
The Effect of Aggregated Alpha Synuclein on Synaptic and Axonal Proteins in Parkinson’s Disease—A Systematic Review



Abstract
:1. Introduction
2. Methods
2.1. Search Strategy
2.2. Study Screening and Selection
2.3. Data Extraction
3. Results
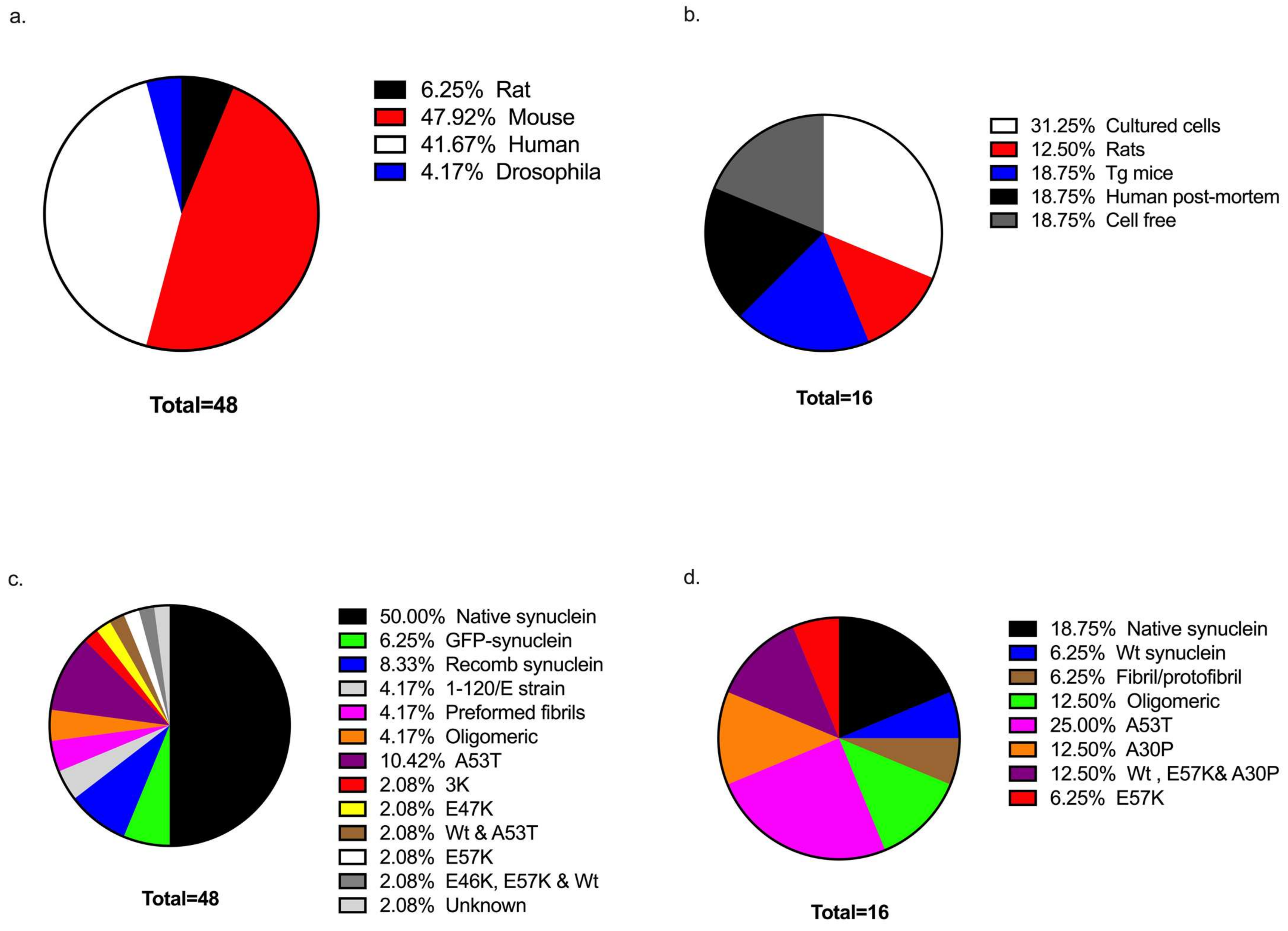
3.1. Study Characteristics
3.2. Sample Sources Used in Investigations
3.3. Forms of α-Synuclein
3.4. Alpha Synuclein Effects on Synaptic Proteins
3.5. Effects of α-Synuclein on Axonal Proteins
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cheng, F.; Vivacqua, G.; Yu, S. The role of alpha-synuclein in neurotransmission and synaptic plasticity. J. Chem. Neuroanat. 2011, 42, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Kiely, A.P.; Asi, Y.T.; Kara, E.; Limousin, P.; Ling, H.; Lewis, P.; Proukakis, C.; Quinn, N.; Lees, A.J.; Hardy, J. α-Synucleinopathy associated with G51D SNCA mutation: A link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol. 2013, 125, 753–769. [Google Scholar] [CrossRef] [PubMed]
- Clayton, D.F.; George, J.M. Synucleins in synaptic plasticity and neurodegenerative disorders. J. Neurosci. Res. 1999, 58, 120–129. [Google Scholar] [CrossRef]
- Ghiglieri, V.; Calabrese, V.; Calabresi, P. Alpha-synuclein: From early synaptic dysfunction to neurodegeneration. Front. Neurol. 2018, 9, 295. [Google Scholar] [CrossRef]
- He, S.; Zhong, S.; Liu, G.; Yang, J. Alpha-synuclein: The interplay of pathology, neuroinflammation, and environmental factors in Parkinson’s disease. Neurodegener. Dis. 2020, 20, 55–64. [Google Scholar] [CrossRef]
- Stefanis, L. α-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef]
- Meade, R.M.; Fairlie, D.P.; Mason, J.M. Alpha-synuclein structure and Parkinson’s disease—Lessons and emerging principles. Mol. Neurodegener. 2019, 14, 29. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.A. Effects of α-synuclein on axonal transport. Neurobiol. Dis. 2017, 105, 321–327. [Google Scholar] [CrossRef]
- Chen, H.; Zhao, Y.-F.; Chen, Y.-X.; Li, Y.-M. Exploring the roles of post-translational modifications in the pathogenesis of Parkinson’s disease using synthetic and semisynthetic modified α-synuclein. ACS Chem. Neurosci. 2019, 10, 910–921. [Google Scholar] [CrossRef]
- Pienaar, I.S.; Burn, D.; Morris, C.; Dexter, D. Synaptic protein alterations in Parkinson’s disease. Mol. Neurobiol. 2012, 45, 126–143. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. α-Synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [PubMed]
- Rizo, J.; Rosenmund, C. Synaptic vesicle fusion. Nat. Struct. Mol. Biol. 2008, 15, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Südhof, T.C.; Rizo, J. Synaptic vesicle exocytosis. Cold Spring Harb. Perspect. Biol. 2011, 3, a005637. [Google Scholar] [CrossRef] [PubMed]
- Calo, L.; Wegrzynowicz, M.; Santivañez-Perez, J.; Grazia Spillantini, M. Synaptic failure and α-synuclein. Mov. Disord. 2016, 31, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Simon-Sanchez, J.; Schulte, C.; Bras, J.M.; Sharma, M.; Gibbs, J.R.; Berg, D.; Paisan-Ruiz, C.; Lichtner, P.; Scholz, S.W.; Hernandez, D.G. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 2009, 41, 1308–1312. [Google Scholar] [CrossRef]
- Chung, C.Y.; Koprich, J.B.; Siddiqi, H.; Isacson, O. Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV α-synucleinopathy. J. Neurosci. 2009, 29, 3365–3373. [Google Scholar] [CrossRef]
- Garcia-Reitböck, P.; Anichtchik, O.; Bellucci, A.; Iovino, M.; Ballini, C.; Fineberg, E.; Ghetti, B.; Della Corte, L.; Spano, P.; Tofaris, G.K. SNARE protein redistribution and synaptic failure in a transgenic mouse model of Parkinson’s disease. Brain 2010, 133, 2032–2044. [Google Scholar] [CrossRef]
- Nemani, V.M.; Lu, W.; Berge, V.; Nakamura, K.; Onoa, B.; Lee, M.K.; Chaudhry, F.A.; Nicoll, R.A.; Edwards, R.H. Increased expression of α-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 2010, 65, 66–79. [Google Scholar] [CrossRef]
- Scott, D.A.; Tabarean, I.; Tang, Y.; Cartier, A.; Masliah, E.; Roy, S. A pathologic cascade leading to synaptic dysfunction in α-synuclein-induced neurodegeneration. J. Neurosci. 2010, 30, 8083–8095. [Google Scholar] [CrossRef]
- Rizo, J.; Südhof, T.C. The membrane fusion enigma: SNAREs, Sec1/Munc18 proteins, and their accomplices—Guilty as charged? Annu. Rev. Cell Dev. Biol. 2012, 28, 279–308. [Google Scholar] [CrossRef]
- Sudhof, T.C. The Presynaptic Active Zone. Neuron 2012, 75, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Bucci, C.; Alifano, P.; Cogli, L. The role of rab proteins in neuronal cells and in the trafficking of neurotrophin receptors. Membranes 2014, 4, 642–677. [Google Scholar] [CrossRef] [PubMed]
- Tagliaferro, P.; Burke, R.E. Retrograde axonal degeneration in Parkinson disease. J. Parkinson’s Dis. 2016, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Millecamps, S.; Julien, J.-P. Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 161–176. [Google Scholar] [CrossRef]
- Chu, Y.; Morfini, G.A.; Langhamer, L.B.; He, Y.; Brady, S.T.; Kordower, J.H. Alterations in axonal transport motor proteins in sporadic and experimental Parkinson’s disease. Brain 2012, 135, 2058–2073. [Google Scholar] [CrossRef]
- Galvin, J.E.; Uryu, K.; Lee, V.M.-Y.; Trojanowski, J.Q. Axon pathology in Parkinson’s disease and Lewy body dementia hippocampus contains α-, β-, and γ-synuclein. Proc. Natl. Acad. Sci. USA 1999, 96, 13450–13455. [Google Scholar] [CrossRef]
- Morfini, G.; Pigino, G.; Opalach, K.; Serulle, Y.; Moreira, J.; Sugimori, M.; Llinas, R.; Brady, S. 1-Methyl-4-phenylpyridinium affects fast axonal transport by activation of caspase and protein kinase C. Proc. Natl. Acad. Sci. USA 2007, 104, 2442–2447. [Google Scholar] [CrossRef]
- Lamberts, J.T.; Hildebrandt, E.N.; Brundin, P. Spreading of α-synuclein in the face of axonal transport deficits in Parkinson’s disease: A speculative synthesis. Neurobiol. Dis. 2015, 77, 276–283. [Google Scholar] [CrossRef]
- Rockenstein, E.; Nuber, S.; Overk, C.R.; Ubhi, K.; Mante, M.; Patrick, C.; Adame, A.; Trejo-Morales, M.; Gerez, J.; Picotti, P. Accumulation of oligomer-prone α-synuclein exacerbates synaptic and neuronal degeneration in vivo. Brain 2014, 137, 1496–1513. [Google Scholar] [CrossRef]
- Scott, D.; Roy, S. α-Synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. J. Neurosci. 2012, 32, 10129–10135. [Google Scholar] [CrossRef] [Green Version]
- Volpicelli-Daley, L.A.; Luk, K.C.; Patel, T.P.; Tanik, S.A.; Riddle, D.M.; Stieber, A.; Meaney, D.F.; Trojanowski, J.Q.; Lee, V.M.-Y. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 2011, 72, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.; Kehm, V.M.; Li, C.; Trojanowski, J.Q.; Lee, V.M.-Y. Forebrain overexpression of α-synuclein leads to early postnatal hippocampal neuron loss and synaptic disruption. Exp. Neurol. 2010, 221, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Bridi, J.C.; Bereczki, E.; Smith, S.K.; Poças, G.M.; Kottler, B.; Domingos, P.M.; Elliott, C.J.; Aarsland, D.; Hirth, F. Presynaptic accumulation of α-synuclein causes synaptopathy and progressive neurodegeneration in Drosophila. Brain Commun. 2021, 3, fcab049. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.-G.; Kim, M.J.; Kim, D.-G.; Yu, R.; Jang, Y.-N.; Oh, W.-J. Sequestration of synaptic proteins by α-synuclein aggregates leading to neurotoxicity is inhibited by small peptide. PLoS ONE 2018, 13, e0195339. [Google Scholar] [CrossRef]
- Lim, Y.; Kehm, V.M.; Lee, E.B.; Soper, J.H.; Li, C.; Trojanowski, J.Q.; Lee, V.M.-Y. α-Syn suppression reverses synaptic and memory defects in a mouse model of dementia with Lewy bodies. J. Neurosci. 2011, 31, 10076–10087. [Google Scholar] [CrossRef] [PubMed]
- Wihan, J.; Grosch, J.; Kalinichenko, L.S.; Müller, C.P.; Winkler, J.; Kohl, Z. Layer-specific axonal degeneration of serotonergic fibers in the prefrontal cortex of aged A53T α-synuclein–expressing mice. Neurobiol. Aging 2019, 80, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Agliardi, C.; Meloni, M.; Guerini, F.R.; Zanzottera, M.; Bolognesi, E.; Baglio, F.; Clerici, M. Oligomeric α-Syn and SNARE complex proteins in peripheral extracellular vesicles of neural origin are biomarkers for Parkinson’s disease. Neurobiol. Dis. 2021, 148, 105185. [Google Scholar] [CrossRef]
- Bereczki, E.; Francis, P.T.; Howlett, D.; Pereira, J.B.; Höglund, K.; Bogstedt, A.; Cedazo-Minguez, A.; Baek, J.-H.; Hortobágyi, T.; Attems, J. Synaptic proteins predict cognitive decline in Alzheimer’s disease and Lewy body dementia. Alzheimer’s Dement. 2016, 12, 1149–1158. [Google Scholar] [CrossRef]
- Kohl, Z.; Abdallah, N.B.; Vogelgsang, J.; Tischer, L.; Deusser, J.; Amato, D.; Anderson, S.; Müller, C.P.; Riess, O.; Masliah, E. Severely impaired hippocampal neurogenesis associates with an early serotonergic deficit in a BAC α-synuclein transgenic rat model of Parkinson’s disease. Neurobiol. Dis. 2016, 85, 206–217. [Google Scholar] [CrossRef]
- Prots, I.; Grosch, J.; Brazdis, R.-M.; Simmnacher, K.; Veber, V.; Havlicek, S.; Hannappel, C.; Krach, F.; Krumbiegel, M.; Schütz, O. α-Synuclein oligomers induce early axonal dysfunction in human iPSC-based models of synucleinopathies. Proc. Natl. Acad. Sci. USA 2018, 115, 7813–7818. [Google Scholar] [CrossRef] [Green Version]
- Tang, Q.; Gao, P.; Arzberger, T.; Höllerhage, M.; Herms, J.; Höglinger, G.; Koeglsperger, T. Alpha-Synuclein defects autophagy by impairing SNAP29-mediated autophagosome-lysosome fusion. Cell Death Dis. 2021, 12, 854. [Google Scholar] [CrossRef] [PubMed]
- Bereczki, E.; Branca, R.M.; Francis, P.T.; Pereira, J.B.; Baek, J.-H.; Hortobágyi, T.; Winblad, B.; Ballard, C.; Lehtiö, J.; Aarsland, D. Synaptic markers of cognitive decline in neurodegenerative diseases: A proteomic approach. Brain 2018, 141, 582–595. [Google Scholar] [CrossRef] [PubMed]
- Fonseca-Ornelas, L.; Viennet, T.; Rovere, M.; Jiang, H.; Liu, L.; Nuber, S.; Ericsson, M.; Arthanari, H.; Selkoe, D.J. Altered conformation of α-synuclein drives dysfunction of synaptic vesicles in a synaptosomal model of Parkinson’s disease. Cell Rep. 2021, 36, 109333. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, A.A.; Ingrassia, A.; de Menezes, R.X.; van Kesteren, R.E.; Rozemuller, A.J.; Heutink, P.; van de Berg, W.D. Evidence for immune response, axonal dysfunction and reduced endocytosis in the substantia nigra in early stage Parkinson’s disease. PLoS ONE 2015, 10, e0128651. [Google Scholar]
- Vallortigara, J.; Whitfield, D.; Quelch, W.; Alghamdi, A.; Howlett, D.; Hortobágyi, T.; Johnson, M.; Attems, J.; O’Brien, J.T.; Thomas, A. Decreased levels of VAMP2 and monomeric alpha-synuclein correlate with duration of dementia. J. Alzheimer’s Dis. 2016, 50, 101–110. [Google Scholar] [CrossRef]
- Kramer, M.L.; Schulz-Schaeffer, W.J. Presynaptic α-synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J. Neurosci. 2007, 27, 1405–1410. [Google Scholar] [CrossRef]
- Spinelli, K.J.; Taylor, J.K.; Osterberg, V.R.; Churchill, M.J.; Pollock, E.; Moore, C.; Meshul, C.K.; Unni, V.K. Presynaptic alpha-synuclein aggregation in a mouse model of Parkinson’s disease. J. Neurosci. 2014, 34, 2037–2050. [Google Scholar] [CrossRef]
- Hoffmann, C.; Sansevrino, R.; Morabito, G.; Logan, C.; Vabulas, R.M.; Ulusoy, A.; Ganzella, M.; Milovanovic, D. Synapsin condensates recruit alpha-synuclein. J. Mol. Biol. 2021, 433, 166961. [Google Scholar] [CrossRef]
- Zaltieri, M.; Grigoletto, J.; Longhena, F.; Navarria, L.; Favero, G.; Castrezzati, S.; Colivicchi, M.A.; Della Corte, L.; Rezzani, R.; Pizzi, M. α-synuclein and synapsin III cooperatively regulate synaptic function in dopamine neurons. J. Cell Sci. 2015, 128, 2231–2243. [Google Scholar] [CrossRef]
- Bate, C.; Gentleman, S.; Williams, A. α-synuclein induced synapse damage is enhanced by amyloid-β1-42. Mol. Neurodegener. 2010, 5, 55. [Google Scholar] [CrossRef]
- Games, D.; Valera, E.; Spencer, B.; Rockenstein, E.; Mante, M.; Adame, A.; Patrick, C.; Ubhi, K.; Nuber, S.; Sacayon, P. Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson’s disease-like models. J. Neurosci. 2014, 34, 9441–9454. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, D.R.; Vallortigara, J.; Alghamdi, A.; Howlett, D.; Hortobágyi, T.; Johnson, M.; Attems, J.; Newhouse, S.; Ballard, C.; Thomas, A.J. Assessment of ZnT3 and PSD95 protein levels in Lewy body dementias and Alzheimer’s disease: Association with cognitive impairment. Neurobiol. Aging 2014, 35, 2836–2844. [Google Scholar] [CrossRef] [PubMed]
- Dawson, T.M.; Ko, H.S.; Dawson, V.L. Genetic animal models of Parkinson’s disease. Neuron 2010, 66, 646–661. [Google Scholar] [CrossRef] [PubMed]
- Warner, T.T.; Schapira, A.H. Genetic and environmental factors in the cause of Parkinson’s disease. Ann. Neurol. 2003, 53, S16–S25. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.; Bemelmans, A.P.; Josephine, C.; Brouillet, E.; McKernan, D.P.; Dowd, E. Time-Course of Alterations in the Endocannabinoid System after Viral-Mediated Overexpression of alpha-Synuclein in the Rat Brain. Molecules 2022, 27, 507. [Google Scholar] [CrossRef] [PubMed]
- McCabe, K.; Concannon, R.M.; McKernan, D.P.; Dowd, E. Time-course of striatal Toll-like receptor expression in neurotoxic, environmental and inflammatory rat models of Parkinson’s disease. J. Neuroimmunol. 2017, 310, 103–106. [Google Scholar] [CrossRef]
- Olsen, L.K.; Cairns, A.G.; Aden, J.; Moriarty, N.; Cabre, S.; Alamilla, V.R.; Almqvist, F.; Dowd, E.; McKernan, D.P. Viral mimetic priming enhances alpha-synuclein-induced degeneration: Implications for Parkinson’s disease. Brain Behav. Immun. 2019, 80, 525–535. [Google Scholar] [CrossRef]
- Cardinale, A.; Calabrese, V.; de Iure, A.; Picconi, B. Alpha-synuclein as a prominent actor in the inflammatory synaptopathy of parkinson’s disease. Int. J. Mol. Sci. 2021, 22, 6517. [Google Scholar] [CrossRef]
- Prots, I.; Veber, V.; Brey, S.; Campioni, S.; Buder, K.; Riek, R.; Böhm, K.J.; Winner, B. α-Synuclein oligomers impair neuronal microtubule-kinesin interplay. J. Biol. Chem. 2013, 288, 21742–21754. [Google Scholar] [CrossRef]
- Liu, Y.; Yuan, Y.-H.; Sun, J.-D.; Li, J.; Li, Z.-P.; Chen, N.-H. Nigrostriatal dynein changes in A53T alpha-synuclein transgenic mice. F1000Research 2014, 3, 68. [Google Scholar] [CrossRef]
- Oikawa, T.; Nonaka, T.; Terada, M.; Tamaoka, A.; Hisanaga, S.-i.; Hasegawa, M. α-Synuclein fibrils exhibit gain of toxic function, promoting tau aggregation and inhibiting microtubule assembly. J. Biol. Chem. 2016, 291, 15046–15056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Hoffman, P.N.; Stirling, W.; Price, D.L.; Lee, M.K. Axonal transport of human α-synuclein slows with aging but is not affected by familial Parkinson’s disease-linked mutations. J. Neurochem. 2004, 88, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Khoshaghideh, F.; Lee, S.; Lee, S.J. Impairment of microtubule-dependent trafficking by overexpression of α-synuclein. Eur. J. Neurosci. 2006, 24, 3153–3162. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Jin, J.; Davis, J.; Zhou, Y.; Wang, Y.; Liu, J.; Lockhart, P.J.; Zhang, J. Oligomeric α-synuclein inhibits tubulin polymerization. Biochem. Biophys. Res. Commun. 2007, 356, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. J. Clin. Epidemiol. 2021, 134, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Pluhackova, K.; Böckmann, R.A. The multifaceted role of SNARE proteins in membrane fusion. Front. Physiol. 2017, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Tarsa, L.; Goda, Y. Synaptophysin regulates activity-dependent synapse formation in cultured hippocampal neurons. Proc. Natl. Acad. Sci. USA 2002, 99, 1012–1016. [Google Scholar] [CrossRef] [PubMed]
- Mirza, F.J.; Zahid, S. The role of synapsins in neurological disorders. Neurosci. Bull. 2018, 34, 349–358. [Google Scholar] [CrossRef]
- Ciruelas, K.; Marcotulli, D.; Bajjalieh, S.M. Synaptic vesicle protein 2: A multi-faceted regulator of secretion. Semin. Cell Dev. Biol. 2019, 95, 130–141. [Google Scholar] [CrossRef]
- Coley, A.A.; Gao, W.-J. PSD95: A synaptic protein implicated in schizophrenia or autism? Prog. Neuro Psychopharmacol. Biol. Psychiatry 2018, 82, 187–194. [Google Scholar] [CrossRef]
- Zhong, L.; Cherry, T.; Bies, C.E.; Florence, M.A.; Gerges, N.Z. Neurogranin enhances synaptic strength through its interaction with calmodulin. EMBO J. 2009, 28, 3027–3039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gitler, A.D.; Bevis, B.J.; Shorter, J.; Strathearn, K.E.; Hamamichi, S.; Su, L.J.; Caldwell, K.A.; Caldwell, G.A.; Rochet, J.-C.; McCaffery, J.M. The Parkinson’s disease protein α-synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Zhang, Y.; Iqbal, J.; Ke, M.; Wang, Y.; Li, Y.; Qing, H.; Deng, Y. Differential expression of synaptic proteins in unilateral 6-OHDA lesioned rat model—A comparative proteomics approach. Proteomics 2014, 14, 1808–1819. [Google Scholar] [CrossRef] [PubMed]
- Diógenes, M.J.; Dias, R.B.; Rombo, D.M.; Miranda, H.V.; Maiolino, F.; Guerreiro, P.; Näsström, T.; Franquelim, H.G.; Oliveira, L.M.; Castanho, M.A. Extracellular alpha-synuclein oligomers modulate synaptic transmission and impair LTP via NMDA-receptor activation. J. Neurosci. 2012, 32, 11750–11762. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, T.J.; Harrison, P.M.; Richardson, M.J.; Pinheiro, T.J.; Wall, M.J. Intracellular soluble α-synuclein oligomers reduce pyramidal cell excitability. J. Physiol. 2016, 594, 2751–2772. [Google Scholar] [CrossRef] [PubMed]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S. In vivo demonstration that α-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef]
- Maheshwary, A.; Mohite, D.; Omole, J.A.; Bhatti, K.S.; Khan, S. Is Deep Brain Stimulation Associated With Detrimental Effects on Cognitive Functions in Patients of Parkinson’s Disease? A Systematic Review. Cureus 2020, 12, e9688. [Google Scholar] [CrossRef]
- Goedert, M. Alpha-synuclein and neurodegenerative diseases. Nat. Rev. Neurosci. 2001, 2, 492–501. [Google Scholar] [CrossRef]
- Burke, R.E.; O’Malley, K. Axon degeneration in Parkinson’s disease. Exp. Neurol. 2013, 246, 72–83. [Google Scholar] [CrossRef]
- Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F. α-Synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chutna, O.; Gonçalves, S.; Villar-Piqué, A.; Guerreiro, P.; Marijanovic, Z.; Mendes, T.; Ramalho, J.; Emmanouilidou, E.; Ventura, S.; Klucken, J. The small GTPase Rab11 co-localizes with α-synuclein in intracellular inclusions and modulates its aggregation, secretion and toxicity. Hum. Mol. Genet. 2014, 23, 6732–6745. [Google Scholar] [CrossRef] [PubMed]
- Yoo, G.; Yeou, S.; Son, J.B.; Shin, Y.-K.; Lee, N.K. Cooperative inhibition of SNARE-mediated vesicle fusion by α-synuclein monomers and oligomers. Sci. Rep. 2021, 11, 10955. [Google Scholar] [CrossRef]
- Das, G.; Shravage, B.V.; Baehrecke, E.H. Regulation and function of autophagy during cell survival and cell death. Cold Spring Harb. Perspect. Biol. 2012, 4, a008813. [Google Scholar] [CrossRef] [PubMed]
- Maxfield, F.R. Role of endosomes and lysosomes in human disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016931. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| Protein | Effect | Quantity of Change | Sample | Model | Technique | Reference |
|---|---|---|---|---|---|---|
| SNAP-25 | No change | Mice | mThy-1 α-syn E57K Tg mouse and mThy-1 α-syn WT mouse models | WB | [29] | |
| Increased ↑ | 12% | Mice | Overexpression of α-syn in cultured mouse hippocampal neurons | IHC | [30] | |
| Reduced ↓ | Rats | Primary hippocampal neuronal culture treated with α-syn-hWT PFFs | WB | [31] | ||
| No change | Mice | Truncated human α-syn, strains (1-120) and (1-120E) expressed in Tg mice | WB | [17] | ||
| Redistributed | Human | Post-mortem tissue from the straitum of PD patients | IHC | [17] | ||
| Redistributed | Mice | Truncated human α-syn, strains (1-120) and (1-120E) expressed in Tg mice | IHC | [17] | ||
| No change | Mice | Forebrain-specific conditional Tg mice overexpressing human WT or A53T | WB | [32] | ||
| No change | Drosophila flies | Drosophila model expressing human α-syn | IHC | [33] | ||
| Reduced ↓ | Human | DA-induced oligomeric α-syn aggregates in human neuroblastoma SH-SY5Y | WB | [34] | ||
| Reduced ↓ | Rats | Primary cultured neurons treated with α-syn aggregates | WB | [34] | ||
| No change | Mice | A53T mutant α-syn were expressed in Tg mice models of PDD and DLB | IHC | [35] | ||
| No change | Mice | Tg mice expressing the A53T mutant α-syn under the PDGFb promoter | WB | [36] | ||
| No change | Human | NDEs from peripheral blood of PD patients | WB | [37] | ||
| Reduced ↓ | 24–42% | Human | Post-mortem tissue from the PFC, BA21, BA24, and BA40 of PDD and DLB patients | WB | [38] | |
| Reduced ↓ | 15–48% | Human | Post-mortem tissue from the PFC, BA21, BA24, and BA40 of PDD and DLB patients | ELISA | [38] | |
| No change | Rats | BAC Tg rat model overexpressing the full-length human SNCA | WB | [39] | ||
| Reduced ↓ | Human | α-syn oligomer-forming mutants E46K and E57K and WT α-syn expressed in human iPSC-derived neurons | WB | [40] | ||
| No change | Mice | Overexpression of human WT α-syn in Tg mice | WB | [18] | ||
| SNAP-29 | Reduced ↓ | Human | Post-mortem tissue from the SNpc of PD patients | IHC | [41] | |
| Reduced ↓ | Human | α-syn transduced LUHMES cells via adenoviruses serotype 5 (AV5) | WB | [41] | ||
| SNAP-23 | Reduced ↓ | Human | α-syn transduced LUHMES cells via adenoviruses serotype 5 (AV5) | WB | [41] | |
| SNAP-47 | Reduced ↓ | 24% | Human | Post-mortem tissue from the PFC of PD patients | ELISA | [42] |
| VAMP2 | Reduced ↓ | 31% | Mice | Overexpression of α-syn in cultured mouse hippocampal neurons | IHC | [30] |
| No change | Mice | Overexpression of human WT α-syn in Tg mice | WB | [18] | ||
| Reduced ↓ | Mice | Primary hippocampal neuronal culture treated with α-syn PPFs | WB | [31] | ||
| Redistributed | Mice | Truncated human α-syn, strains (1-120) and (1-120E) expressed in Tg mice | IHC | [17] | ||
| Reduced ↓ | Mice | A53T mutant α-syn were expressed in Tg mice models of PDD and DLB using the CaMKII promoter | IHC | [35] | ||
| Undetectable | Mice | Hippocampal neurons of Tg mice overexpressing human α-syn:GFP | IHC | [19] | ||
| Reduced ↓ | 30% | Mice | Hippocampal neurons of Tg mice overexpressing human α-syn:GFP | IHC | [19] | |
| Reduced ↓ | 30% | Human | NDEs from peripheral blood of PD patients | WB | [37] | |
| No change | Drosophila flies | Drosophila model expressing human α-syn | IHC | [33] | ||
| Reduced ↓ | Human | DA-induced oligomeric α-syn aggregates in human neuroblastoma SH-SY5Y | WB | [34] | ||
| Reduced ↓ | Rats | Primary cultured neurons treated with α-syn aggregates | WB | [34] | ||
| Reduced ↓ | Mice | Tg mice expressing 3K α-syn mutation | WB | [43] | ||
| Reduced ↓ | Human | Post-mortem tissue from the SN of PD patients (Braak stages 5–6) | QPCR | [44] | ||
| Reduced ↓ | Human | Post-mortem tissue from the PFC of DLB patients | WB | [45] | ||
| Syntaxin | Reduced ↓ | Rats | Nigral injection of AAv2-A53T α-syn | WB | [16] | |
| Reduced ↓ | Mice | Forebrain-specific conditional Tg mice overexpressing human WT or A53T mutant α-syn | WB | [32] | ||
| Redistributed | Mice | A53T mutant α-syn were expressed in Tg mice models of PDD and DLB using the CaMKII promoter | IHC | [35] | ||
| No change | Human | α-syn was overexpressed in LUHMES cells via adenoviruses serotype 5 (AV5) | WB | [41] | ||
| Reduced ↓ | Drosophila flies | Drosophila model expressing human α-syn | WB | [33] | ||
| Reduced ↓ | 57% | Human | iPSCs from patients with the A53T α-syn mutation | WB | [46] | |
| Synataxin 1 | No change | Mice | Truncated human α-syn, strains (1-120) and (1-120E) expressed in Tg mice | WB | [17] | |
| Reduced ↓ | Human | Post-mortem tissue from the PFC of DLB patients | WB | [45] | ||
| Redistributed | Human | Post-mortem tissue from the striatum of PD patients | IHC | [17] | ||
| Redistributed | Mice | Truncated human α-syn, strains (1-120) and (1-120E) expressed in Tg mice | IHC | [17] | ||
| Reduced | Mice | Tg mice expressing 3K α-syn mutation | WB | [43] | ||
| Reduced | Human | Post-mortem tissue from the SN of PD patients (Braak stages 5-6) | QPCR | [44] | ||
| No change | Mice | Primary hippocampal neuronal culture treated with α-syn PFFs | WB | [31] | ||
| No change | Mice | Overexpression of human WT α-syn in Tg mice | WB | [18] | ||
| Syntaxin 1a | No change | Human | DA-induced oligomeric α-syn aggregates in human neuroblastoma SH-SY5Y | WB | [34] | |
| No change | Rats | Primary cultured neurons treated with α-syn aggregates | WB | [34] | ||
| Reduced ↓ | 20% | Human | NDEs from peripheral blood of PD patients | WB | [37] | |
| Synaptotagmin | No change | Mice | Overexpression of human WT α-syn in Tg mice | WB | [18] | |
| Reduced ↓ | Mice | A53T mutant α-syn were expressed in Tg mice models of PDD and DLB using the CaMKII promoter | IHC | [35] | ||
| No change | Drosophila flies | Drosophila model expressing human α-syn | IHC | [33] | ||
| Synaptotagmin 1 | No change | Human | DA-induced oligomeric α-syn aggregates in human neuroblastoma SH-SY5Y | WB | [34] | |
| Synaptotagmin 2 | Reduced ↓ | 19% | Human | Post-mortem tissue from the PFC of PD patients | WB | [42] |
| Reduced ↓ | 24% | Human | Post-mortem tissue from the PFC of DLB patients | WB | [42] | |
| Increased ↑ | Human | Post-mortem tissue from the SN of PD patients (Braak stages 1–2) | QPCR | [44] | ||
| SM proteins | No change | Mice | Overexpression of human WT α-syn in Tg mice | WB | [18] | |
| Complexin | Reduced ↓ | Mice | Tg mice expressing 3K α-syn mutation | WB | [43] | |
| Complexin 2 | Reduced ↓ | Human | Post-mortem tissue from the SN of PD patients (Braak stages 5–6) | QPCR | [44] | |
| Reduced ↓ | Mice | Overexpression of human WT α-syn in Tg mice | WB | [18] | ||
| Synapsin | Undetectable | Human | Post-mortem tissue from the frontal cortical sections of the brain from DLB patients | IHC | [19] | |
| Reduced ↓ | Drosophila flies | Drosophila model expressing human α-syn | IHC | [33] | ||
| Reduced ↓ | Drosophila flies | Drosophila model expressing human α-syn | WB | [33] | ||
| Reduced ↓ | Mice | GFP-tagged human α-syn was overexpressed in Tg mice | IHC | [47] | ||
| Reduced ↓ | Mice | Forebrain-specific conditional Tg mice overexpressing human WT or A53T mutant α-syn | WB | [32] | ||
| Reduced rate ↓ | Mice | Human α-syn transduced via pET17b vector into hippocampal neurons to assess synapsin condensate formation | WB | [48] | ||
| Synapsins | Reduced ↓ | Mice | Overexpression of human WT α-syn in Tg mice | WB | [18] | |
| Synapsin I | Reduced ↓ | 30–50% | Mice | mThy-1 α-syn E57K Tg mouse and mThy-1 α-syn WT mouse model | WB | [29] |
| No change | Human | Human neuroblastoma SH-SY5Y cells expressing human α-syn | WB | [49] | ||
| No change | Mice | Primary embryonic mouse ventral mesencephalic cells expressing aggregated α-syn via glucose deprivation | WB | [49] | ||
| Reduced ↓ | Mice | A53T mutant α-syn expressed in Tg mice models of PDD and DLB using the CaMKII promoter | IHC | [35] | ||
| Undetectable | Mice | Hippocampal neurons of Tg mice overexpressing human α-syn:GFP | IHC | [19] | ||
| Reduced ↓ | 51% | Mice | Hippocampal neurons of Tg mice overexpressing human WT α-syn:GFP | IHC | [19] | |
| Reduced ↓ | Human | α-syn oligomer-forming mutants E46K and E57K and WT α-syn expressed in human iPSC-derived neurons | WB | [40] | ||
| Reduced ↓ | 43% | Mice | Overexpression of α-syn in cultured mouse hippocampal neurons | IHC | [30] | |
| No change | Mice | Tg mice expressing the A53T mutant α-syn under the PDGFb promoter | WB | [36] | ||
| Reduced ↓ | Human | Post-mortem tissue from the SN of PD patients (Braak stages 5–6) | QPCR | [44] | ||
| Reduced ↓ | Rats | BAC Tg rat model overexpressing the full-length human SNCA | WB | [39] | ||
| Synapsin II | Reduced ↓ | Mice | Primary hippocampal neuronal culture treated with α-syn PFFs | WB | [31] | |
| Synapsin III | Increased ↑ | Human | Post-mortem tissue from the caudate and putamen of PD patients | IHC | [49] | |
| Increased ↑ | Mice | Primary embryonic mouse ventral mesencephalic cells expressing aggregated α-syn via glucose deprivation | WB | [49] | ||
| Increased ↑ | Human | Human neuroblastoma SH-SY5Y cells expressing human α-syn | WB | [49] | ||
| Redistributed | Mice | Expression of a C-terminally truncated form of human α-syn (1–120) in Tg mice | IHC | [49] | ||
| Redistributed | Mice | Primary embryonic mouse ventral mesencephalic cells expressing aggregated α-syn via glucose deprivation | IHC | [49] | ||
| Increased ↑ | Human | Post-mortem tissue from the SN of PD patients (Braak stages 1–2) | QPCR | [44] | ||
| Synaptophysin | Reduced ↓ | 45% | Mice | Overexpression of α-syn in cultured mouse hippocampal neurons | IHC | [30] |
| No change | mice | Overexpression of human WT α-syn in Tg mice | WB | [18] | ||
| Reduced ↓ | Mice | A53T mutant α-syn were expressed in Tg mice models of PDD and DLB using the CaMKII promoter | IHC | [35] | ||
| Reduced ↓ | Mice | Tg mice expressing 3K α-syn mutation | WB | [43] | ||
| Reduced ↓ | 50% | Mice | Cultured cortical neurons incubated with α-syn (500 nM) | ELISA | [50] | |
| Reduced ↓ | 80% | Mice | Cultured cortical neurons incubated with α-syn (10 uM) | ELISA | [50] | |
| Reduced ↓ | Mice | Hippocampal neurons with added α-syn | ELISA | [50] | ||
| Reduced ↓ | Mice | Forebrain-specific conditional Tg mice overexpressing human WT or A53T mutant α-syn | WB | [32] | ||
| Reduced ↓ | Human | α-syn oligomer-forming mutants E46K and E57K and WT α-syn expressed in human iPSC-derived neurons | WB | [40] | ||
| No change | Mice | Tg mice expressing the A53T mutant α-syn under the PDGFb promoter | WB | [36] | ||
| Increased ↑ | Human | Post-mortem tissue from the SN of PD patients (Braak stages 5–6) | QPCR | [44] | ||
| Increased ↑ | Human | Post-mortem tissue from the SN of PD patients (Braak stages 1–2) | QPCR | [44] | ||
| Reduced ↓ | Mice | Overexpression of human α-syn under the mThy1 promoter | IHC | [51] | ||
| No change | Rats | BAC Tg rat model overexpressing the full-length human SNCA | WB | [39] | ||
| Reduced ↓ | 52% | Human | iPSCs from patients with the A53T α-syn mutation | WB | [46] | |
| Reduced ↓ | 20% | Mice | mThy-1 α-syn E57K Tg mouse model | WB | [29] | |
| Reduced ↓ | Human | Post-mortem tissue from the PFC of PDD patients | WB | [45] | ||
| No change | Mice | Primary hippocampal neuronal culture treated with α-syn PFFS | WB | [31] | ||
| SV2C | Reduced ↓ | 24% | Human | Post-mortem tissue from the PFC of DLB patients | ELISA | [42] |
| SV2 | No change | Mice | Overexpression of human WT α-syn in Tg mice | WB | [18] | |
| Amphiphysin | Reduced ↓ | 30% | Mice | Overexpression of α-syn in cultured mouse hippocampal neurons | IHC | [30] |
| Reduced ↓ | 17% | Mice | Hippocampal neurons of Tg mice overexpressing human WT α-syn:GFP | IHC | [19] | |
| Undetectable | Mice | Hippocampal neurons of Tg mice overexpressing human WT α-syn:GFP | IHC | [19] | ||
| Reduced ↓ | Mice | Tg mice expressing 3K α-syn mutation | WB | [43] | ||
| GAP43 | Reduced ↓ | 20% | Human | Post-mortem tissue from the PFC of PDD patients | ELISA | [42] |
| Rabphilin 3A | Reduced ↓ | Rats | Nigral injection of AAv2-A53T α-syn | WB | [16] | |
| PSD-95 * | Reduced ↓ | Mice | Forebrain-specific conditional Tg mice overexpressing the A53T mutant α-syn | WB | [32] | |
| Reduced ↓ | Mice | Tg mice expressing 3K α-syn mutation | WB | [43] | ||
| No change | Mice | Tg mice expressing the A53T mutant α-syn under the PDGFb promoter | WB | [36] | ||
| Reduced ↓ | Mice | Overexpression of human α-syn under the mThy1 promoter | IHC | [51] | ||
| Increased ↑ | Rats | BAC Tg rat model overexpressing the full-length human SNCA | WB | [39] | ||
| Reduced ↓ | 48% | Human | iPSCs from patients with the A53T α-syn mutation | WB | [46] | |
| Reduced ↓ | 28% | Human | Post-mortem tissue from the PFC, BA24, and BA40 of PDD patients | WB | [52] | |
| Reduced ↓ | 17% | Human | Post-mortem tissue from the PFC, BA24, and BA40 of DLB patients | WB | [52] | |
| Drebrin * | Undetectable | Human | iPSCs from patients with the A53T α-syn mutation | WB | [46] | |
| Reduced ↓ | Mice | Forebrain-specific conditional Tg mice overexpressing the A53T mutant α-syn | WB | [32] | ||
| Neurogranin * | Reduced ↓ | 21–38% | Human | Post-mortem tissue from the PFC, BA21, BA24, and BA40 of PDD and DLB patients | ELISA | [38] |
| Reduced ↓ | 30–51% | Human | Post-mortem tissue from the PFC, BA21, BA24, and BA40 of PDD and DLB patients | WB | [38] |
| Protein | Effect | Sample | Model | Technique | Authors |
|---|---|---|---|---|---|
| Kinesin | Reduced | Human | Post-mortem tissue from the SN of PD patients | IHC | [25] |
| Reduced | Rat | Overexpression via injection of rAAV-h-A30P α-syn in SN of rats | IHC | [25] | |
| Kinesin 1 | Reduced | Human | E46K and E57K oligomer forming mutant α-syn expressed in human iPSC-derived neurons | WB | [40] |
| Kinesin light chain | Reduced | Human | Post-mortem tissue from the SN of PD and iLBD patients (Braak stage 1–2) | QPCR | [44] |
| Kinesin Family 20A | Reduced | Human | Post-mortem tissue from the SN of PD and iLBD patients (Braak stage 1–2) | QPCR | [44] |
| KIF1A | Reduced | Rat | Injection of AAv2-A53T α-syn in striatum of rats (8 weeks) | WB | [16] |
| Increased | Rat | Injection of AAv2-A53T α-syn in SN of rats | WB | [16] | |
| Increased | Mice | A53T mutant α-syn expressed in Tg under the PDGFb promoter | WB | [36] | |
| KIF1B | Reduced | Rat | Injection of AAv2-A53T α-syn in striatum of rats (8 weeks) | WB | [16] |
| KIF2A | Reduced | Rat | Injection of AAv2-A53T α-syn in striatum of rats (8 weeks) | WB | [16] |
| Increased | Rat | Injection of AAv2-A53T α-syn in SN of rats | WB | [16] | |
| KIF3A | Increased | Rat | Injection of AAv2-A53T α-syn in striatum of rats (4 weeks) | WB | [16] |
| Reduced | Rat | Injection of AAv2-A53T α-syn in striatum of rats (8 weeks) | WB | [16] | |
| KIF5 | Reduced | Human | α-syn E57K mutant oligomers overexpressed in LUHMES cells via lentiviral construct | IHC | [59] |
| Reduced | Human | WT α-syn seeds overexpressed in LUHMES cells via lentiviral construct | IHC | [59] | |
| No change | Rat | Injection of AAv2-A53T α-syn in striatum of rats (8 weeks) | WB | [16] | |
| KIF17 | Reduced | Rat | Injection of AAv2-A53T α-syn in striatum of rats (4 weeks) | WB | [16] |
| Dynactin1 | Increased | Rat | Injection of AAv2-A53T α-syn in striatum of rats (8 weeks) | WB | [16] |
| Dynamitin | No change | Rat | Injection of AAv2-A53T α-syn in striatum of rats (4 weeks) | WB | [16] |
| Increased | Rat | Injection of AAv2-A53T α-syn in striatum of rats (8 weeks) | WB | [16] | |
| Dynein | Reduced | Rat | Injection of AAv2-A53T α-syn in striatum of rats (4 weeks) | WB | [16] |
| Increased | Rat | Injection of AAv2-A53T α-syn in striatum of rats (8 weeks) | WB | [16] | |
| Reduced | Rat | Overexpression via injection of rAAV-h-A30P α-syn in SN of rats | IHC | [25] | |
| Unchanged | Mice | A53T mutant α-syn expressed in Tg mice under the PDGFb promoter | WB | [36] | |
| Decreased | Mice | Overexpression of A53T α-syn in striatum of Tg mice | WB | [60] | |
| Increased | Mice | Overexpression of A53T α-syn in SNpc of Tg mice | WB | [60] | |
| DYNLT3 | Reduced | Human | Post-mortem tissue from the SN of PD patients | IHC | [25] |
| Myosin Va | Increased | Rat | Injection of AAv2-A53T α-syn in striatum of rats (4 weeks) | WB | [16] |
| Reduced | Rat | Injection of AAv2-A53T α-syn in striatum of rats (8 weeks) | WB | [16] | |
| MT assembly (tau promoted) | Reduced | Cell free system | WT α-syn expressed in a cell free system (Escherichia coli). | EM | [59] |
| Cell free system | C-terminally truncated α-syn fibrils and protofibrils subcloned into pRK172 and expressed in a cell-free system (Escherichia coli BL21 (DE3)) | WB | [61] | ||
| MT gliding | Reduced | Cell free system | E57K and A30P oligomers of α-syn expressed in cell-free system (Escherichia coli). | M | [59] |
| Tubulin | Reduced | Human | α-synuclein E57K mutant oligomers overexpressed in LUHMES cells via lentiviral construct | IHC | [59] |
| Reduced | Human | Post-mortem tissue from the SN of PD and iLBD patients (Braak stage 5–6) | QPCR | [44] | |
| Reduced | Human | WT α-syn seeds overexpressed in LUHMES cells via lentiviral construct | IHC | [59] | |
| No change | Mice | Tg mice expressing mutant A53T α-syn | WB | [62] | |
| Redistributed | Human | Overexpression of α-syn via recombinant adenoviral vector in SH-SY5Y human neuroblastoma cells | IHC | [63] | |
| Tubulin (non-polymerised) | Increased | Mice | MES cell treated with oligomeric α-syn | WB | [64] |
| Neurofilament triplets * | No change | Mice | Tg mice expressing mutant A53T α-syn | WB | [62] |
| Actin | No change | Mice | Tg mice expressing mutant A53T α-syn | WB | [62] |
| Increased | Rat | Injection of AAv2-A53T α-syn in striatum of rats (4 weeks) | WB | [16] | |
| Increased | Rat | Injection of Aav2-A53T α-syn in striatum of rats (8 weeks) | WB | [16] | |
| Actin microfilaments | No change | Human | Overexpression of α-syn via recombinant adenoviral vector in SH-SY5Y human neuroblastoma cells | IHC | [63] |
| MTs (filamentous) | Reduced | Human | Overexpression of α-syn via recombinant adenoviral vector in SH-SY5Y human neuroblastoma cells | IHC | [63] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murphy, J.; McKernan, D.P. The Effect of Aggregated Alpha Synuclein on Synaptic and Axonal Proteins in Parkinson’s Disease—A Systematic Review. Biomolecules 2022, 12, 1199. https://doi.org/10.3390/biom12091199
Murphy J, McKernan DP. The Effect of Aggregated Alpha Synuclein on Synaptic and Axonal Proteins in Parkinson’s Disease—A Systematic Review. Biomolecules. 2022; 12(9):1199. https://doi.org/10.3390/biom12091199
Chicago/Turabian StyleMurphy, Jennifer, and Declan P. McKernan. 2022. "The Effect of Aggregated Alpha Synuclein on Synaptic and Axonal Proteins in Parkinson’s Disease—A Systematic Review" Biomolecules 12, no. 9: 1199. https://doi.org/10.3390/biom12091199
APA StyleMurphy, J., & McKernan, D. P. (2022). The Effect of Aggregated Alpha Synuclein on Synaptic and Axonal Proteins in Parkinson’s Disease—A Systematic Review. Biomolecules, 12(9), 1199. https://doi.org/10.3390/biom12091199