,7]. Injury to the ventral root induces biochemical cascades in spinal motoneurons, resulting in glutamate-mediated excitotoxic events [8]. Injury upregulates the subtypes of NMDA and AMPA/kainate receptors which play a key role in glutamate-mediated excitotoxicity [9]. Activation of NMDA and AMPA receptors leads to the influx of Ca2+ ions into neurons and the increased cytoplasmatic concentration of Ca2+ induces a cascade of secondary processes, resulting in cell death. Glutamate release and thus excitotoxicity can be effectively blocked by presynaptic glutamate release inhibitors—for example, riluzole (2-amino-6-trifluoromethoxybenzothiazole) [10].
Koliatsos et al. [3] have demonstrated that avulsion injuries result in the retrograde death of the majority of the affected motoneurons in the first 2 weeks after injury. The outcome of motoneuron damage depends on the distance of the implied lesion from the cell body of the injured motoneurons. If the axotomy is performed close to the perikaryon most of the affected motoneurons die. On the other hand, motoneurons survive after axotomy of a peripheral nerve, if at least a 5 mm segment of the nerve remains proximal to the injury site and thus provides trophic support for the damaged motoneurons [11]. If the avulsed ventral root is surgically re-implanted into the spinal cord segment soon after injury, some of the injured motoneurons survive and maintain their capacity for axonal regrowth [5,10,11]. Cervical motoneurons have a greater capacity to survive and re-innervate target muscles than motoneurons localized in the lumbar spinal cord segments. This phenomenon is likely due to (a) the shorter distance that axons of the surviving neurons have to bridge to reach their targets and (b) the fact that the distal degenerated peripheral nerve segments may undergo “predegeneration”; an effect considerably slowing down or prohibiting the growth of regenerating axons [12,13,14]. It has been earlier recognized that successful reinnervation may occur only if a satisfactory number of surviving motoneurons remains in the spinal cord after avulsion injury. Therefore, different strategies to prevent the death of the injured motoneurons have been developed in the last two decades including various growth factors [15,16,17].
Neurotrophic factors are essential proteins for neuronal differentiation during development [18] and they also support neuronal maintenance in the adult central nervous system (CNS), modulate neuronal plasticity and synaptic transmission [13]. The neurotrophic factor family has several members and many of these have more or less neuroregenerative effects. An expanded repertoire of neurotrophic factors is now available for improving the outcome of motoneuron injury, especially the administration of brain- or glial cell line-derived neurotrophic factors (BDNF and GDNF, respectively).
3. Molecular Pathways of BDNF GDNF and NGF
Brain-derived neurotrophic factor BDNF was described in the 1980s by Barde et al. and belongs to the family of neurotrophins [19]. BDNF acts on certain neurons, helping to support the survival and differentiation of distinct neurons and synaptic plasticity in the CNS [18,20]. Its active form can be found in various parts of the brain and is also expressed in the retina, motoneurons, and skeletal muscle [21,22,23]. BDNF is first synthesized as a precursor, pro-BDNF in the endoplasmic reticulum, that acts as a biologically active factor, different from mature BDNF. Pro-BDNF can reduce dendritic complexity and synaptic plasticity in the hippocampus, while mature BDNF exhibits an opposing function in the CNS [24,25]. Pro-BDNF predominates in developmental stages, whereas mature BDNF is expressed in considerable amounts in adulthood [25].
BDNF mediates its effect through the TrkB (pronounced “Track B”) and the pan-neurotrophin receptor p75NTR. The latter was initially identified as the Low-affinity Nerve Growth Factor Receptor (LNGFR) and belongs to the tumor necrosis factor (TNF) receptor family [26]. TrkB receptors are present both on the presynaptic and postsynaptic membranes and are ~145 kDa membrane-bound glycoproteins. They bind with high affinity to BDNF, whereupon they dimerize and transphosphorylate each other. These post-translational modifications further induce phosphorylation of other tyrosine residues that operate as a docking site for adaptor proteins which launch further intracellular signaling pathways [18]. The cascades activate protein kinase B (AKT) and mitogen-activated protein kinase (MAPK) pathways that regulate several gene expression processes via transcription factor cAMP response element binding protein (CREB) and modulate apoptotic, survival mechanism and mammalian target of rapamycin (mTor) [27,28].
GDNF was first described as a neurotrophic factor in 1991 by Engele et al. [29]. GDNF is mainly expressed by neurons in the developing and adult CNS but is released from glial cells, too [30,31]. Numerous studies have shown that GDNF promotes differentiation of neurons, prevents apoptosis and enhances survival of injured motoneurons induced by ventral root avulsion injury [15,16]. GDNF preferentially forms a complex with the GDNF family receptor α1 (GFR α1, a glycosylphosphatidylinositol [GPI]-linked cell surface receptor), which is anchored to the plasma membrane of neurons. The GDNF-GFR α1 complex interacts with a receptor tyrosine kinase, RET receptors, resulting in activation of the intracellular kinase domain to induce multiple intracellular signaling pathways [32]. RET is mainly expressed in sensory neurons, dopaminergic and noradrenergic neurons and plays an essential role in the development of the sympathetic, parasympathetic and enteric nervous systems [33,34,35]. Activation of RET receptors induces various signaling pathways such as MAPK, phosphoinositide 3-kinase, and the phosphoinositide phospholipase C-γ pathway, which regulate cell survival, differentiation, proliferation, migration, neurite outgrowth, and synaptic plasticity [36].
Nerve growth factor (NGF) was first described in chicken embryo by Levi-Montalcini and Hamburger [37]. NGF binds to the tropomyosin receptor kinase A (trkA) and the p75 neurotrophin receptor (p75NTR). TrkA has a high affinity for NGF. The NGF–trkA interaction activates various molecular pathways including the phospholipase C-γ (PLCγ), the mitogen-activated protein kinase (MAPK)/Erk and the phosphoinositide 3-kinase (PI3K) pathways [38]. While NGF has affinity to both receptors, p75 NTR shows higher affinity for pro-NGF than NGF itself. p75NTR is a Trk co-receptor that can activate signaling cascades such as the NF-κB, Akt, and JNK pathways, resulting in the induction of apoptosis or in the promotion of survival of neurons [39]. As far as we are concerned, extraocular motoneurons are sensitive and responsive to NGF treatment [40].
4. Therapeutic Potential of BDNF, GDNF and NGF following Motoneuron Injury
Most of the studies concerning motoneuron survival and regeneration have focused on the neuroprotective role of different neurotrophic factors, such as BDNF and GDNF, known to promote neuronal survival and axonal growth both in vitro and in vivo [15,16,41]. These factors have been shown to induce extensive survival of neurons and partial or aberrant axonal regeneration after injuries, but the efficacy of these factors depends on their continuous supply. The side effect of this uncontrolled and persistent expression of neurotrophic factors at the site of injury was the formation of irregular axon coils, likely due to the trapping effect of neurotrophic factors on the injured axons [42]. A study by Novikov et al. demonstrated that exogenous treatment with BDNF rescued axotomized spinal motoneurons and also promoted partial recovery of the afferents [38]. However, long-term infusion of BDNF has other disadvantages, too, e.g., loss of S-type boutons on the injured and intact motoneurons [42,43]. A serious side effect of intraspinal axonal sprouting induced by BDNF treatment is spasticity, which may seriously jeopardize the positive therapeutic effects [44]. One of the promising methods to overcome these problems is the use of transplanted cells with neurotrophic factor expression capacity for more localized and regulated effects. Different groups have provided evidence that BDNF-expressing cells support extensive axonal growth at sites of spinal cord injury [15,45].
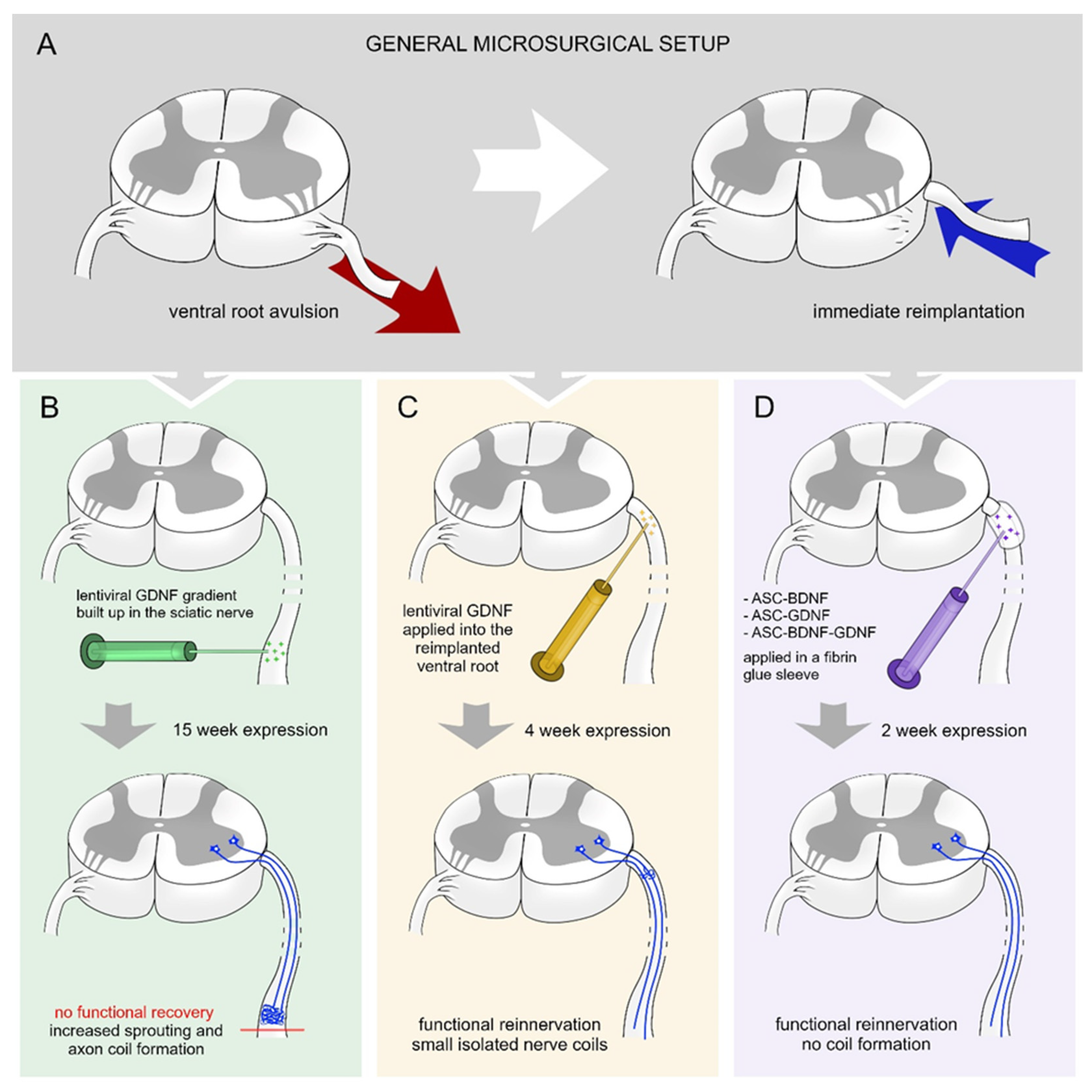
Eggers et al. created gradients of GDNF in the sciatic nerve 2 weeks after a ventral root avulsion (Figure 1A,B) [41]. The gradually increasing lentiviral-mediated GDNF expression results in increased axon numbers and formation of nerve coils. Moreover, the density of Schwann cells and motor axon sprouting were increased. The continuous expression of GDNF did not enhance the survival of motoneurons or their regeneration [41].
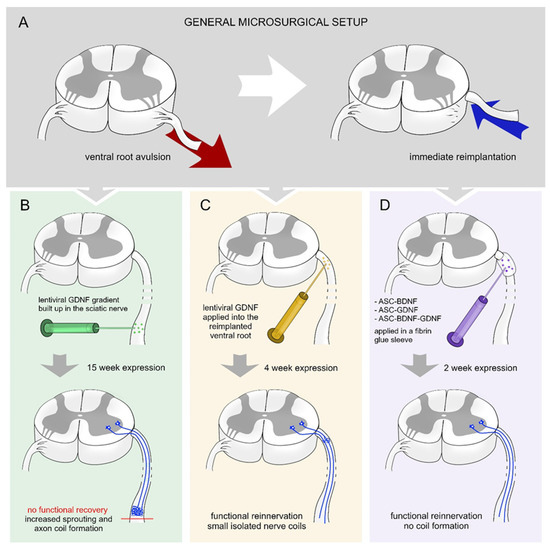
Figure 1.
Various treatment strategies with BDNF and GDNF following ventral root injury. (A) Schematic overview of experimental ventral root avulsion and reimplantation. (B) Building up a proximo-distal gradient of GDNF [41] (15-week-long effect) in the sciatic nerve leads to robust axon coil formation that hinders the regeneration of the injured motoneurons and functional reinnervation of denervated hind limb muscles. (C) Shorter (4-week-long) expression period of time of GDNF [16] in the reimplanted ventral root results in the appearance of small isolated axon coils and functional recovery. (D) Transfected rat adipose tissue-derived stem cells (rASCs) grafted around the reimplanted ventral root producing GDNF and/or BDNF for 2 weeks [15] enhances elongative axon growth without coil formation and results in functional recovery.
In other approaches, a viral or non-viral vector system was used to assess the efficacy of time-restricted BDNF and GDNF expression on motor neuron survival and axon regeneration following ventral root injury [15,46]. Time-restricted expression of GDNF hinders the axon coil formation and a considerable number of regenerating axons showed elongative growth pattern (Figure 1C) [16]. The time-restricted expression of GDNF enhanced the motoneuron survival in the lumbar segment and their axons reinnervated the nerve. The temporally expressed GDNF enhanced motoneuron survival in cervical segments following ventral root avulsion injury and promoted axon regeneration and muscle reinnervation. The expression of BDNF was capable of itself to induce as high amount of motoneuron survival and regeneration as individual production of GDNF (Figure 1D) [15]. However, it is important to note that the combined action of BDNF and GDNF had no synergistic effect on the regeneration of injured motoneurons and did not induce significantly improved functional recovery [15]. Further studies are required to be performed to investigate the time-restricted sequential expression effect of BDNF and GDNF on motoneuron survival and regeneration.
NGF is also a promising neurotrophic factor that is mainly used to induce nerve regeneration following peripheral nerve injury. Kemp and his colleagues have shown in an elegant study that NGF has a bell-shaped dose response curve for axonal regeneration. The optimal dose of NGF is 80 ng/day for 3 weeks that induced the best sensorimotor recovery compared to all other treatment groups [47].
To reach the potential neuroregenerative effect of NGF, NGF-loaded delivery systems have been used to enhance axon regeneration following peripheral nerve injury. These nerve conduits provide a suitable alternative to autologous nerve grafts and can release NGF for extended periods of time [48]. Several studies have reported that nerve conduits treated by NGF promote peripheral nerve regeneration efficiently [49,50,51]. A longitudinally oriented collagen conduit combined with NGF was able to reconstruct a long distance (35 mm) nerve gap via axonal regeneration following sciatic nerve injury in dog [50]. Similarly, thermo-sensitive heparin-poloxamer hydrogel co-delivered with NGF facilitated Schwann cell proliferation, enhanced axonal regeneration and remyelination, and improved recovery of motor function following sciatic nerve crush injury in diabetic rats [52]. Enzymatically cross-linked silk fibroin-based conduits were also used as a platform for the controlled delivery of NGF [53]. In another experimental set up NGF was loaded in the size-tunable microfluidic hollow fibers, which was released gradually and promoted a rapid morphological and functional axon regeneration in rats with 5-mm sciatic nerve defects [54]. All together, these results show that NGF loaded in nerve conduit possesses the capacity of promoting nerve regeneration after peripheral nerve injury; however, further studies are required to achieve the best and the perfectly safe therapeutic approach.
5. Neurotrophins and Metals
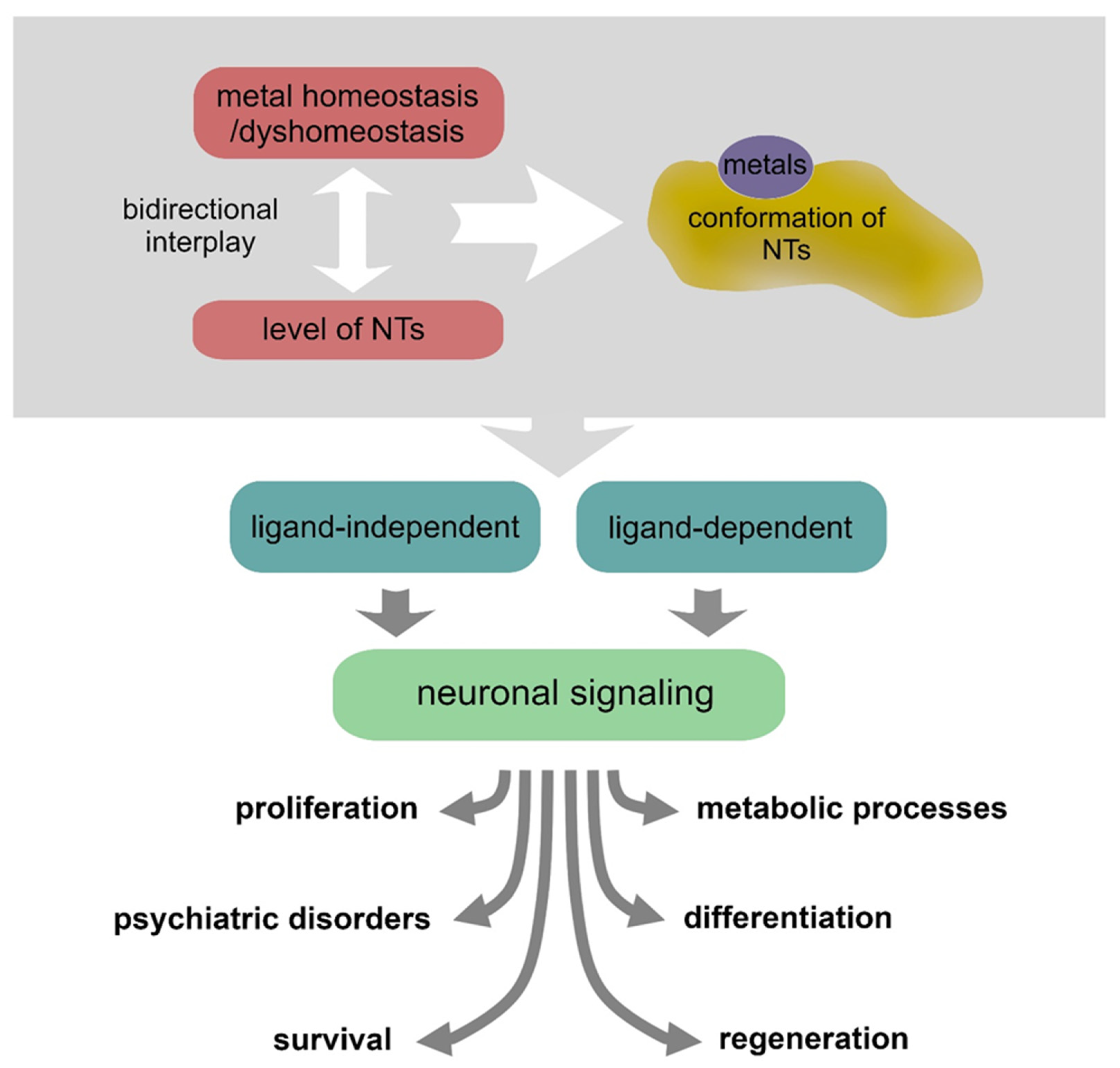
In the last two decades, it has emerged that there is a strict interplay between NTs and various metals. Metals can induce conformational changes by direct interaction with NTs, hence influencing the receptor recognition processes, or can contribute to their expression/secretion [55]. Intricate mechanisms of metallostasis have a critical impact on the NT activities in supporting neuronal physiology and also play a significant role in pathological conditions [56,57] (Figure 2 and Table 1).
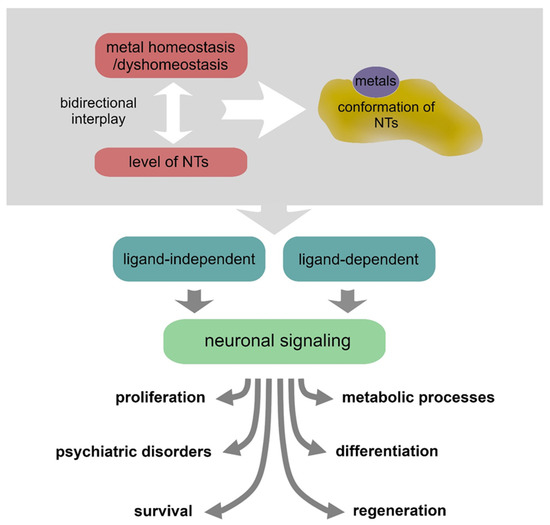
Figure 2.
Metal homeostasis/dyshomeostasis can affect the expression, conformation and signaling of various neurotrophins, thus producing various effects on neuronal pathophysiology.
Environmental metal neurotoxicity is not addressed in this review; however, considerable evidence suggests that targeting the metal homeostasis represents a new challenge in the exploration of alternative ways to gain neuronal functional recovery [58,59,60]. While these studies do not specifically target motor neurons, they can provide useful information to better understand motor neuron physiology and discover new therapeutic approaches for neural recovery.
The activity of NTs can be modulated differently by copper and zinc, two transition metals highly implicated in neuronal physiology and pathology. Intriguingly, their direct interaction with these metals can exert different and sometimes even opposite effects. In neuronal cell cultures, Zn2+ treatment and increased binding to BDNF have been associated with proliferative activity. Conversely, the effect of Cu2+ addition on the BDNF activity is the opposite. Interestingly, the effects of these metal ions are reversed in the case of cell culture treatment with NGF: the presence of Cu2+ exerts proliferative activity, whereas cell proliferation is repressed after Zn2+ addition [61,62]. Noteworthy, in vitro evidence indicates that high concentrations of Zn2+ and Cu2+ can produce conformational changes of different NTs and, in turn, ineffective interaction with specific receptors that affect trophic and antioxidant properties with consequent detrimental effect on motoneuron viability [63], or blocking of the NGF-mediated neurite outgrowth [64,65]. However, Zn2+ has been indicated as a key cofactor for the protective activity of NGF, that could be related to either metals inhibition of p75-mediated apoptotic cascades, or metals participation in neurotrophic signaling via the TrkA receptor [66,67,68]. Although the signaling induced by NTs is well characterized, ligand-independent activation of TrkB receptor has been also observed as solely mediated by zinc [69,70]. Zinc promotes a signaling event critical for transactivation of TrkB by preferential phosphorylation of Tyr-705/Tyr-706 of TrkB by a Src family kinase (SFK)-dependent, but TrkB kinase-independent mechanism, that implicates a regulatory role of SFKs in TrkB activation by BDNF [71]. Similarly, zinc induces transactivation of the EGF receptor (EGFR) by Src-dependent phosphorylation of Tyr-845 of EGFR [72]. These events are of particular relevance to neural activity involved in long-term potentiation (LTP), mediated by the release of zinc stored in secretory vesicles [73]. In this respect, it is worth mentioning the metal ions effects on glutamatergic synapses in the hippocampus, especially considering that this brain area is involved in learning and memory and is the brain area with the highest levels of Zn2+ and Cu2+, and NT activity [74,75]. Other evidence supports the hypothesis that also released Cu2+ ions can transactivate TrkB through extracellular signal-regulated kinase 1/2 and Src tyrosine kinase. This Cu-mediated pathway seems associated with increased activity of matrix metalloproteinase 2 and 9, which contributes to increased secretion of pro-BDNF and mature BDNF in cortical neurons and during wound healing [76,77].
A bidirectional interplay between metals and NTs should be also taken into consideration. BDNF and GDNF, as well as other factors including CNTF (Ciliary neurotrophic factor) and PEDF (pigment epithelium-derived factor), have been shown to modulate the expression of zinc(II) influx transporter, ZIP2, which increases metal ion uptake and intracellular Zn2+ level in RPE (retinal pigment epithelium) cells. However, differential effects on other zinc transporters are observed with other NTs: CNTF downregulates the expression of ZIP4, ZIP14, and ZnT6; PEDF downregulates ZIP4 and ZIP14; GDNF acts similarly on ZnT6; both PEDF and GDNF promote higher levels of ZnT2; PEDF upregulates the expression of ZnT3, the primary zinc loader in the brain [78].
Various NTs, including BDNF, NGF, and GDNF, as well as their receptors can also be upregulated by lithium, a metal with a long-lasting history of use as first-line drug for treating bipolar disorder and depression [79,80]. This effect explains the neuroprotective properties of lithium against injuries and in neuroregeneration.
On account of the aforementioned points, it is evident that transition metal–NT interactions could trigger various signaling pathways, suggesting that any intervention on transition metals homeostasis can produce relevant effects not restricted to various metabolic processes, including enzyme activities and gene expression, but nervous system physiology and many pathologic statuses (e.g., Alzheimer’s diseases) can be strongly challenged as well [81,82]. This issue is still debated but could deserve deeper understanding to highlight specific pathways and cell-specific targeting. This can open the way to gain valuable therapeutic tools for tissue-directed neuronal protection and recovery.

Table 1.
Neurotrophin–metal interplay and metal-related neuromodulatory effects.
Table 1.
Neurotrophin–metal interplay and metal-related neuromodulatory effects.
| NTs | Metal | Mechanism | Effect | Ref. |
|---|---|---|---|---|
| n.a. | Various | Metal dysregulation | Motoneuron pathology | [83,84,85,86] |
| Various | Copper or Zinc | Protein conformational changes | Protein-misfolding diseases | [58,59,60,87,88] |
| NGF | Copper or Zinc | Neurotrophic effects | Neuronal cell culture changes in proliferation | [62] |
| BDNF | [63] | |||
| BDNF | Copper or Zinc | Direct interaction and NT conformational changes | Altered motoneuron trophic signaling | [63] |
| NGF | Altered neuronal trophic signaling | [64,65] | ||
| NGF | Zinc | Direct interaction and receptor transactivation | Neuroprotective outcome | [66,67,68] |
| BDNF | Zinc | Receptor transactivation | Tyr phosphorylation cascade (SFK) | [69,70,71] |
| EGF | [60,72] | |||
| BDNF | Copper | NT level changes | Increased secretion of pro-BDNF and mature BDNF | [76,77] |
| BDNF | Zinc | NT-mediated changes of metal homeostasis | Modulation of zinc transporters | [78] |
| GDNF | ||||
| Other NTs | ||||
| BDNF, GDNF, NGF | Lithium | NT upregulation | Neuroprotection, neuroregeneration, and axons remyelination | [79,80,89,90] |
| BDNF | Zinc | [91,92] | ||
| n.a. | Lithium | Others | Antidepressant | [86,89,93,94] |
| Induction of autophagy | protection from spinal cord injury | [90,95,96,97,98] |
n.a. = not applicable.
Table 1 shows that a functional bidirectional interplay between NTs and essential metals is emerging as a relevant mechanism in the processes that sustain neuronal protection and recovery from various injuries. NT levels can be modulated by the presence of various metals, but NTs can also modulate metals homeostasis, and metals per se can also trigger neurotrophic signaling. However, metal dyshomeostasis is known to produce neurotoxic conditions as well.
6. Current Insight into the Potential of Regenerative Medicine Combining Metal Modulation and Neurotrophins
Considerable efforts have been dedicated to the search for effective treatments of injured neurons either during a degenerative disease or after traumatic events. The study of the role of NTs in the recovery of nerve function and nerve regeneration is one the most important issues to achieve this goal. Many growth factors and signaling pathways are known to be involved to various extents in axonal maturation as well as restoration after injury [82,99]. Noteworthy, emerging evidence highlights the role of metals in NT upregulation or modulation of specific signaling cascades and provides new perspectives in the search for advanced therapeutic tools. The importance of metals for neuronal pathophysiology is currently mainly studied in the function of neurodegenerative diseases. The molecular events described to date, however, could also be of interest to understand the molecular pathways involved in motoneuron recovery, and this review highlights the need to expand studies in particular on neurotrophin–metal correlations.
Despite conflicting results that have been reported so far, the study of the mimetic activity of NGF peptides showed that copper and zinc ions exert modulatory effects through conformational changes, and/or indirectly by activating their downstream signaling in a neurotrophin-independent mode [100]. Zinc and copper ions seem to induce restoring outcomes in several in vitro and in vivo models. Both metals rescued the level of NGF in zinc(II)-deficient mice, counteracted p75-driven apoptosis in the chick neural retina, and blocked the binding of NGF to p75, attenuating the triggered pro-apoptotic signaling cascade in chick embryo cell cultures [67]. Further, copper and zinc play a prominent role in the signaling during memory formation [87,101,102,103].
Accordingly, zinc treatment (micromolar concentrations) can boost BDNF mRNA expression in cortical cell cultures and promotes BDNF release and maturation [91,92,104]. A well-known pathway of synaptic plasticity. For these properties, zinc has been included in the integrative therapy for some psychiatric illnesses [105]. However, more generally, these metals can influence the pathways involved in CREB expression and phosphorylation [100,106,107], thus sustaining the multiple paths of CREB activity that have been shown to induce successful regeneration into the lesion site in spinal cord injury models [108,109].
The constant expansion in understanding the roles of zinc in normal human physiology is also accompanied by a new perspective on the role of zinc homeostasis management as a potential therapeutic tool in neuroregeneration. In the case of optic nerve damage, the death of retinal ganglion cells (RGCs) has been observed to occur in association with a rapid increase in mobile zinc in the retina, particularly in synaptic vesicles of amacrine cell processes, after optic nerve injury. In this study, by removing the Zn2+ transporter ZnT-3, or chelating the Zn2+ ions, the authors were successful in rescuing many injured CGRs and regenerating axons. This is a clear demonstration of the importance of Zn2+ dyshomeostasis in both RGC death and regenerative failure, and the clinical feasibility of zinc homeostasis management [110].
An increase in intracellular copper can modulate many proteins associated with the regulated secretory vesicle pathways, including synaptophysin, syntaxin-1, SNAP-25, and the LDCV proteins chromogranins A and C [111]. This agrees with the observations that copper internalization is associated with molecular steps essential in neuronal differentiation and neurite branching [112,113]. Remarkably, copper interplay with prion protein (PrPC) and binding within the physiological concentration range displays a functional role of PrPC in normal copper homeostasis in brain metabolism, whereas Cu2+ rapidly and reversibly stimulates the internalization of PrPC [114,115]. Hence, suggesting a copper contribution to PrPC-mediated survival signal and the promotion of neuritogenesis [116,117].
A large body of evidence associates metal dysregulation with motoneuron diseases [118,119]. Many studies have been focused on the role of aberrant metal homeostasis and binding in the aggregation process of SOD1 that has been shown to trigger SOD1 toxic deposition in amyotrophic lateral sclerosis (ALS) [83]. Furthermore, mutations in the copper transport gene ATP7A cause, among another two distinct human diseases, X-linked distal hereditary motor neuropathy, a condition that affects ATP7A intracellular trafficking and alters Cu levels within the nervous system. [84]. Based on the discovery that a CCS/G93A-SOD1 dual transgenic mice model of ALS develop accelerated neurological deficits, consistent with an apparent copper deficiency within the spinal cord, Williams et al. tested a therapeutic approach based on copper replenishment. Using the copper complex CuATSM, a safe vehicle of copper selective in cells with damaged mitochondria, they demonstrated that early CuATSM treatment of pups expressing SODG93A with CCS avoided the early mortality and allowed a normal grow [118]. According to this study, direct evidence reveals that altered metal uptake during specific early lifetime windows is associated with adult-onset ALS [85]. Thus, copper homeostasis is undeniably crucial in the maintenance and function of motor neurons, and therefore in the pathogenic mechanisms underlying motoneuron degeneration. This question deserves further examination with a view to possible connections with the functions of NTs.
Lithium is a metal with a long-lasting history of use as first-line drug for treating bipolar disorder, as well as recurrent depression [86]. Lithium-induced pharmacological effects were explained by replenishment with BDNF and NGF in patients with depression. Indeed, lithium treatment can upregulate expression and secretion of various NTs, including BDNF, NGF, and GDNF, as well as their receptors [79,80,89]. In addition, lithium’s effects are linked with its inhibitory activity on several cell signaling-related key enzymes, including glycogen synthase kinase-3β (GSK3β), inositol monophosphatase, phosphoadenosylphosphate phosphatase, and Akt/beta-arrestin 2 [93]. Nevertheless, accumulating evidence also attributes lithium’s properties to many other effects on neurotransmitter release/signaling, oxidative stress, apoptosis, hormonal modulation, upregulation, and secretion of neurotrophic factor, enhancement of neurogenesis and differentiation, learning and memory improvement [80,94,95,96,97,98].
These effects confirm the high potential of lithium to prevent neuronal degeneration [94] and to facilitate nerve regeneration, also enhancing the speed and extent of axons remyelination [90,94]. Conversely, the benefits of lithium treatment for bipolar disorders do not appear to be related to both NT-3 and NT-4/5 expression [88].
Moreover, lithium’s ability in reducing neuronal damage after acute spinal cord injury (SCI) by inducing autophagy is of great importance [120,121,122]. Lithium competition with magnesium (Mg2+) can depress the phosphoinositide cycle and leas to a net reduction in intracellular 1,4,5 inositol triphosphate (IP3), a known autophagy suppressor [123,124]. The overall mechanisms triggered by lithium make this metal a powerful and safe tool against many neuronal pathologies, where neuronal regeneration appears to be more challenging. Much evidence demonstrated that this avenue is open and waiting for more extensive applications for humans [125,126]. As a consequence of genetic background or environmental factors, deviation of metal homeostasis from the normal state may occur, ultimately resulting in functional changes in NT activities that can lead to an increased risk for disease onset and poor prognosis. Hence, interventions on the quality and quantity of metal–NT functional interplay can be prominent tools in the treatments of various neurological diseases that require neuroregenerative support.
7. Concluding Remarks and Future Perspectives
BDNF, GNDF, and NGF have long been considered to be the major regulators of motoneuron physiology and recovery from various insults. NTs play key roles in regenerative medicine because they are required not only for the restoration of trophic circuitry but also for triggering differentiation and axon targeting. However, NT activities are complex processes, and regulatory mechanisms of their expression, secretion, and binding to receptors need further understanding, also considering the more recent knowledge on metal participation in neurorecovery.
The progress in mechanistic investigations in the field of neurotrophin-mediated neuroprotection and neuroregeneration, aiming at a complete understanding of integrated pathways, offers possibilities for the development of novel neuroregenerative therapeutic approaches. This is an opportunity that warrants further attention.
Author Contributions
Conceptualization, V.G.N. and A.N.; software, K.P.; validation, D.C. and G.P.; formal analysis, D.C. and G.P.; writing—original draft preparation, V.G.N. and A.N.; writing—review and editing, V.G.N., A.N. and K.P.; supervision, A.N. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Research program PIACERI (UNICT 2020-22) line #2.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rasmussen, R.; Carlsen, E.M. Spontaneous functional recovery from incomplete spinal cord injury. J. Neurosci. 2016, 36, 8535–8537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shadrach, J.L.; Stansberry, W.M.; Milen, A.M.; Ives, R.E.; Fogarty, E.A.; Antonellis, A.; Pierchala, B.A. Translatomic analysis of regenerating and degenerating spinal motor neurons in injury and ALS. iScience 2021, 24, 102700. [Google Scholar] [CrossRef] [PubMed]
- Koliatsos, V.E.; Price, W.L.; Pardo, C.A.; Price, D.L. Ventral root avulsion: An experimental model of death of adult motor neurons. J. Comp. Neurol. 1994, 342, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Nógrádi, A.; Vrbová, G. Improved motor function of denervated rat hindlimb muscles induced by embryonic spinal cord grafts. Eur. J. Neurosci. 1996, 8, 2198–2203. [Google Scholar] [CrossRef]
- Pajer, K.; Nemes, C.; Berzsenyi, S.; Kovács, K.A.; Pirity, M.K.; Pajenda, G.; Nógrádi, A.; Dinnyés, A. Grafted murine induced pluripotent stem cells prevent death of injured rat motoneurons otherwise destined to die. Exp. Neurol. 2015, 269, 188–201. [Google Scholar] [CrossRef] [Green Version]
- Nissl, F. Über die sogenannten Granula der Nervenzellen. Neurol. Centrbl. 1894, 29, 127–128. [Google Scholar]
- Lenhossek, M.V. Ueber den bau der spinalganglienzellen des menschen. Arch. Psychiat. Nervenkrankh. 1897, 29, 345–380. [Google Scholar] [CrossRef]
- Mills, C.D.; Fullwood, S.D.; Hulsebosch, C.E. Changes in metabotropic glutamate receptor expression following spinal cord injury. Exp. Neurol. 2001, 170, 244–257. [Google Scholar] [CrossRef]
- Beattie, M.S.; Ferguson, A.R.; Bresnahan, J.C. AMPA-receptor trafficking and injury-induced cell death. Eur. J. Neurosci. 2010, 32, 290–297. [Google Scholar] [CrossRef] [Green Version]
- Gloviczki, B.; Török, D.G.; Márton, G.; Gál, L.; Bodzay, T.; Pintér, S.; Nógrádi, A. Delayed spinal cord-brachial plexus reconnection after C7 ventral root avulsion: The effect of reinnervating motoneurons rescued by riluzole treatment. J. Neurotrauma 2017, 34, 2364–2374. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Chai, H.; Zhang, J.; Gu, H.; Xie, Y.; Zhou, L. Delayed implantation of a peripheral nerve graft reduces motoneuron survival but does not affect regeneration following spinal root avulsion in adult rats. J. Neurotrauma 2004, 21, 1050–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyd, J.G.; Gordon, T. Glial cell line-derived neurotrophic factor and brain-derived neurotrophic factor sustain the axonal regeneration of chronically axotomized motoneurons in vivo. Exp. Neurol. 2003, 183, 610–619. [Google Scholar] [CrossRef]
- Boyd, J.G.; Gordon, T. The neurotrophin receptors, trkB and p75, differentially regulate motor axonal regeneration. J. Neurobiol. 2001, 49, 314–325. [Google Scholar] [CrossRef]
- Eggers, R.; Tannemaat, M.R.; Ehlert, E.M.; Verhaagen, J. A spatio-temporal analysis of motoneuron survival, axonal regeneration and neurotrophic factor expression after lumbar ventral root avulsion and implantation. Exp. Neurol. 2010, 223, 207–220. [Google Scholar] [CrossRef]
- Pajenda, G.; Hercher, D.; Márton, G.; Pajer, K.; Feichtinger, G.A.; Maléth, J.; Redl, H.; Nógrádi, A. Spatiotemporally limited BDNF and GDNF overexpression rescues motoneurons destined to die and induces elongative axon growth. Exp. Neurol. 2014, 261, 367–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggers, R.; de Winter, F.; Arkenaar, C.; Tannemaat, M.R.; Verhaagen, J. Enhanced regeneration and reinnervation following timed GDNF gene therapy in a cervical ventral root avulsion. Exp. Neurol 2019, 321, 113037. [Google Scholar] [CrossRef] [PubMed]
- Santos, D.; González-Pérez, F.; Giudetti, G.; Micera, S.; Udina, E.; Del Valle, J.; Navarro, X. Preferential enhancement of sensory and motor axon regeneration by combining extracellular matrix components with neurotrophic factors. Int. J. Mol. Sci. 2016, 18, 65. [Google Scholar] [CrossRef] [Green Version]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in neuronal development and function. Annu. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef] [Green Version]
- Barde, Y.A.; Edgar, D.; Thoenen, H. Purification of a new neurotrophic factor from mammalian brain. EMBO J. 1982, 1, 549–553. [Google Scholar] [CrossRef]
- Acheson, A.; Conover, J.C.; Fandl, J.P.; DeChiara, T.M.; Russell, M.; Thadani, A.; Squinto, S.P.; Yancopoulos, G.D.; Lindsay, R.M. A BDNF autocrine loop in adult sensory neurons prevents cell death. Nature 1995, 374, 450–453. [Google Scholar] [CrossRef]
- Yamada, K.; Nabeshima, T. Brain-derived neurotrophic factor/TrkB signaling in memory processes. J. Pharmacol. Sci. 2003, 91, 267–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandel, A.L.; Ozdener, H.; Utermohlen, V. Identification of pro- and mature brain-derived neurotrophic factor in human saliva. Arch. Oral. Biol. 2009, 54, 689–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delezie, J.; Handschin, C. Endocrine Crosstalk between skeletal muscle and the brain. Front. Neurol. 2018, 9, 698. [Google Scholar] [CrossRef] [PubMed]
- Orefice, L.L.; Waterhouse, E.G.; Partridge, J.G.; Lalchandani, R.R.; Vicini, S.; Xu, B. Distinct roles for somatically and dendritically synthesized brain-derived neurotrophic factor in morphogenesis of dendritic spines. J. Neurosci. 2013, 33, 11618–11632. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Harte-Hargrove, L.C.; Siao, C.J.; Marinic, T.; Clarke, R.; Ma, Q.; Jing, D.; Lafrancois, J.J.; Bath, K.G.; Mark, W.; et al. proBDNF negatively regulates neuronal remodeling, synaptic transmission, and synaptic plasticity in hippocampus. Cell Rep. 2014, 7, 796–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dechant, G.; Barde, Y.A. The neurotrophin receptor p75(NTR): Novel functions and implications for diseases of the nervous system. Nat. Neurosci. 2002, 5, 1131–1136. [Google Scholar] [CrossRef]
- Hermida, M.A.; Dinesh Kumar, J.; Leslie, N.R. GSK3 and its interactions with the PI3K/AKT/mTOR signalling network. Adv. Biol. Regul. 2017, 65, 5–15. [Google Scholar] [CrossRef]
- Rantamäki, T. TrkB neurotrophin receptor at the core of antidepressant effects, but how? Cell Tissue Res. 2019, 377, 115–124. [Google Scholar] [CrossRef] [Green Version]
- Engele, J.; Schubert, D.; Bohn, M.C. Conditioned media derived from glial cell lines promote survival and differentiation of dopaminergic neurons in vitro: Role of mesencephalic glia. J. Neurosci. Res. 1991, 30, 359–371. [Google Scholar] [CrossRef]
- Bresjanac, M.; Antauer, G. Reactive astrocytes of the quinolinic acid-lesioned rat striatum express GFRalpha1 as well as GDNF in vivo. Exp. Neurol. 2000, 164, 53–59. [Google Scholar] [CrossRef]
- Lee, J.; Hyeon, S.J.; Im, H.; Ryu, H.; Kim, Y. Astrocytes and microglia as non-cell autonomous players in the pathogenesis of ALS. Exp. Neurobiol. 2016, 25, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Wang, X. Structural studies of GDNF family ligands with their receptors-Insights into ligand recognition and activation of receptor tyrosine kinase RET. Biochim. Biophys. Acta 2013, 1834, 2205–2212. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Lindahl, M.; Hyvönen, M.E.; Parvinen, M.; de Rooij, D.G.; Hess, M.W.; Raatikainen-Ahokas, A.; Sainio, K.; Rauvala, H.; Lakso, M.; et al. Regulation of cell fate decision of undifferentiated spermatogonia by GDNF. Science 2000, 287, 1489–1493. [Google Scholar] [CrossRef] [PubMed]
- Schuchardt, A.; D’Agati, V.; Larsson-Blomberg, L.; Costantini, F.; Pachnis, V. Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature 1994, 367, 380–383. [Google Scholar] [CrossRef]
- Durbec, P.L.; Larsson-Blomberg, L.B.; Schuchardt, A.; Costantini, F.; Pachnis, V. Common origin and developmental dependence on c-ret of subsets of enteric and sympathetic neuroblasts. Development 1996, 122, 349–358. [Google Scholar] [CrossRef]
- Naoi, M.; Inaba-Hasegawa, K.; Shamoto-Nagai, M.; Maruyama, W. Neurotrophic function of phytochemicals for neuroprotection in aging and neurodegenerative disorders: Modulation of intracellular signaling and gene expression. J. Neural. Transm. 2017, 124, 1515–1527. [Google Scholar] [CrossRef]
- Rita, L.-M.; Viktor, H. Selective growth stimulating effects of mouse sarcoma on the sensory and sympathetic nervous system of the chick embryo. J. Exp. Zool. 1951, 116, 321–361. [Google Scholar] [CrossRef]
- Marlin, M.C.; Li, G. Biogenesis and function of the NGF/TrkA signaling endosome. Int. Rev. Cell Mol. Biol. 2015, 314, 239–257. [Google Scholar] [CrossRef] [Green Version]
- Roux, P.P.; Barker, P.A. Neurotrophin signaling through the p75 neurotrophin receptor. Prog. Neurobiol. 2002, 67, 203–233. [Google Scholar] [CrossRef]
- Benítez-Temiño, B.; Davis-López de Carrizosa, M.A.; Morcuende, S.; Matarredona, E.R.; de la Cruz, R.R.; Pastor, A.M. Functional diversity of neurotrophin actions on the oculomotor system. Int. J. Mol. Sci. 2016, 17, 2016. [Google Scholar] [CrossRef] [Green Version]
- Eggers, R.; de Winter, F.; Hoyng, S.A.; Roet, K.C.; Ehlert, E.M.; Malessy, M.J.; Verhaagen, J.; Tannemaat, M.R. Lentiviral vector-mediated gradients of GDNF in the injured peripheral nerve: Effects on nerve coil formation, Schwann cell maturation and myelination. PLoS ONE 2013, 8, e71076. [Google Scholar] [CrossRef] [PubMed]
- Eggers, R.; Hendriks, W.T.; Tannemaat, M.R.; van Heerikhuize, J.J.; Pool, C.W.; Carlstedt, T.P.; Zaldumbide, A.; Hoeben, R.C.; Boer, G.J.; Verhaagen, J. Neuroregenerative effects of lentiviral vector-mediated GDNF expression in reimplanted ventral roots. Mol. Cell Neurosci. 2008, 39, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Novikov, L.N.; Novikova, L.N.; Holmberg, P.; Kellerth, J. Exogenous brain-derived neurotrophic factor regulates the synaptic composition of axonally lesioned and normal adult rat motoneurons. Neuroscience 2000, 100, 171–181. [Google Scholar] [CrossRef]
- Weishaupt, N.; Blesch, A.; Fouad, K. BDNF: The career of a multifaceted neurotrophin in spinal cord injury. Exp. Neurol. 2012, 238, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Jones, L.L.; Tuszynski, M.H. BDNF-expressing marrow stromal cells support extensive axonal growth at sites of spinal cord injury. Exp. Neurol. 2005, 191, 344–360. [Google Scholar] [CrossRef]
- Eggers, R.; de Winter, F.; Hoyng, S.A.; Hoeben, R.C.; Malessy, M.J.A.; Tannemaat, M.R.; Verhaagen, J. Timed GDNF gene therapy using an immune-evasive gene switch promotes long distance axon regeneration. Brain 2019, 142, 295–311. [Google Scholar] [CrossRef]
- Kemp, S.W.; Webb, A.A.; Dhaliwal, S.; Syed, S.; Walsh, S.K.; Midha, R. Dose and duration of nerve growth factor (NGF) administration determine the extent of behavioral recovery following peripheral nerve injury in the rat. Exp. Neurol. 2011, 229, 460–470. [Google Scholar] [CrossRef]
- Labroo, P.; Shea, J.; Edwards, K.; Ho, S.; Davis, B.; Sant, H.; Goodwin, I.; Gale, B.; Agarwal, J. Novel drug delivering conduit for peripheral nerve regeneration. J. Neural. Eng. 2017, 14, 066011. [Google Scholar] [CrossRef]
- Li, R.; Wu, J.; Lin, Z.; Nangle, M.R.; Li, Y.; Cai, P.; Liu, D.; Ye, L.; Xiao, Z.; He, C.; et al. Single injection of a novel nerve growth factor coacervate improves structural and functional regeneration after sciatic nerve injury in adult rats. Exp. Neurol. 2017, 288, 1–10. [Google Scholar] [CrossRef]
- Yao, Y.; Cui, Y.; Zhao, Y.; Xiao, Z.; Li, X.; Han, S.; Chen, B.; Fang, Y.; Wang, P.; Pan, J.; et al. Efect of longitudinally oriented collagen conduit combined with nerve growth factor on nerve regeneration after dog sciatic nerve injury. J. Biomed. Mater. Res. B Appl. Biomater. 2018, 106, 2131–2139. [Google Scholar] [CrossRef]
- Ma, S.; Peng, C.; Wu, S.; Wu, D.; Gao, C. Sciatic nerve regeneration using a nerve growth factor-containing fibrin glue membrane. Neural. Regen. Res. 2013, 8, 3416–3422. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Li, Y.; Wu, Y.; Zhao, Y.; Chen, H.; Yuan, Y.; Xu, K.; Zhang, H.; Lu, Y.; Wang, J.; et al. Heparin-poloxamer thermosensitive hydrogel loaded with bFGF and NGF enhances peripheral nerve regeneration in diabetic rats. Biomaterials 2018, 168, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.R.; Chang, W.; Silva-Correia, J.; Reis, R.L.; Oliveira, J.M.; Kohn, J. Engineering silk fibroin-based nerve conduit with neurotrophic factors for proximal protection after peripheral nerve injury. Adv. Healthc. Mater. 2021, 10, e2000753. [Google Scholar] [CrossRef] [PubMed]
- Jiao, J.; Feng, W.; Jie-Jie, H.; Jin-Jian, H.; Zong-An, L.; Yan, K.; Zhi-Jun, Z. Microfluidic hollow fiber with improved stiffness repairs peripheral nerve injury through non-invasive electromagnetic induction and controlled release of NGF. Chem. Eng. J. 2021, 426, 131826. [Google Scholar] [CrossRef]
- Travaglia, A.; Pietropaolo, A.; La Mendola, D.; Nicoletti, V.G.; Rizzarelli, E. The inorganic perspectives of neurotrophins and Alzheimer’s disease. J. Inorg. Biochem. 2012, 111, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Tamano, H.; Takeda, A. Dynamic action of neurometals at the synapse. Metallomics 2011, 3, 656–661. [Google Scholar] [CrossRef]
- Finney, L.A.; O’Halloran, T.V. Transition metal speciation in the cell: Insights from the chemistry of metal ion receptors. Science 2003, 300, 931–936. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Jiao, Q.; Xu, H.; Du, X.; Shi, L.; Jia, F.; Jiang, H. Biometal Dyshomeostasis and Toxic Metal Accumulations in the Development of Alzheimer’s Disease. Front Mol. Neurosci. 2017, 10, 339. [Google Scholar] [CrossRef]
- Toni, M.; Massimino, M.L.; De Mario, A.; Angiulli, E.; Spisni, E. Metal dyshomeostasis and their pathological role in prion and prion-like diseases: The basis for a nutritional approach. Front. Neurosci. 2017, 11, 3. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Xiao, G.; Liu, L.; Lang, M. Zinc transporters in Alzheimer’s disease. Mol. Brain 2019, 12, 106. [Google Scholar] [CrossRef] [Green Version]
- Travaglia, A.; Arena, G.; Fattorusso, R.; Isernia, C.; La Mendola, D.; Malgieri, G.; Nicoletti, V.G.; Rizzarelli, E. The inorganic perspective of nerve growth factor: Interactions of Cu2+ and Zn2+ with the N-terminus fragment of nerve growth factor encompassing the recognition domain of the TrkA receptor. Chemistry 2011, 17, 3726–3738. [Google Scholar] [CrossRef] [PubMed]
- Travaglia, A.; La Mendola, D.; Magrì, A.; Nicoletti, V.G.; Pietropaolo, A.; Rizzarelli, E. Copper, BDNF and Its N-terminal domain: Inorganic features and biological perspectives. Chemistry 2012, 18, 15618–15631. [Google Scholar] [CrossRef] [PubMed]
- Post, J.I.; Eibl, J.K.; Ross, G.M. Zinc induces motor neuron death via a selective inhibition of brain-derived neurotrophic factor activity. Amyotroph. Lateral Scler. 2008, 9, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Travaglia, A.; La Mendola, D.; Magrì, A.; Pietropaolo, A.; Nicoletti, V.G.; Grasso, G.; Malgieri, G.; Fattorusso, R.; Isernia, C.; Rizzarelli, E. Zinc(II) interactions with brain-derived neurotrophic factor N-terminal peptide fragments: Inorganic features and biological perspectives. Inorg. Chem. 2013, 52, 11075–11083. [Google Scholar] [CrossRef]
- Wang, W.; Post, J.I.; Dow, K.E.; Shin, S.H.; Riopelle, R.J.; Ross, G.M. Zinc and copper inhibit nerve growth factor-mediated protection from oxidative stress-induced apoptosis. Neurosci. Lett. 1999, 259, 115–118. [Google Scholar] [CrossRef]
- Zhao, G.H.; Yu, P.; Hu, X.S.; Zhao, L. Effect of Zn(II) on the structure and biological activity of natural beta-NGF. Acta Biochim. Biophys. Sin. 2004, 36, 99–104. [Google Scholar] [CrossRef]
- Allington, C.; Shamovsky, I.L.; Ross, G.M.; Riopelle, R.J. Zinc inhibits p75NTR-mediated apoptosis in chick neural retina. Cell Death Differ. 2001, 8, 451–456. [Google Scholar] [CrossRef] [Green Version]
- Ross, G.M.; Shamovsky, I.L.; Woo, S.B.; Post, J.I.; Vrkljan, P.N.; Lawrance, G.; Solc, M.; Dostaler, S.M.; Neet, K.E.; Riopelle, R.J. The binding of zinc and copper ions to nerve growth factor is differentially affected by pH: Implications for cerebral acidosis. J. Neurochem. 2001, 78, 515–523. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.Z.; Pan, E.; Xiong, Z.Q.; McNamara, J.O. Zinc-mediated transactivation of TrkB potentiates the hippocampal mossy fiber-CA3 pyramid synapse. Neuron 2008, 57, 546–558. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.Z.; McNamara, J.O. Neuroprotective effects of reactive oxygen species mediated by BDNF-independent activation of TrkB. J. Neurosci. 2012, 32, 15521–15532. [Google Scholar] [CrossRef]
- Huang, Y.Z.; McNamara, J.O. Mutual regulation of Src family kinases and the neurotrophin receptor TrkB. J. Biol. Chem. 2010, 285, 8207–8217. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Graves, L.M.; Gill, G.N.; Parsons, S.J.; Samet, J.M. Src-dependent phosphorylation of the epidermal growth factor receptor on tyrosine 845 is required for zinc-induced Ras activation. J. Biol. Chem. 2002, 277, 24252–24257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamano, H.; Koike, Y.; Nakada, H.; Shakushi, Y.; Takeda, A. Significance of synaptic Zn. J. Trace Elem. Med. Biol. 2016, 38, 93–98. [Google Scholar] [CrossRef] [PubMed]
- McAllister, B.B.; Dyck, R.H. Zinc transporter 3 (ZnT3) and vesicular zinc in central nervous system function. Neurosci. Biobehav. Rev. 2017, 80, 329–350. [Google Scholar] [CrossRef] [PubMed]
- Tóth, K. Zinc in neurotransmission. Annu. Rev. Nutr. 2011, 31, 139–153. [Google Scholar] [CrossRef]
- Hwang, J.J.; Park, M.H.; Koh, J.Y. Copper activates TrkB in cortical neurons in a metalloproteinase-dependent manner. J. Neurosci. Res. 2007, 85, 2160–2166. [Google Scholar] [CrossRef]
- Kornblatt, A.P.; Nicoletti, V.G.; Travaglia, A. The neglected role of copper ions in wound healing. J. Inorg. Biochem. 2016, 161, 1–8. [Google Scholar] [CrossRef]
- Leung, K.W.; Liu, M.; Xu, X.; Seiler, M.J.; Barnstable, C.J.; Tombran-Tink, J. Expression of ZnT and ZIP zinc transporters in the human RPE and their regulation by neurotrophic factors. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1221–1231. [Google Scholar] [CrossRef]
- Simões, L.R.; Abreu, R.R.E.S.; Generoso, J.S.; Goularte, J.A.; Collodel, A.; Giridharan, V.V.; Arumanayagam, A.C.S.; Valvassori, S.S.; Quevedo, J.; Barichello, T. Prevention of memory impairment and neurotrophic factors increased by lithium in wistar rats submitted to pneumococcal meningitis model. Mediat. Inflamm. 2017, 2017, 6490652. [Google Scholar] [CrossRef] [Green Version]
- Dal-Pont, G.C.; Jório, M.T.S.; Resende, W.R.; Gava, F.F.; Aguiar-Geraldo, J.M.; Possamai-Della, T.; Peper-Nascimento, J.; Quevedo, J.; Valvassori, S.S. Effects of lithium and valproate on behavioral parameters and neurotrophic factor levels in an animal model of mania induced by paradoxical sleep deprivation. J. Psychiatr. Res. 2019, 119, 76–83. [Google Scholar] [CrossRef]
- Squitti, R.; Ventriglia, M.; Simonelli, I.; Bonvicini, C.; Costa, A.; Perini, G.; Binetti, G.; Benussi, L.; Ghidoni, R.; Koch, G.; et al. Copper imbalance in Alzheimer’s disease: Meta-analysis of serum, plasma, and brain specimens, and replication study evaluating ATP7B gene variants. Biomolecules 2021, 11, 960. [Google Scholar] [CrossRef] [PubMed]
- Adlard, P.A.; Bush, A.I. Metals and Alzheimer’s disease: How far have we come in the clinic? J. Alzheimers Dis. 2018, 62, 1369–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirangelo, I.; Iannuzzi, C. The role of metal binding in the amyotrophic lateral sclerosis-related aggregation of copper-zinc superoxide dismutase. Molecules 2017, 22, 1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, L.; Kaler, S. ATP7A trafficking and mechanisms underlying the distal motor neuropathy induced by mutations in ATP7A. Ann. N. Y. Acad. Sci. 2014, 1314, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Figueroa-Romero, C.; Mikhail, K.A.; Gennings, C.; Curtin, P.; Bello, G.A.; Botero, T.M.; Goutman, S.A.; Feldman, E.L.; Arora, M.; Austin, C. Early life metal dysregulation in amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2020, 7, 872–882. [Google Scholar] [CrossRef]
- Rakofsky, J.J.; Lucido, M.J.; Dunlop, B.W. Lithium in the treatment of acute bipolar depression: A systematic review and meta-analysis. J Affect Disord 2022, 308, 268–280. [Google Scholar] [CrossRef]
- Squitti, R.; Pal, A.; Picozza, M.; Avan, A.; Ventriglia, M.; Rongioletti, M.C.; Hoogenraad, T. Zinc Therapy in early Alzheimer’s disease: Safety and potential therapeutic efficacy. Biomolecules 2020, 10, 1164. [Google Scholar] [CrossRef]
- Loch, A.A.; Zanetti, M.V.; de Sousa, R.T.; Chaim, T.M.; Serpa, M.H.; Gattaz, W.F.; Teixeira, A.L.; Machado-Vieira, R. Elevated neurotrophin-3 and neurotrophin 4/5 levels in unmedicated bipolar depression and the effects of lithium. Prog. Neuropsychopharmacol. Biol. Psychiatry 2015, 56, 243–246. [Google Scholar] [CrossRef]
- Chiou, Y.J.; Huang, T.L. Brain-derived neurotrophic factor (BDNF) and bipolar disorder. Psychiatry Res. 2019, 274, 395–399. [Google Scholar] [CrossRef]
- Fang, X.Y.; Zhang, W.M.; Zhang, C.F.; Wong, W.M.; Li, W.; Wu, W.; Lin, J.H. Lithium accelerates functional motor recovery by improving remyelination of regenerating axons following ventral root avulsion and reimplantation. Neuroscience 2016, 329, 213–225. [Google Scholar] [CrossRef]
- Hwang, I.Y.; Sun, E.S.; An, J.H.; Im, H.; Lee, S.H.; Lee, J.Y.; Han, P.L.; Koh, J.Y.; Kim, Y.H. Zinc-triggered induction of tissue plasminogen activator by brain-derived neurotrophic factor and metalloproteinases. J. Neurochem. 2011, 118, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Poddar, R.; Rajagopal, S.; Shuttleworth, C.W.; Paul, S. Zn2+-dependent activation of the Trk signaling pathway induces phosphorylation of the brain-enriched tyrosine phosphatase step: Molecular basis for ZN2+-induced ERK mapk activation. J. Biol. Chem. 2016, 291, 813–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajmone-Cat, M.A.; D’Urso, M.C.; di Blasio, G.; Brignone, M.S.; De Simone, R.; Minghetti, L. Glycogen synthase kinase 3 is part of the molecular machinery regulating the adaptive response to LPS stimulation in microglial cells. Brain Behav. Immun. 2016, 55, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Won, E.; Kim, Y.K. An Oldie but Goodie: Lithium in the treatment of bipolar disorder through neuroprotective and neurotrophic mechanisms. Int. J. Mol. Sci. 2017, 18, 2679. [Google Scholar] [CrossRef] [Green Version]
- Su, H.; Chu, T.H.; Wu, W. Lithium enhances proliferation and neuronal differentiation of neural progenitor cells in vitro and after transplantation into the adult rat spinal cord. Exp. Neurol. 2007, 206, 296–307. [Google Scholar] [CrossRef]
- Yasuda, S.; Liang, M.H.; Marinova, Z.; Yahyavi, A.; Chuang, D.M. The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol. Psychiatry 2009, 14, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Rybakowski, J.K.; Suwalska, A.; Hajek, T. Clinical perspectives of lithium’s neuroprotective effect. Pharmacopsychiatry 2018, 51, 194–199. [Google Scholar] [CrossRef]
- Qi, L.; Tang, Y.; He, W.; Pan, H.; Jiang, W.; Wang, L.; Deng, W. Lithium chloride promotes neuronal differentiation of rat neural stem cells and enhances neural regeneration in Parkinson’s disease model. Cytotechnology 2017, 69, 277–287. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, M.; Garate, A.; Delgado, D.; Padilla, S. Platelet-rich plasma, an adjuvant biological therapy to assist peripheral nerve repair. Neural. Regen. Res. 2017, 12, 47–52. [Google Scholar] [CrossRef]
- Pandini, G.; Satriano, C.; Pietropaolo, A.; Gianì, F.; Travaglia, A.; La Mendola, D.; Nicoletti, V.G.; Rizzarelli, E. The inorganic side of NGF: Copper(II) and Zinc(II) affect the NGF mimicking signaling of the N-terminus peptides encompassing the recognition domain of TrkA receptor. Front. Neurosci. 2016, 10, 569. [Google Scholar] [CrossRef] [Green Version]
- Behzadfar, L.; Abdollahi, M.; Sabzevari, O.; Hosseini, R.; Salimi, A.; Naserzadeh, P.; Sharifzadeh, M.; Pourahmad, J. Potentiating role of copper on spatial memory deficit induced by beta amyloid and evaluation of mitochondrial function markers in the hippocampus of rats. Metallomics 2017, 9, 969–980. [Google Scholar] [CrossRef]
- Sandusky-Beltran, L.A.; Manchester, B.L.; McNay, E.C. Supplementation with zinc in rats enhances memory and reverses an age-dependent increase in plasma copper. Behav. Brain Res. 2017, 333, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Corona, C.; Masciopinto, F.; Silvestri, E.; Viscovo, A.D.; Lattanzio, R.; Sorda, R.L.; Ciavardelli, D.; Goglia, F.; Piantelli, M.; Canzoniero, L.M.; et al. Dietary zinc supplementation of 3xTg-AD mice increases BDNF levels and prevents cognitive deficits as well as mitochondrial dysfunction. Cell Death Dis. 2010, 1, e91. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.J.; Park, M.H.; Choi, S.Y.; Koh, J.Y. Activation of the Trk signaling pathway by extracellular zinc. Role of metalloproteinases. J. Biol. Chem. 2005, 280, 11995–12001. [Google Scholar] [CrossRef] [Green Version]
- Sarris, J.; Ravindran, A.; Yatham, L.N.; Marx, W.; Rucklidge, J.J.; McIntyre, R.S.; Akhondzadeh, S.; Benedetti, F.; Caneo, C.; Cramer, H.; et al. Clinician guidelines for the treatment of psychiatric disorders with nutraceuticals and phytoceuticals: The World Federation of Societies of Biological Psychiatry (WFSBP) and Canadian Network for Mood and Anxiety Treatments (CANMAT) Taskforce. World J. Biol. Psychiatry 2022, 1–32. [Google Scholar] [CrossRef]
- Liu, J.; Jiang, Y.G.; Huang, C.Y.; Fang, H.Y.; Fang, H.T.; Pang, W. Depletion of intracellular zinc down-regulates expression of Uch-L1 mRNA and protein, and CREB mRNA in cultured hippocampal neurons. Nutr. Neurosci. 2008, 11, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Naletova, I.; Satriano, C.; Pietropaolo, A.; Gianì, F.; Pandini, G.; Triaca, V.; Amadoro, G.; Latina, V.; Calissano, P.; Travaglia, A.; et al. The copper(II)-assisted connection between NGF and BDNF by means of nerve growth factor-mimicking short peptides. Cells 2019, 8, 301. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.; Yang, H.; Jones, L.L.; Filbin, M.T.; Tuszynski, M.H. Combinatorial therapy with neurotrophins and cAMP promotes axonal regeneration beyond sites of spinal cord injury. J. Neurosci. 2004, 24, 6402–6409. [Google Scholar] [CrossRef] [Green Version]
- Teng, F.Y.; Tang, B.L. Axonal regeneration in adult CNS neurons—Signaling molecules and pathways. J. Neurochem. 2006, 96, 1501–1508. [Google Scholar] [CrossRef]
- Li, Y.; Andereggen, L.; Yuki, K.; Omura, K.; Yin, Y.; Gilbert, H.Y.; Erdogan, B.; Asdourian, M.S.; Shrock, C.; de Lima, S.; et al. Mobile zinc increases rapidly in the retina after optic nerve injury and regulates ganglion cell survival and optic nerve regeneration. Proc. Natl. Acad. Sci. USA 2017, 114, E209–E218. [Google Scholar] [CrossRef] [Green Version]
- Duncan, C.; Bica, L.; Crouch, P.J.; Caragounis, A.; Lidgerwood, G.E.; Parker, S.J.; Meyerowitz, J.; Volitakis, I.; Liddell, J.R.; Raghupathi, R.; et al. Copper modulates the large dense core vesicle secretory pathway in PC12 cells. Metallomics 2013, 5, 700–714. [Google Scholar] [CrossRef] [PubMed]
- Hatori, Y.; Lutsenko, S. The role of copper chaperone Atox1 in coupling redox homeostasis to intracellular copper distribution. Antioxidants 2016, 5, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birkaya, B.; Aletta, J.M. NGF promotes copper accumulation required for optimum neurite outgrowth and protein methylation. J. Neurobiol. 2005, 63, 49–61. [Google Scholar] [CrossRef]
- Kretzschmar, H.A.; Tings, T.; Madlung, A.; Giese, A.; Herms, J. Function of PrP(C) as a copper-binding protein at the synapse. Arch. Virol. Suppl. 2000, 16, 239–249. [Google Scholar] [CrossRef]
- Haigh, C.L.; Edwards, K.; Brown, D.R. Copper binding is the governing determinant of prion protein turnover. Mol. Cell Neurosci. 2005, 30, 186–196. [Google Scholar] [CrossRef]
- Steele, A.D.; Emsley, J.G.; Ozdinler, P.H.; Lindquist, S.; Macklis, J.D. Prion protein (PrPc) positively regulates neural precursor proliferation during developmental and adult mammaliwan neurogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 3416–3421. [Google Scholar] [CrossRef] [Green Version]
- Loubet, D.; Dakowski, C.; Pietri, M.; Pradines, E.; Bernard, S.; Callebert, J.; Ardila-Osorio, H.; Mouillet-Richard, S.; Launay, J.M.; Kellermann, O.; et al. Neuritogenesis: The prion protein controls β1 integrin signaling activity. FASEB J. 2012, 26, 678–690. [Google Scholar] [CrossRef]
- Williams, J.R.; Trias, E.; Beilby, P.R.; Lopez, N.I.; Labut, E.M.; Bradford, C.S.; Roberts, B.R.; McAllum, E.J.; Crouch, P.J.; Rhoads, T.W.; et al. Copper delivery to the CNS by CuATSM effectively treats motor neuron disease in SOD(G93A) mice co-expressing the Copper-Chaperone-for-SOD. Neurobiol. Dis. 2016, 89, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Sheykhansari, S.; Kozielski, K.; Bill, J.; Sitti, M.; Gemmati, D.; Zamboni, P.; Singh, A.V. Redox metals homeostasis in multiple sclerosis and amyotrophic lateral sclerosis: A review. Cell Death Dis. 2018, 9, 348. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, F.; Zhai, X.; Li, X.H.; He, X.J. Lithium promotes recovery of neurological function after spinal cord injury by inducing autophagy. Neural. Regen. Res. 2018, 13, 2191–2199. [Google Scholar] [CrossRef]
- Hou, L.; Xiong, N.; Liu, L.; Huang, J.; Han, C.; Zhang, G.; Li, J.; Xu, X.; Lin, Z.; Wang, T. Lithium protects dopaminergic cells from rotenone toxicity via autophagy enhancement. BMC Neurosci. 2015, 16, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, L.; Komatsu, D.E.; Gurevich, M.; Hurst, L.C. Emerging strategies on adjuvant therapies for nerve recovery. J. Hand. Surg. Am. 2018, 43, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Machado-Vieira, R. Lithium, stress, and resilience in bipolar disorder: Deciphering this key homeostatic synaptic plasticity regulator. J. Affect. Disord. 2018, 233, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Thellung, S.; Corsaro, A.; Nizzari, M.; Barbieri, F.; Florio, T. Autophagy activator drugs: A new opportunity in neuroprotection from misfolded protein toxicity. Int. J. Mol. Sci. 2019, 20, 901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leeds, P.R.; Yu, F.; Wang, Z.; Chiu, C.T.; Zhang, Y.; Leng, Y.; Linares, G.R.; Chuang, D.M. A new avenue for lithium: Intervention in traumatic brain injury. ACS Chem. Neurosci. 2014, 5, 422–433. [Google Scholar] [CrossRef]
- Ala, M.; Mohammad Jafari, R.; Nematian, H.; Ganjedanesh, M.R.; Naderi, A.; Akbariani, M.; Sanatkar, M.; Satarian, L.; Aghsaei Fard, M.; Dehpour, A.R. Neuroprotective effect of intravitreal single-dose lithium chloride after optic nerve injury in rats. Curr. Eye Res. 2021, 46, 558–567. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).