
Structure–Activity Relationship of the Dimeric and Oligomeric Forms of a Cytotoxic Biotherapeutic Based on Diphtheria Toxin



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Plasmid and Host Strain
2.3. Transgene Expression and Purification of the Recombinant Cytotoxin from Inclusion Bodies
2.4. Reducing and Non-Reducing SDS-Polyacrylamide Gel Electrophoresis (PAGE)
2.5. Molecular Dynamic Simulations
2.6. Batch Dynamic Light Scattering (DLS)
2.7. Size-Exclusion Chromatography Coupled to Multi-Angle Light Scattering and DLS (SEC-MALS-DLS)
2.8. Size-Exclusion Chromatography Coupled to Small-Angle X-ray Scattering (SEC-SAXS)
2.9. SAXS Structural Modeling
2.10. SAXS Data Processing
2.11. Disulfide Bonds Mapping and Structural Analysis
2.12. Titration Enzyme-Linked Immunosorbent Assay (ELISA)
2.13. In Vitro Functional Assay
3. Results and Discussion
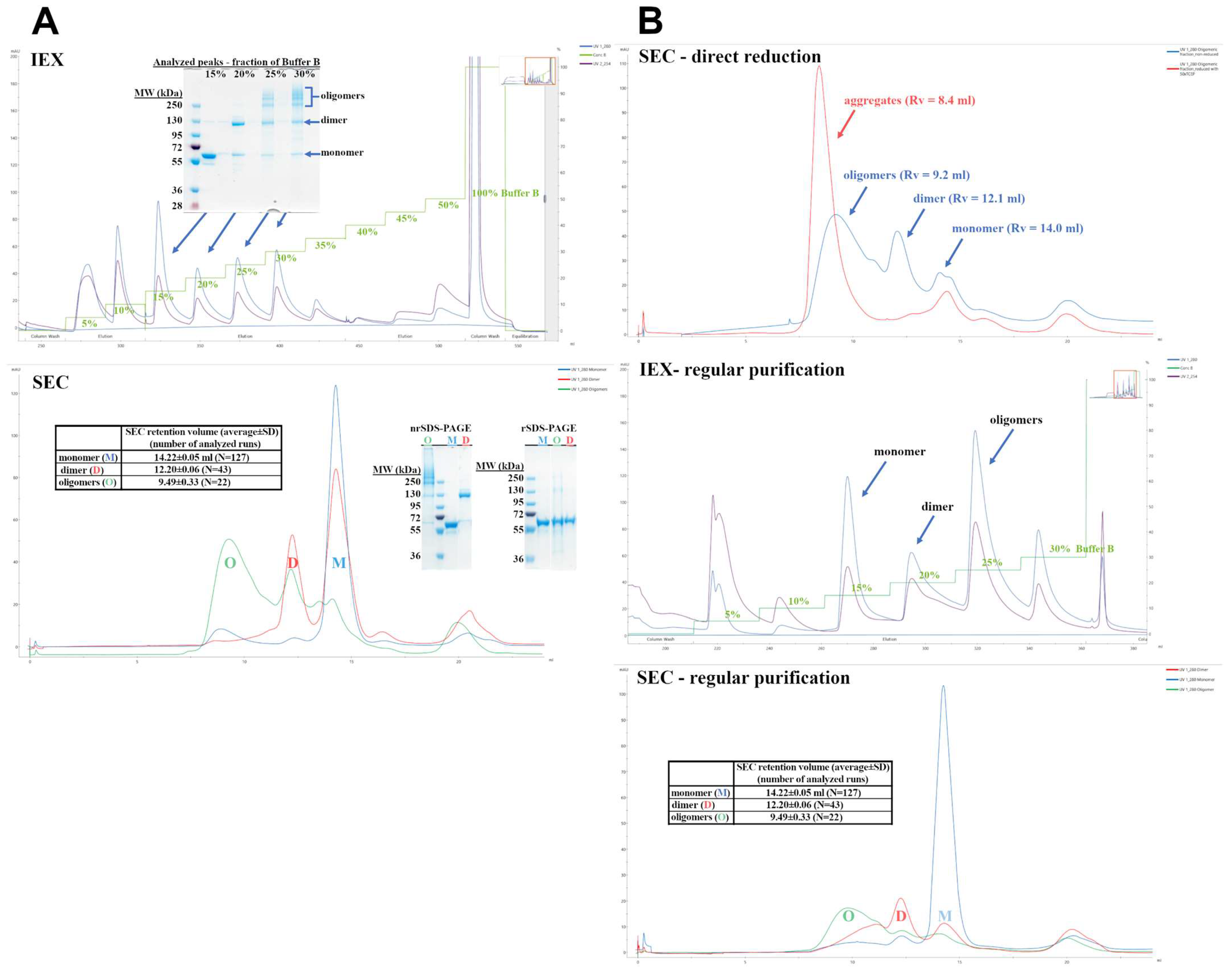
3.1. Preparation and Purification of the Cytotoxin
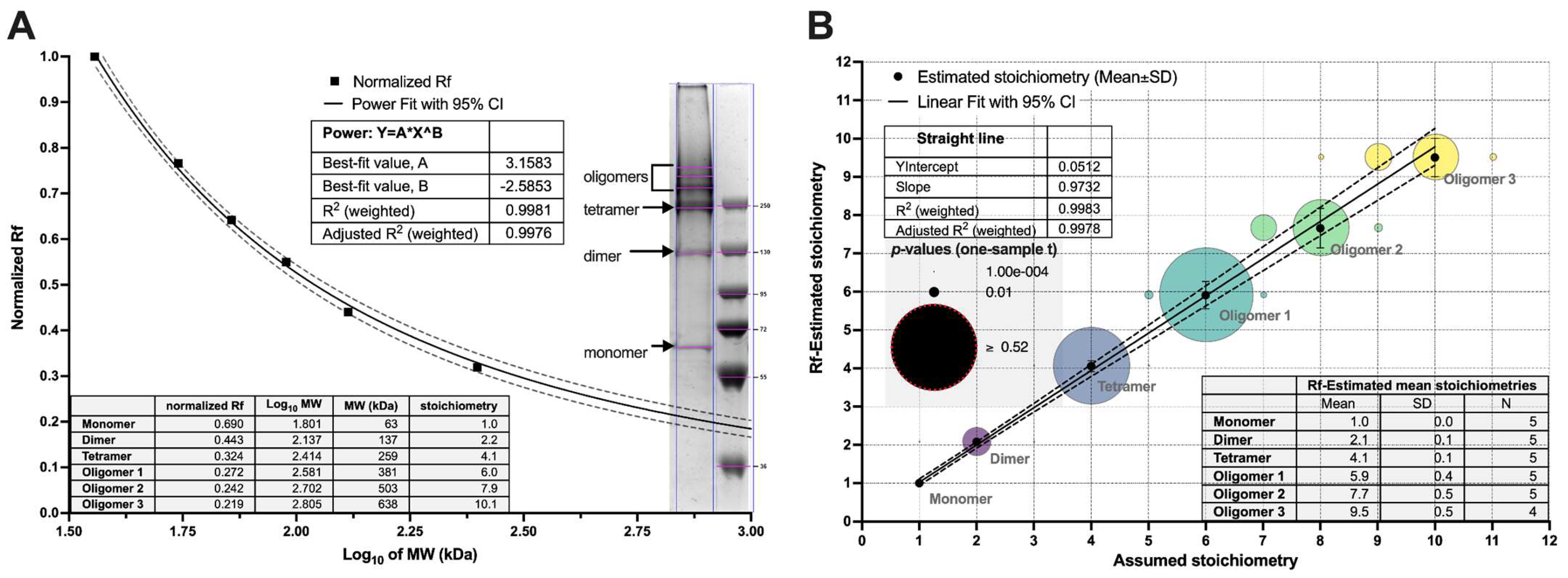
3.2. Stoichiometry of the Oligomers
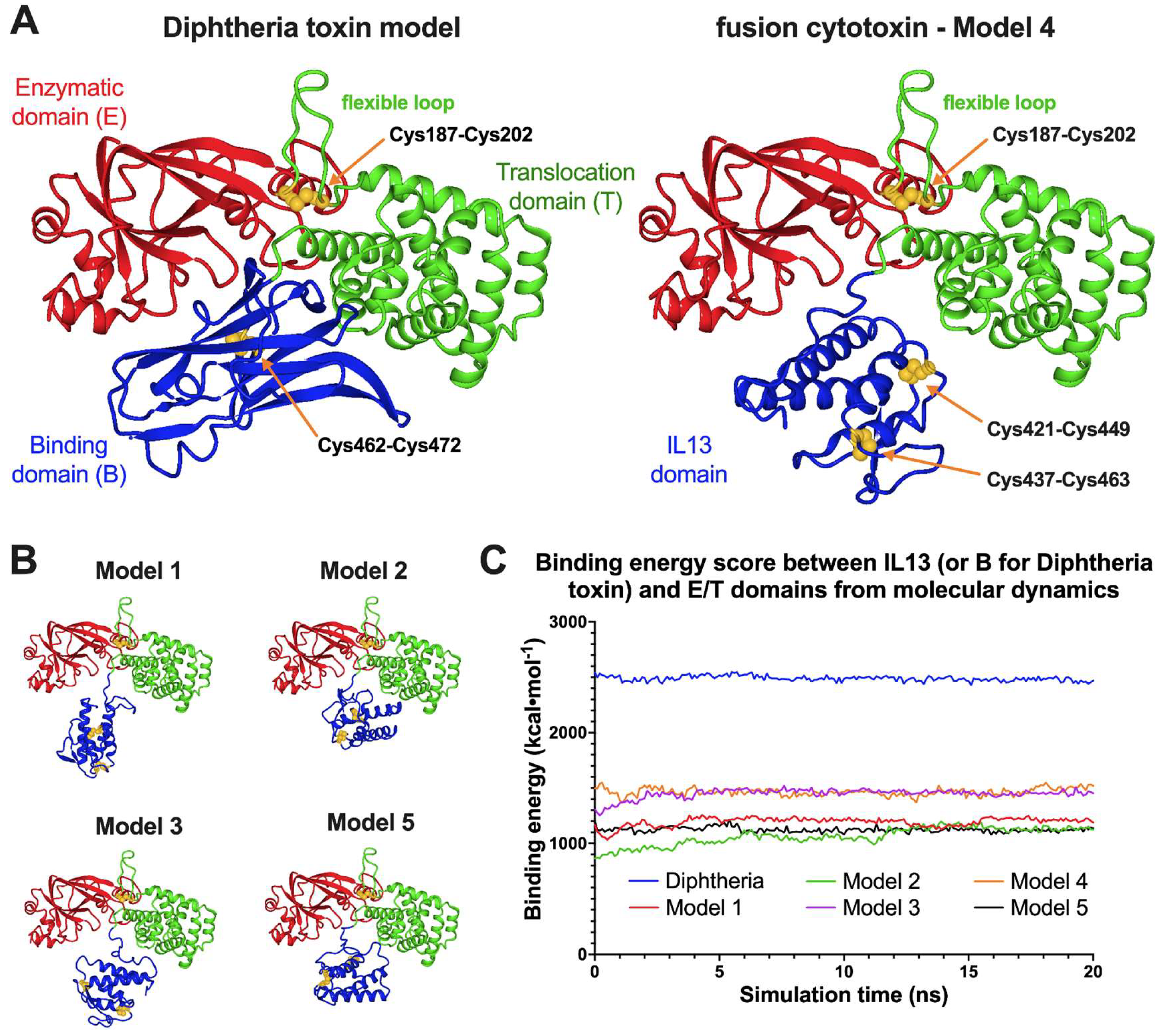
3.3. Computational Structural Studies
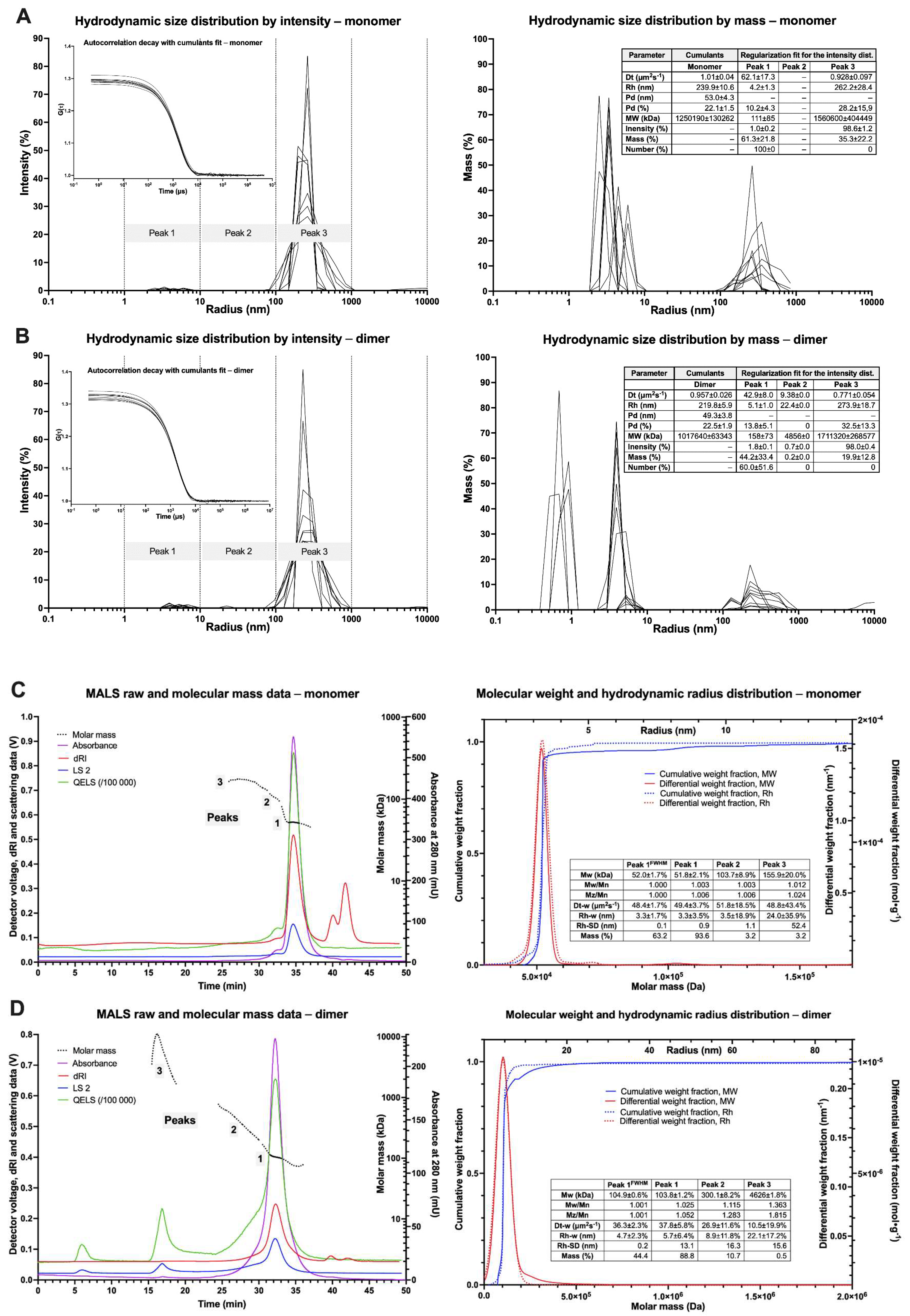
3.4. Light Scattering Spectroscopy—DLS
3.5. Light Scattering Spectroscopy—MALS
3.6. Small-Angle X-ray Scattering
3.6.1. Decomposition of SAXS-SEC Data
3.6.2. Selection of Suitable Datasets
3.6.3. Further Analysis of SAXS Data for Selected Datasets
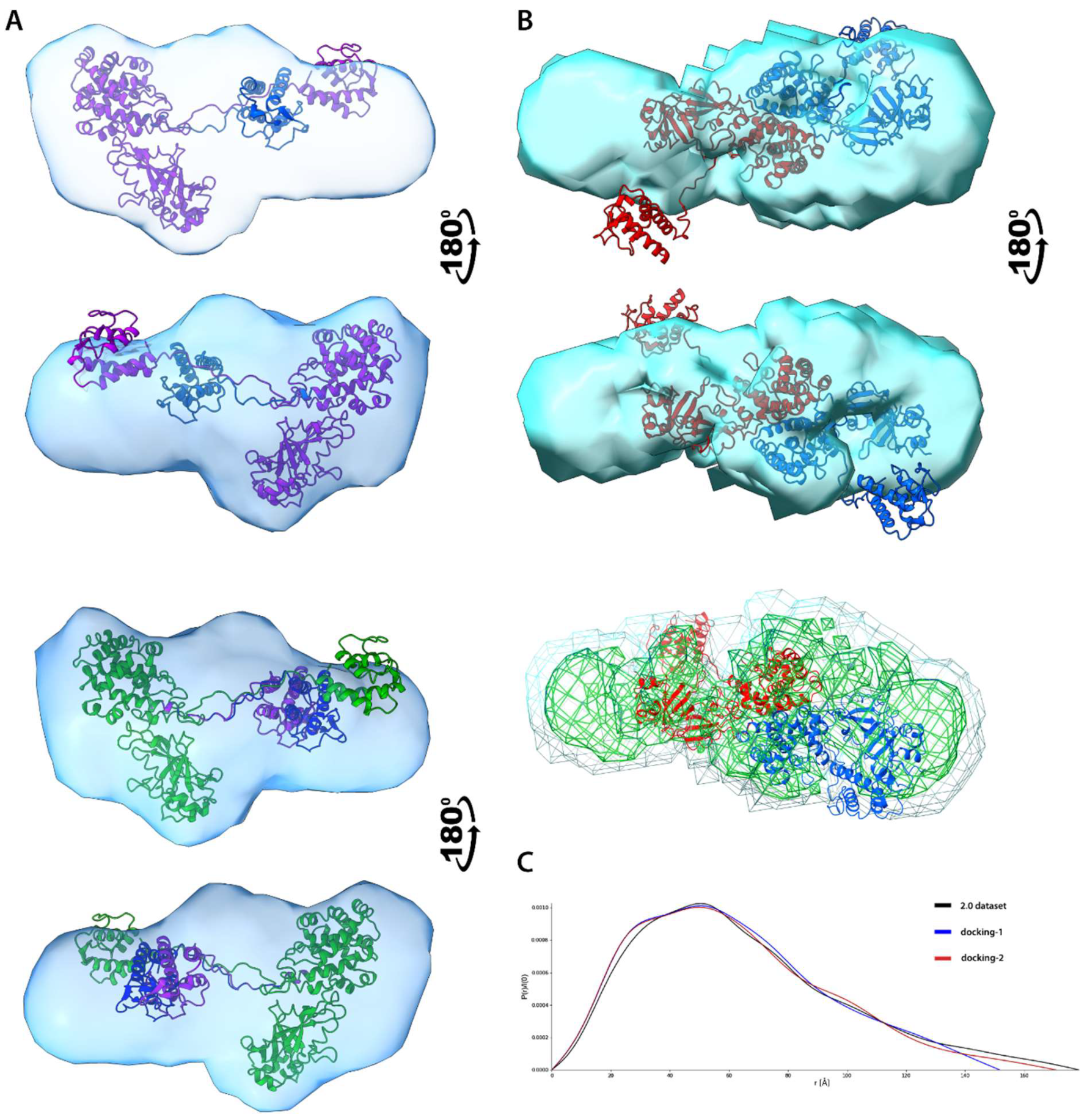
3.6.4. Electron Density Reconstruction and Ensemble Analysis
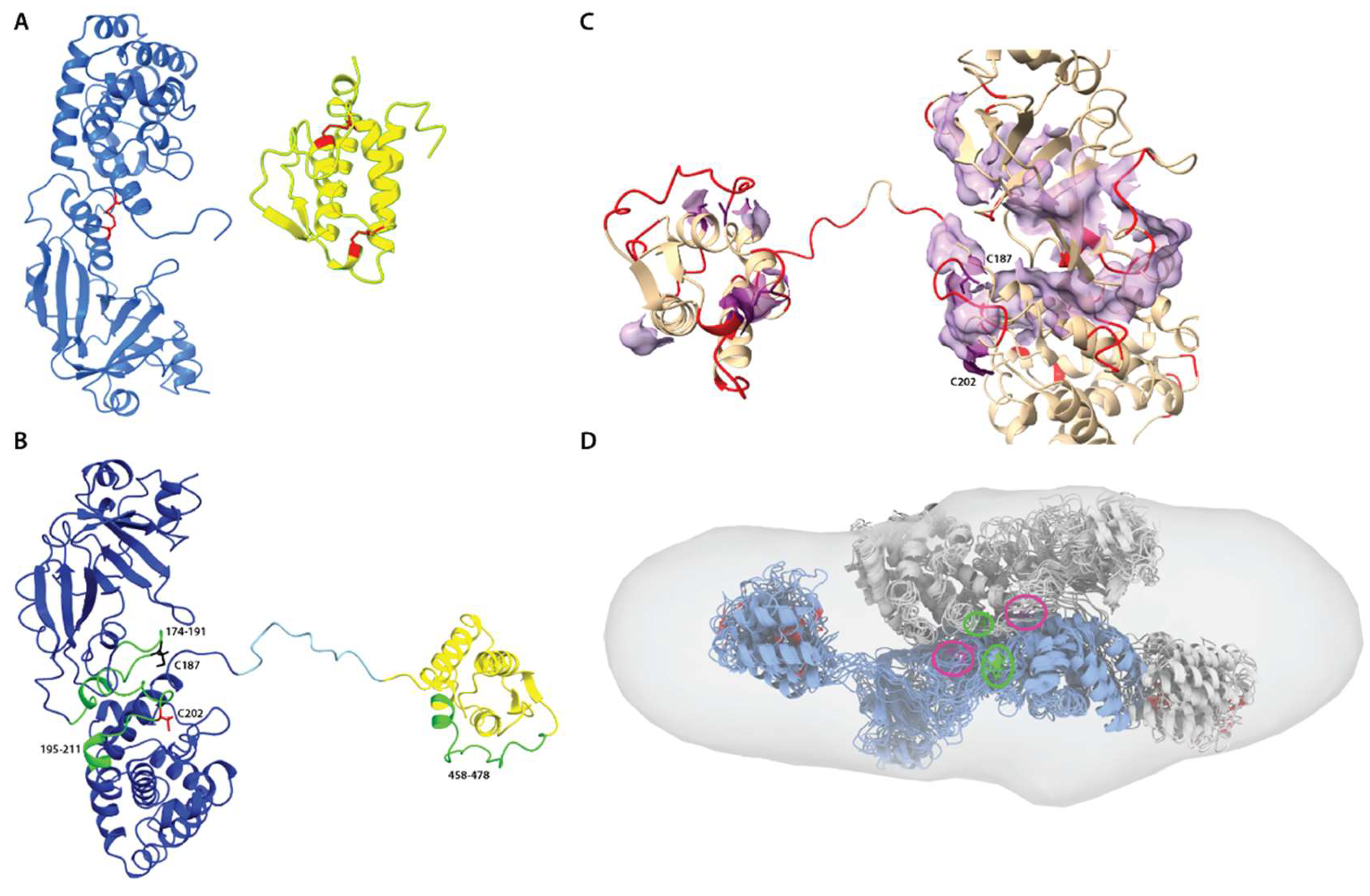
3.7. Disulfide Mapping and Structural Analysis
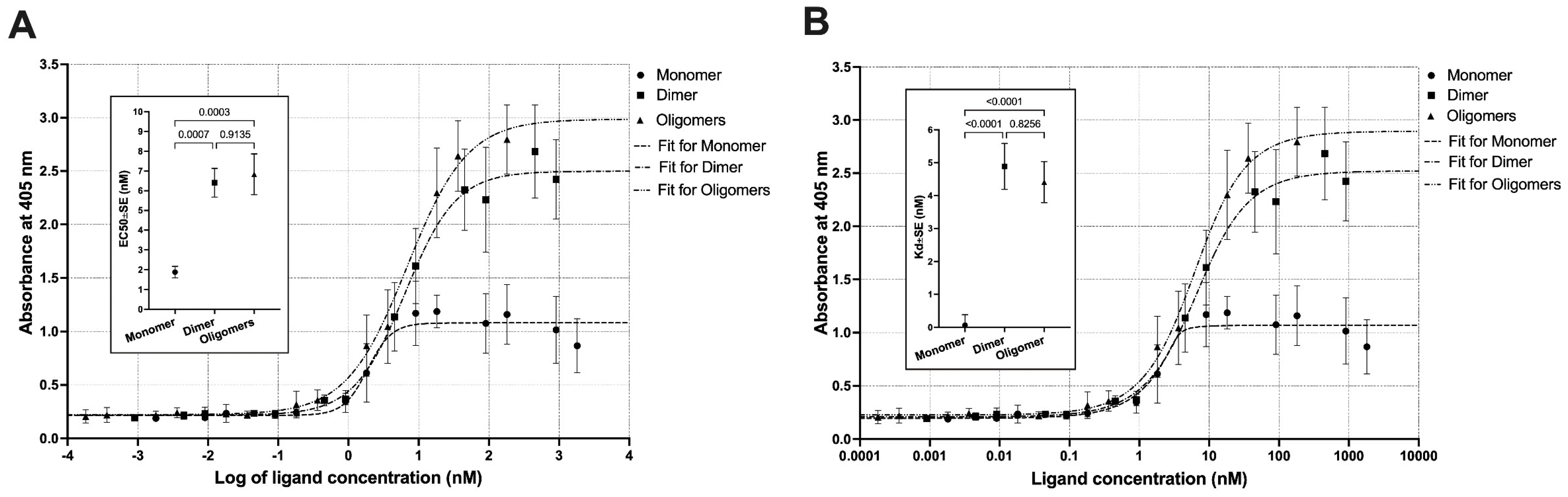
3.8. Bioactivity Functional Assays—ELISA and MTS
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zahaf, N.-I.; Schmidt, G. Bacterial Toxins for Cancer Therapy. Toxins 2017, 9, 236. [Google Scholar] [CrossRef] [PubMed]
- Peigneur, S.; Tytgat, J. Toxins and drug discovery. Toxins 2018, 10, 126. [Google Scholar] [CrossRef] [PubMed]
- Garashchenko, B.L.; Korsakova, V.A.; Yakovlev, R.Y. Radiopharmaceuticals Based on Alpha Emitters: Preparation, Properties, and Application. Phys. At. Nucl. 2018, 81, 1515–1525. [Google Scholar] [CrossRef]
- Pagliaro, L.C.; Liu, B.; Munker, R.; Andreeff, M.; Freireich, E.J.; Scheinberg, D.A.; Rosenblum, M.G. Humanized M195 monoclonal antibody conjugated to recombinant gelonin: An anti-CD33 immunotoxin with antileukemic activity. Clin. Cancer Res. 1998, 4, 1971–1976. [Google Scholar] [PubMed]
- Chandramohan, V.; Sampson, J.H.; Pastan, I.; Bigner, D.D. Toxin-Based Targeted Therapy for Malignant Brain Tumors. Clin. Dev. Immunol. 2012, 2012, 480429. [Google Scholar] [CrossRef] [PubMed]
- Sedighi, M.; Zahedi Bialvaei, A.; Hamblin, M.R.; Ohadi, E.; Asadi, A.; Halajzadeh, M.; Lohrasbi, V.; Mohammadzadeh, N.; Amiriani, T.; Krutova, M.; et al. Theraputic bacteria to combat cancer; current advances, challenges, and opportunities. Cancer Med. 2019, 8, 3167–3181. [Google Scholar] [PubMed]
- Kaminetzky, D.; Hymes, K.B. Denileukin diftitox for the treatment of cutaneous T-cell lymphoma. Biol. Targets Ther. 2008, 2, 717–724. [Google Scholar]
- Madhumathi, J.; Sridevi, S.; Verma, R.S. Novel TNF-related Apoptotic-inducing Ligand-based Immunotoxin for Therapeutic Targeting of CD25 Positive Leukemia. Target. Oncol. 2016, 11, 535–547. [Google Scholar] [CrossRef]
- Frankel, A.E.; Woo, J.H.; Ahn, C.; Foss, F.M.; Duvic, M.; Neville, P.H.; Neville, D.M. Resimmune, an anti-CD3e recombinant immunotoxin, induces durable remissions in patients with cutaneous T-cell lymphoma. Haematologica 2015, 100, 794–800. [Google Scholar] [CrossRef]
- Shan, L.; Liu, Y.; Wang, P. Recombinant Immunotoxin Therapy of Solid Tumors: Challenges and Strategies. J. Basic Clin. Med. 2013, 2, 1–6. [Google Scholar]
- Zhu, S.; Liu, Y.; Wang, P.C.; Gu, X.; Shan, L. Recombinant immunotoxins therapy of glioblastoma: Smart design, key findings and specific challenges. Biomed. Res. Int. 2017, 2017, 7929286. [Google Scholar] [CrossRef]
- Saito, R.; Tominaga, T. Convection-enhanced Delivery of Therapeutics for Malignant Gliomas. Neurol. Med.-Chir. 2017, 57, 8–16. [Google Scholar] [CrossRef]
- Rossmeisl, J.H.; Herpai, D.; Quigley, M.; Cecere, T.E.; Robertson, J.L.; D’Agostino, R.B.; Hinckley, J.; Tatter, S.B.; Dickinson, P.J.; Debinski, W. Phase I trial of convection-enhanced delivery of IL13RA2 and EPHA2 receptor targeted cytotoxins in dogs with spontaneous intracranical gliomas. Neuro-Oncol. 2021, 23, 422–434. [Google Scholar] [CrossRef]
- Zapadka, K.L.; Becher, F.J.; Gomes Dos Santos, A.L.; Jackson, S.E. Factors affecting the physical stability (aggregation) of peptide therapeutics. Interface Focus 2017, 7, 20170030. [Google Scholar] [CrossRef]
- Vazquez, E.; Corchero, J.L.; Villaverde, A. Post-production protein stability: Trouble beyond the cell factory. Microb. Cell Factories 2011, 10, 60. [Google Scholar] [CrossRef]
- Cromwell, M.E.M.; Hilario, E.; Jacobson, F. Protein aggregation and bioprocessing. AAPS J. 2006, 8, E572–E579. [Google Scholar] [CrossRef]
- Wang, W.; Nema, S.; Teagarden, D. Protein aggregation—Pathways and influencing factors. Int. J. Pharm. 2010, 390, 89–99. [Google Scholar] [CrossRef]
- Munishkina, L.A.; Ahmad, A.; Fink, A.L.; Uversky, V.N. Guiding Protein Aggregation with Macromolecular Crowding. Biochemistry 2008, 47, 8993–9006. [Google Scholar] [CrossRef]
- Musiani, F.; Giorgetti, A. Chapter two—Protein aggregation and molecular crowding: Perspectives from multiscale simulations. Int. Rev. Cell Mol. Biol. 2017, 329, 49–77. [Google Scholar]
- Roberts, C.J. Therapeutic protein aggregation: Mechanisms, design, and control. Trends Biotechnol. 2014, 32, 372–380. [Google Scholar] [CrossRef]
- Lebendiker, M.; Danieli, T. Production of prone-to-aggregate proteins. FEBS Lett. 2014, 588, 236–246. [Google Scholar] [CrossRef]
- Brange, J.; Andersen, L.; Laursen, E.D.; Meyn, G.; Rasmussen, E. Toward Understanding Insulin Fibrillation. J. Pharm. Sci. 1997, 86, 517–525. [Google Scholar] [CrossRef]
- Rouby, G.; Tran, N.T.; Leblanc, Y.; Taverna, M.; Bihoreau, N. Investigation of monoclonal antibody dimers in a final formulated drug by separation techniques coupled to native mass spectrometry. mAbs 2020, 12, 1781743. [Google Scholar] [CrossRef]
- Kijanka, G.; Bee, J.S.; Schenerman, M.A.; Korman, S.A.; Wu, Y.; Slutter, B.; Jiskoot, W. Monoclonal antibody dimers induced by low pH, heat or light exposure are immunogenic upon subcutaneous administration in a mouse model. J. Pharm. Sci. 2020, 109, 730–738. [Google Scholar] [CrossRef]
- Weinfurtner, D. Analysis of disulfide bond formation in therapeutic proteins. In Oxidative Folding of Proteins: Basic Principles, Cellular Regulation and Engineering; Feige, M.J., Ed.; Royal Society of Chemistry: Cambridge, UK, 2018; pp. 81–98. [Google Scholar]
- Rabdano, S.O.; Izmailov, S.A.; Luzik, D.A.; Groves, A.; Podkorytov, I.S.; Skrynnikov, N.R. Onset of disorder and protein aggregation due to oxidation-induced intermolecular disulfide bonds: Case study of RRM2 domain from TDP-43. Sci. Rep. 2017, 7, 11161. [Google Scholar] [CrossRef]
- Rombouts, I.; Lagrin, K.A.; Scherf, K.A.; Lambrecht, M.A.; Koehler, P.; Decour, J.A. Formation and reshuffling of disulfide bonds in bovine serum albumin demonstrated using tandem mass spectrometry with collision-induced and electron-transfer dissociation. Sci. Rep. 2015, 5, 12210. [Google Scholar] [CrossRef]
- Montoliu-Gaya, L.; Esquerda-Canals, G.; Bronsoms, S.; Villegas, S. Production of an anti-Ab antibody fragment in Pichia pastoris and in vitro and in vivo validation of its therapeutic. PLoS ONE 2017, 12, e0181480. [Google Scholar] [CrossRef]
- Sharma, P.; Sonawane, P.; Herpai, D.; D’Agostino, R.; Rossmeisl, J.; Tatter, S.; Debinski, W. Multireceptor targeting of glioblastoma. Neuro-Oncol. Adv. 2020, 2, vdaa107. [Google Scholar] [CrossRef]
- Krieger, E.; Nielsen, J.E.; Spronk, C.A.; Vriend, G. Fast empirical pKa prediction by Ewald summation. J. Mol. Graph. Model. 2006, 25, 481–486. [Google Scholar] [CrossRef]
- Krieger, E.; Darden, T.; Nabuurs, S.B.; Finkelstein, A.; Vriend, G. Making optimal use of empirical energy functions: Force-field parametrization in crystal space. Proteins 2004, 54, 678–683. [Google Scholar] [CrossRef]
- Fleming, P.J.; Fleming, K.G. HullRad: Fast Calculations of Folded and Disordered Protein and Nucleic Acid Hydrodynamic Properties. Biophys. J. 2018, 114, 856–869. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.S.; Toft, K.N.; Snakenborg, D.; Jeppesen, M.G.; Jacobsen, J.K.; Vestergaard, B.; Kutter, J.P.; Arleth, L. BioXTAS RAW, a software program for high-throughput automated small-angle X-ray scattering data reduction and preliminary analysis. J. Appl. Crystallogr. 2009, 42, 959–964. [Google Scholar] [CrossRef]
- Hopkins, J.B.; Gillilan, R.E.; Skou, S. BioXTAS RAW: Improvements to a free open-source program for small-angle X-ray scattering data reduction and analysis. J. Appl. Crystallogr. 2017, 50, 1545–1553. [Google Scholar] [CrossRef] [PubMed]
- Meisburger, S.P.; Taylor, A.B.; Khan, C.A.; Zhang, S.; Fitzpatrick, P.F.; Ando, N. Domain Movements upon Activation of Phenylalanine Hydroxylase Characterized by Crystallography and Chromatography-Coupled Small-Angle X-ray Scattering. J. Am. Chem. Soc. 2016, 138, 6506–6516. [Google Scholar] [CrossRef]
- Manalastas-Cantos, K.; Konarev, P.V.; Hajizadeh, N.R.; Kikhney, A.G.; Petoukhov, M.V.; Molodenskiy, D.S.; Panjkovich, A.; Mertens, H.D.; Gruzinov, A.; Borges, C.; et al. ATSAS 3.0: Expanded functionality and new tools for small-angle scattering data analysis. J. Appl. Crystallogr. 2021, 54, 343–355. [Google Scholar] [CrossRef]
- Hansen, S. Bayesian estimation of hyperparameters for indirect Fourier transformation in small-angle scattering. J. Appl. Crystallogr. 2000, 33, 1415–1421. [Google Scholar] [CrossRef]
- Piiadov, V.; Ares de Araujo, E.; Neto, M.O.; Craievich, A.F.; Polikarpov, I. SAXSMoW 2.0: Online calculator of the molecular weight of proteins in dilute solution from experimental SAXS data measured on a relative scale. Protein Sci. 2019, 28, 454–463. [Google Scholar] [CrossRef]
- Rambo, R.P.; Tainer, J. Accurate assessment of mass, models and resolution by small-angle scattering. Nature 2013, 496, 477–481. [Google Scholar] [CrossRef]
- Franke, D.; Jeffries, C.M.; Svergun, D.I. Machine Learning Methods for X-ray Scattering Data Analysis from Biomacromolecular Solutions. Biophys. J. 2018, 114, 2485–2492. [Google Scholar] [CrossRef]
- Hajizadeh, N.R.; Franke, D.; Jeffries, C.M.; Svergun, D.I. Consensus Bayesian assessment of protein molecular mass from solution X-ray scattering data. Sci. Rep. 2018, 8, 7204. [Google Scholar] [CrossRef]
- Grant, T.D. Ab initio electron density determination directly from solution scattering data. Nat. Methods 2018, 15, 191–193. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Šali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–781. [Google Scholar] [CrossRef]
- Shapovalov, M.V.; Dunbrack, R.L., Jr. A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 2011, 19, 844–858. [Google Scholar] [CrossRef]
- Feig, M. Local Protein Structure Refinement via Molecular Dynamics Simulations with locPREFMD. J. Chem. Inf. Model. 2016, 56, 1304–1312. [Google Scholar] [CrossRef]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Hammel, M.; Tainer, J.; Sali, A. FoXS, FoXSDock and MultiFoXS: Single-state and multi-state structural modeling of proteins and their complexes based on SAXS profiles. Nucleic Acids Res. 2016, 44, W424–W429. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Hammel, M.; Sali, A. FoXS: A web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res. 2010, 38, W540–W544. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A Peptide Search Engine Integrated into the MaxQuant Environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- Kuriata, A.; Iglesias, V.; Pujols, J.; Kurcinski, M.; Kmiecik, S.; Ventura, S. Aggrescan3D (A3D) 2.0: Prediction and engineering of protein solubility. Nucleic Acids Res. 2019, 47, W300–W307. [Google Scholar] [CrossRef] [PubMed]
- Kuriata, A.; Gierut, A.M.; Oleniecki, T.; Ciemny, M.P.; Kolinski, A.; Kurcinski, M.; Kmiecik, S. CABS-flex 2.0: A web server for fast simulations of flexibility of protein structures. Nucleic Acids Res. 2018, 46, W338–W343. [Google Scholar] [CrossRef] [PubMed]
- Eble, J.A. Titration ELISA as a method to determine the dissociation constant of receptor ligand interaction. J. Vis. Exp. 2018, 15, 57334. [Google Scholar] [CrossRef] [PubMed]
- Pham, N.B.; Meng, W.S. Protein aggregation and immunogenicity of biotherapeutics. Int. J. Pharm. 2020, 585, 119523. [Google Scholar] [CrossRef]
- Owczarek, B.; Gerszberg, A.; Hnatuszko-Konka, K. A Brief Reminder of Systems of Production and Chromatography-Based Recovery of Recombinant Protein Biopharmaceuticals. BioMed Res. Int. 2019, 2019, 4216060. [Google Scholar] [CrossRef]
- Humer, D.; Spadiut, O. Wanted: More monitoring and control during inclusion body processing. World J. Microbiol. Biotechnol. 2018, 34, 158. [Google Scholar] [CrossRef]
- Klema, V.; Laub, A. The singular value decomposition: Its computation and some applications. IEEE Trans. Autom. Control 1980, 25, 164–176. [Google Scholar] [CrossRef]
- Maeder, M. Evolving factor analysis for the resolution of overlapping chromatographic peaks. Anal. Chem. 1987, 59, 527–530. [Google Scholar] [CrossRef]
- Kikhney, A.G.; Svergun, D.I. A practical guide to small angle X-ray scattering (SAXS) of flexible and intrinsically disordered proteins. FEBS Lett. 2015, 589, 2570–2577. [Google Scholar] [CrossRef]
- Rambo, R.P.; Tainer, J.A. Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers 2011, 95, 559–571. [Google Scholar] [CrossRef]
- Pelikan, M.; Hura, G.L.; Hammel, M. Structure and flexibility within proteins as identified through small angle X-ray scattering. Gen. Physiol. Biophys. 2009, 28, 174–189. [Google Scholar] [CrossRef]
- Panjkovich, A.; Svergun, D.I. Deciphering conformational transitions of proteins by small angle X-ray scattering and normal mode analysis. Phys. Chem. Chem. Phys. 2016, 18, 5707–5719. [Google Scholar] [CrossRef]
- Madhankumar, A.B.; Mintz, A.; Debinski, W. Interleukin 13 mutants of enhanced avidity toward the glioma-associated receptor, IL13Ralpha2. Neoplasia 2004, 6, 15–22. [Google Scholar] [CrossRef]
- Lupardus, P.J.; Birnbaum, M.E.; Garcia, K.C. Molecular basis for shared cytokine recognition revealed in the structure of an unusually high affinity complex between IL-13 and IL-13Ralpha2. Structure 2010, 18, 332–342. [Google Scholar] [CrossRef]
- Jarmoskaite, I.; Alsadhan, I.; Vaidyanathan, P.P.; Herschlag, D. How to measure and evaluate binding affinities. eLife 2020, 9, e57264. [Google Scholar] [CrossRef]
- Nash, K.T.; Thompson, J.P.; Debinski, W. Molecular targeting of malignant gliomas with novel multiply-mutated interleukin 13-based cytotoxins. Crit. Rev. Oncol. Hematol. 2001, 39, 87–98. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mielecki, M.; Ziemniak, M.; Ozga, M.; Borowski, R.; Antosik, J.; Kaczyńska, A.; Pająk, B. Structure–Activity Relationship of the Dimeric and Oligomeric Forms of a Cytotoxic Biotherapeutic Based on Diphtheria Toxin. Biomolecules 2022, 12, 1111. https://doi.org/10.3390/biom12081111
Mielecki M, Ziemniak M, Ozga M, Borowski R, Antosik J, Kaczyńska A, Pająk B. Structure–Activity Relationship of the Dimeric and Oligomeric Forms of a Cytotoxic Biotherapeutic Based on Diphtheria Toxin. Biomolecules. 2022; 12(8):1111. https://doi.org/10.3390/biom12081111
Chicago/Turabian StyleMielecki, Marcin, Marcin Ziemniak, Magdalena Ozga, Radosław Borowski, Jarosław Antosik, Angelika Kaczyńska, and Beata Pająk. 2022. "Structure–Activity Relationship of the Dimeric and Oligomeric Forms of a Cytotoxic Biotherapeutic Based on Diphtheria Toxin" Biomolecules 12, no. 8: 1111. https://doi.org/10.3390/biom12081111
APA StyleMielecki, M., Ziemniak, M., Ozga, M., Borowski, R., Antosik, J., Kaczyńska, A., & Pająk, B. (2022). Structure–Activity Relationship of the Dimeric and Oligomeric Forms of a Cytotoxic Biotherapeutic Based on Diphtheria Toxin. Biomolecules, 12(8), 1111. https://doi.org/10.3390/biom12081111