
Structural Comparison of hMDH2 Complexed with Natural Substrates and Cofactors: The Importance of Phosphate Binding for Active Conformation and Catalysis

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cloning, Protein Expression, and Purification
2.2. Crystallization and Data Collection
2.3. Structure Determination and Refinement
2.4. Enzymatic Assay
2.5. ITC Measurement
3. Results
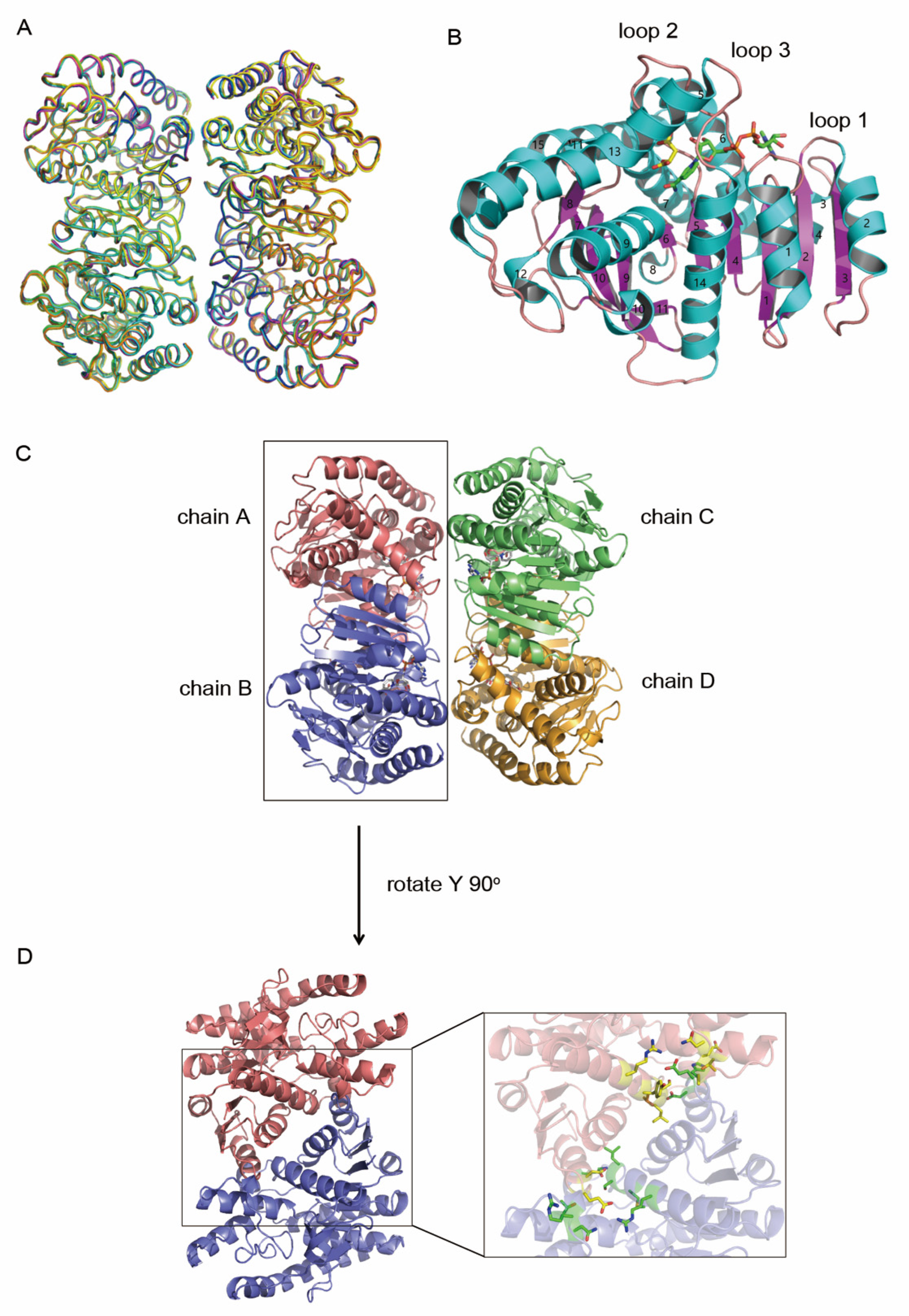
3.1. Overall Structure of hMDH2
3.2. Obtaining Ligand Bound Structures
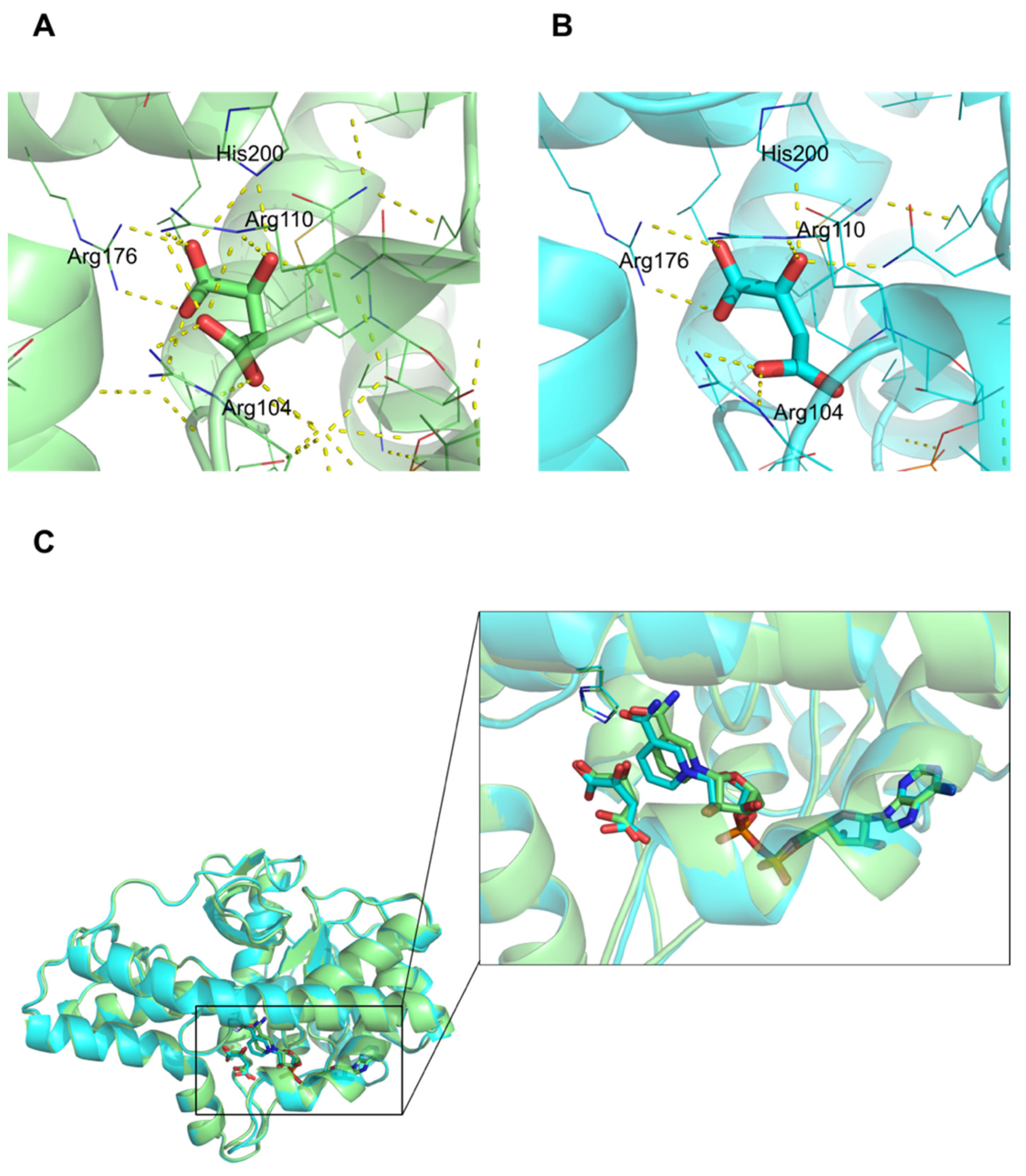
3.3. Comparison of L-Malate and Citrate Binding
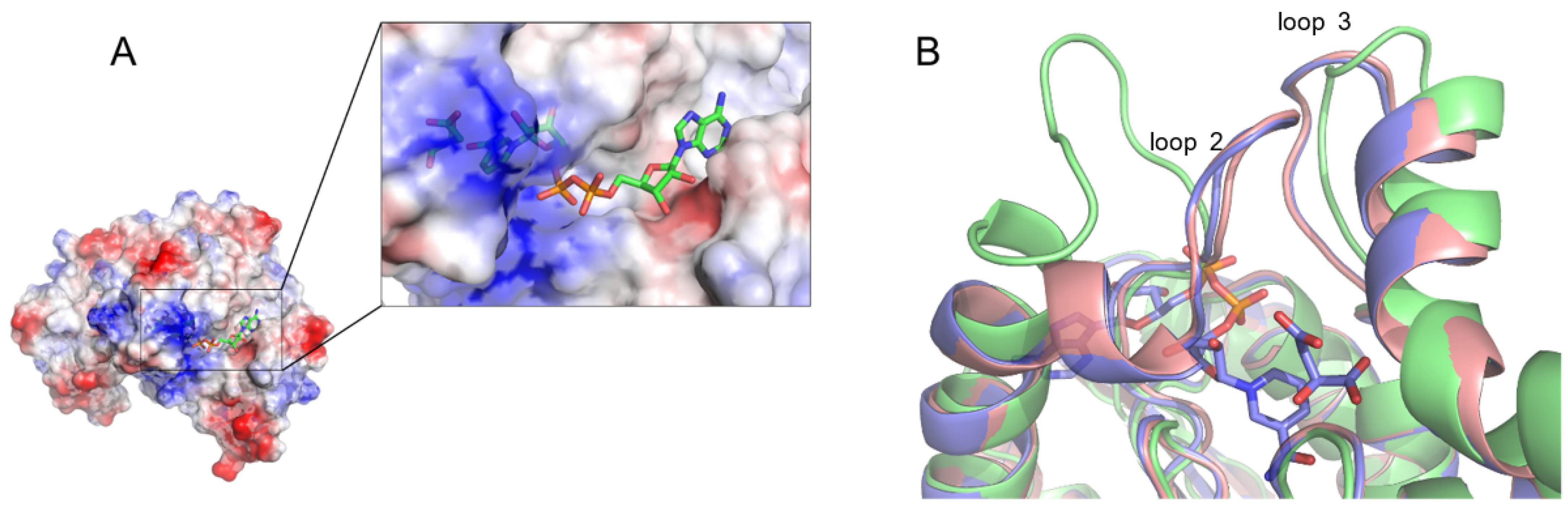
3.4. Comparison of Active Site
3.5. NAD, L-Malate vs. NADH, Oxaloacetate
3.6. Cofactor Binding Affinity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goward, C.R.; Nicholls, D.J. Malate dehydrogenase: A model for structure, evolution, and catalysis. Protein Sci. 1994, 3, 1883–1888. [Google Scholar] [CrossRef] [PubMed]
- Thorne, C.J.; Kaplan, N.O. Physicochemical properties of pig and horse heart mitochondrial malate dehydrogenase. J. Biol. Chem. 1963, 238, 1861–1868. [Google Scholar] [CrossRef]
- Beeckmans, S.; Kanarek, L. Demonstration of physical interactions between consecutive enzymes of the citric acid cycle and of the aspartate-malate shuttle. A study involving fumarase, malate dehydrogenase, citrate synthesis and aspartate aminotransferase. Eur. J. Biochem. 1981, 117, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Minard, K.I.; McAlister-Henn, L. Isolation, nucleotide sequence analysis, and disruption of the MDH2 gene from Saccharomyces cerevisiae: Evidence for three isozymes of yeast malate dehydrogenase. Mol. Cell. Biol. 1991, 11, 370–380. [Google Scholar] [CrossRef] [PubMed]
- McAlister-Henn, L.; Blaber, M.; Bradshaw, R.A.; Nisco, S.J. Complete nucleotide sequence of the Escherichia coli gene encoding malate dehydrogenase. Nucleic Acids Res. 1987, 15, 4993. [Google Scholar] [CrossRef]
- Nishiyama, M.; Matsubara, N.; Yamamoto, K.; Iijima, S.; Uozumi, T.; Beppu, T. Nucleotide sequence of the malate dehydrogenase gene of Thermus flavus and its mutation directing an increase in enzyme activity. J. Biol. Chem. 1986, 261, 14178–14183. [Google Scholar] [CrossRef]
- Minarik, P.; Tomaskova, N.; Kollarova, M.; Antalik, M. Malate dehydrogenases—Structure and function. Gen. Physiol. Biophys. 2002, 21, 257–265. [Google Scholar]
- Madern, D. Molecular evolution within the L-malate and L-lactate dehydrogenase super-family. J. Mol. Evol. 2002, 54, 825–840. [Google Scholar] [CrossRef]
- Nicholls, D.J.; Miller, J.; Scawen, M.D.; Clarke, A.R.; Holbrook, J.J.; Atkinson, T.; Goward, C.R. The importance of arginine 102 for the substrate specificity of Escherichia coli malate dehydrogenase. Biochem. Biophys. Res. Commun. 1992, 189, 1057–1062. [Google Scholar] [CrossRef]
- Wigley, D.B.; Gamblin, S.J.; Turkenburg, J.P.; Dodson, E.J.; Piontek, K.; Muirhead, H.; Holbrook, J.J. Structure of a ternary complex of an allosteric lactate dehydrogenase from Bacillus stearothermophilus at 2.5 A resolution. J. Mol. Biol. 1992, 223, 317–335. [Google Scholar] [CrossRef]
- Zhao, S.; Xu, W.; Jiang, W.; Yu, W.; Lin, Y.; Zhang, T.; Yao, J.; Zhou, L.; Zeng, Y.; Li, H.; et al. Regulation of cellular metabolism by protein lysine acetylation. Science 2010, 327, 1000–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelpi, J.L.; Dordal, A.; Montserrat, J.; Mazo, A.; Cortes, A. Kinetic studies of the regulation of mitochondrial malate dehydrogenase by citrate. Biochem. J. 1992, 283 Pt 1, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.D.; Levitt, D.G.; Banaszak, L.J. Crystal structure of Escherichia coli malate dehydrogenase. A complex of the apoenzyme and citrate at 1.87 A resolution. J. Mol. Biol. 1992, 226, 867–882. [Google Scholar] [CrossRef]
- Matsuda, T.; Takahashi-Yanaga, F.; Yoshihara, T.; Maenaka, K.; Watanabe, Y.; Miwa, Y.; Morimoto, S.; Kubohara, Y.; Hirata, M.; Sasaguri, T. Dictyostelium differentiation-inducing factor-1 binds to mitochondrial malate dehydrogenase and inhibits its activity. J. Pharmacol. Sci. 2010, 112, 320–326. [Google Scholar] [CrossRef]
- Lee, K.; Ban, H.S.; Naik, R.; Hong, Y.S.; Son, S.; Kim, B.K.; Xia, Y.; Song, K.B.; Lee, H.S.; Won, M. Identification of malate dehydrogenase 2 as a target protein of the HIF-1 inhibitor LW6 using chemical probes. Angew. Chem. Int. Ed. Engl. 2013, 52, 10286–10289. [Google Scholar] [CrossRef]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar]
- Collaborative Computational Project, N. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994, 50, 760–763. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Skubak, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Schuttelkopf, A.W.; van Aalten, D.M. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Schrödinger, L.; DeLano, W. PyMOL. Available online: http://www.pymol.org/pymol (accessed on 25 July 2022).
- Breiter, D.R.; Resnik, E.; Banaszak, L.J. Engineering the quaternary structure of an enzyme: Construction and analysis of a monomeric form of malate dehydrogenase from Escherichia coli. Protein Sci. 1994, 3, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [PubMed]
- Gleason, W.B.; Fu, Z.; Birktoft, J.; Banaszak, L. Refined crystal structure of mitochondrial malate dehydrogenase from porcine heart and the consensus structure for dicarboxylic acid oxidoreductases. Biochemistry 1994, 33, 2078–2088. [Google Scholar] [CrossRef]
- Shore, J.D.; Evans, S.A.; Holbrook, J.J.; Parker, D.M. NADH binding to porcine mitochondrial malate dehydrogenase. J. Biol. Chem. 1979, 254, 9059–9062. [Google Scholar] [CrossRef]
- Zaitseva, J.; Meneely, K.M.; Lamb, A.L. Structure of Escherichia coli malate dehydrogenase at 1.45 A resolution. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2009, 65, 866–869. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MDH2_CIT a | MDH2_PO4 | MDH2_LMR a | MDH2_NAD | MDH2_LMR, NAD | MDH2_OAA a, NAI a | |
|---|---|---|---|---|---|---|
| PDB ID | 4WLE | 4WLN | 4WLF | 4WLV | 4WLU | 4WLO |
| Data collection | ||||||
| Space group | p212121 | p212121 | p212121 | p212121 | p212121 | p212121 |
| Cell dimensions | ||||||
| a, b, c (Å) | 59.70, 151.22, 154.65 | 59.96, 151.94, 155.05 | 60.25, 152.18, 155.03 | 58.86, 152.02, 155.67 | 59.98, 152.14, 155.04 | 59.99, 152.11, 155.87 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) * | 45.10-1.90 (1.93-1.90) | 32.56-2.28 (2.32-2.28) | 34.86-2.20 (2.24-2.20) | 30.86-2.40 (2.44-2.40) | 30.79-2.14 (2.19-2.15) | 28.35-2.50 (2.54-2.50) |
| Rmerge b | 9.2 (80.9) | 13.1 (58.1) | 6.8 (33.3) | 7.9 (46.1) | 7.4 (38.7) | 7.5 (41.8) |
| I/σI | 31.3 (3.3) | 34.04 (6.0) | 54.0 (10.0) | 51.2 (9.08) | 55.5 (10.5) | 47.8 (7.6) |
| Completeness (%) * | 99.8 (99.9) | 99.8 (100.0) | 95.9 (91.6) | 99.9 (100.0) | 99.8 (100.0) | 99.0 (98.9) |
| Redundancy * | 7.1 (6.8) | 14.2 (4.7) | 11.1 (10.2) | 14.3 (14.3) | 13.6 (13.5) | 12.8 (11.4) |
| Refinement | ||||||
| Resolution (Å) * | 45.10-1.90 (1.93-1.90) | 32.56-2.28 (2.32-2.28) | 34.86-2.20 (2.24-2.20) | 30.86-2.40 (2.44-2.40) | 30.79-2.14 (2.19-2.15) | 28.35-2.50 (2.54-2.50) |
| No. reflections | 104,532 | 62,209 | 66,329 | 53,618 | 74,182 | 47,440 |
| Rwork/Rfree (%) c | 21.8/23.8 | 21.7/24.6 | 22.7/24.7 | 20.8/24.8 | 21.6/24.4 | 21.1/25.5 |
| No. atoms | 9417 | 9430 | 9351 | 9514 | 9562 | 9488 |
| protein | 9258 | 9248 | 9252 | 9248 | 9248 | 9251 |
| ligand | 52 (CIT: 52) | 60 (PO4: 60) | 56 (L-MLT: 36, PO4: 20) | 216 (NAD: 176, PO4: 40) | 212 (L-MLT: 36, NAD: 176) | 212 (NADH: 176, OAA: 36) |
| water | 107 | 122 | 43 | 50 | 102 | 25 |
| Mean B-factors (Å2) | 35.047 | 33.354 | 37.71 | 38.97 | 35.08 | 45.593 |
| protein | 35.37 | 33.68 | 38.10 | 39.31 | 35.43 | 45.91 |
| ligand | 36.93 (CIT: 36.93) | 34.21 (PO4: 34.21) | L-MLT: 37.52, PO4: 54.87 | NAD: 37.32, PO4: 54.76 | L-MLT: 35.06, NAD: 34.80 | NADH: 54.55, OAA: 50.81 |
| water | 31.73 | 30.53 | 35.50 | 30.82 | 31.88 | 35.68 |
| R.m.s. deviations | ||||||
| Bond lengths (Å) | 0.004 | 0.005 | 0.005 | 0.005 | 0.005 | 0.006 |
| Bond angle (°) | 0.898 | 1.226 | 1.026 | 1.190 | 0.984 | 1.066 |
| Cofactor | ΔH (kcal mol−1) | TΔS (kcal mol−1) | ΔG (kcal mol−1) | N | Kd (μM) |
|---|---|---|---|---|---|
| NADH | −7.29 ± 0.43 | −0.23 | −7.52 | 0.83 ± 0.06 | 3.05 ± 0.06 |
| NAD | −3.65 ± 3.20 | −0.94 | −4.59 | N.A. a | 2306 ± 1733 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eo, Y.; Duong, M.T.H.; Ahn, H.-C. Structural Comparison of hMDH2 Complexed with Natural Substrates and Cofactors: The Importance of Phosphate Binding for Active Conformation and Catalysis. Biomolecules 2022, 12, 1175. https://doi.org/10.3390/biom12091175
Eo Y, Duong MTH, Ahn H-C. Structural Comparison of hMDH2 Complexed with Natural Substrates and Cofactors: The Importance of Phosphate Binding for Active Conformation and Catalysis. Biomolecules. 2022; 12(9):1175. https://doi.org/10.3390/biom12091175
Chicago/Turabian StyleEo, Yumi, Men Thi Hoai Duong, and Hee-Chul Ahn. 2022. "Structural Comparison of hMDH2 Complexed with Natural Substrates and Cofactors: The Importance of Phosphate Binding for Active Conformation and Catalysis" Biomolecules 12, no. 9: 1175. https://doi.org/10.3390/biom12091175