
Structural Basis of a Novel Agonistic Anti-OX40 Antibody

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Antibody Generation
2.3. Protein Expression and Purification
2.4. BLI Assay
2.5. Enzyme-Linked Immunosorbent Assay (ELISA) Assay
2.6. Luciferase Reporter Assay
2.7. CD4+ T Cell Proliferation Assay
2.8. MC38 Tumor Model in Mice
2.9. Structure Determination
2.10. Quantification and Statistical Analysis
3. Results
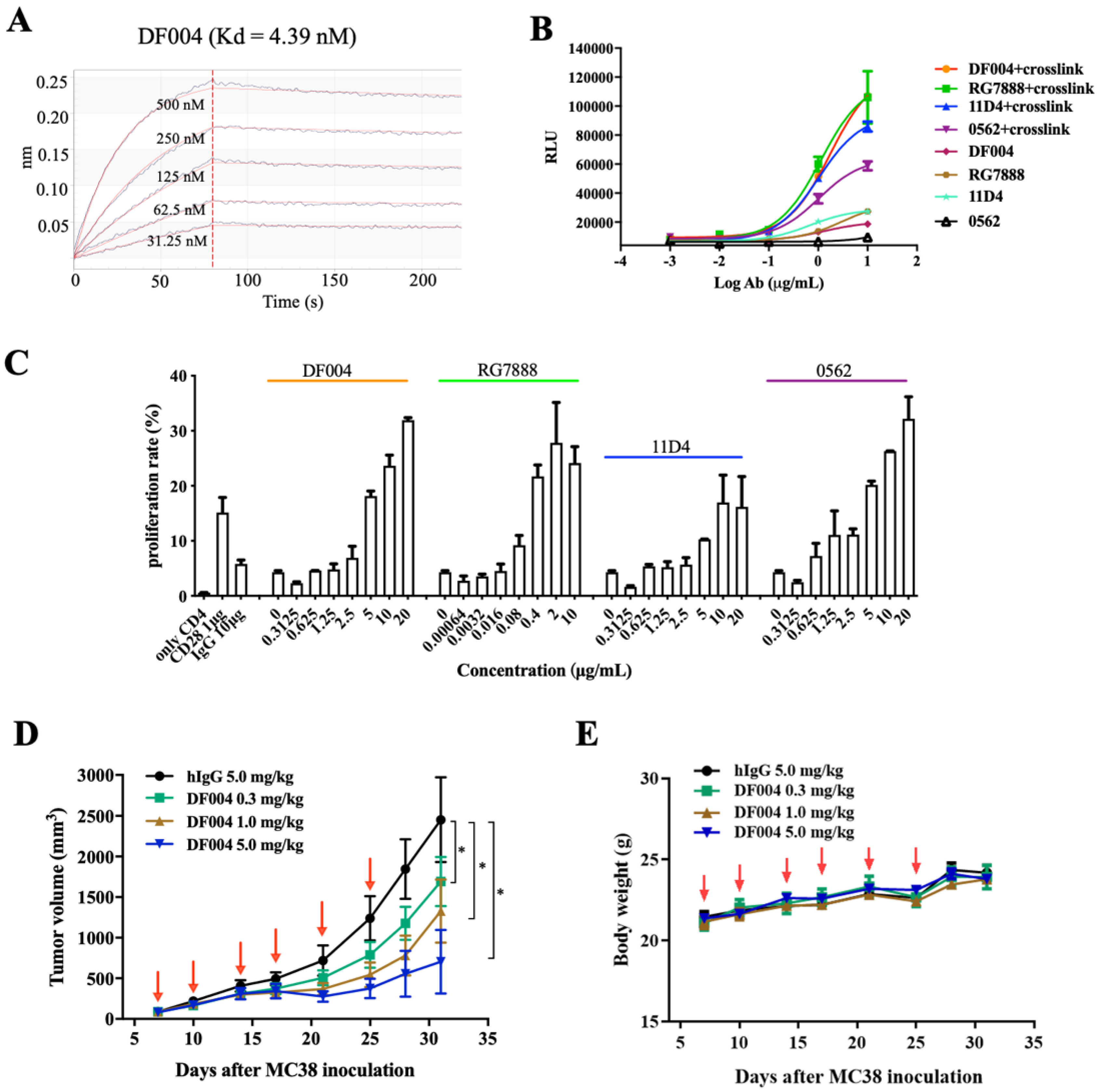
3.1. Generation and Characterization of An OX40 Antibody DF004
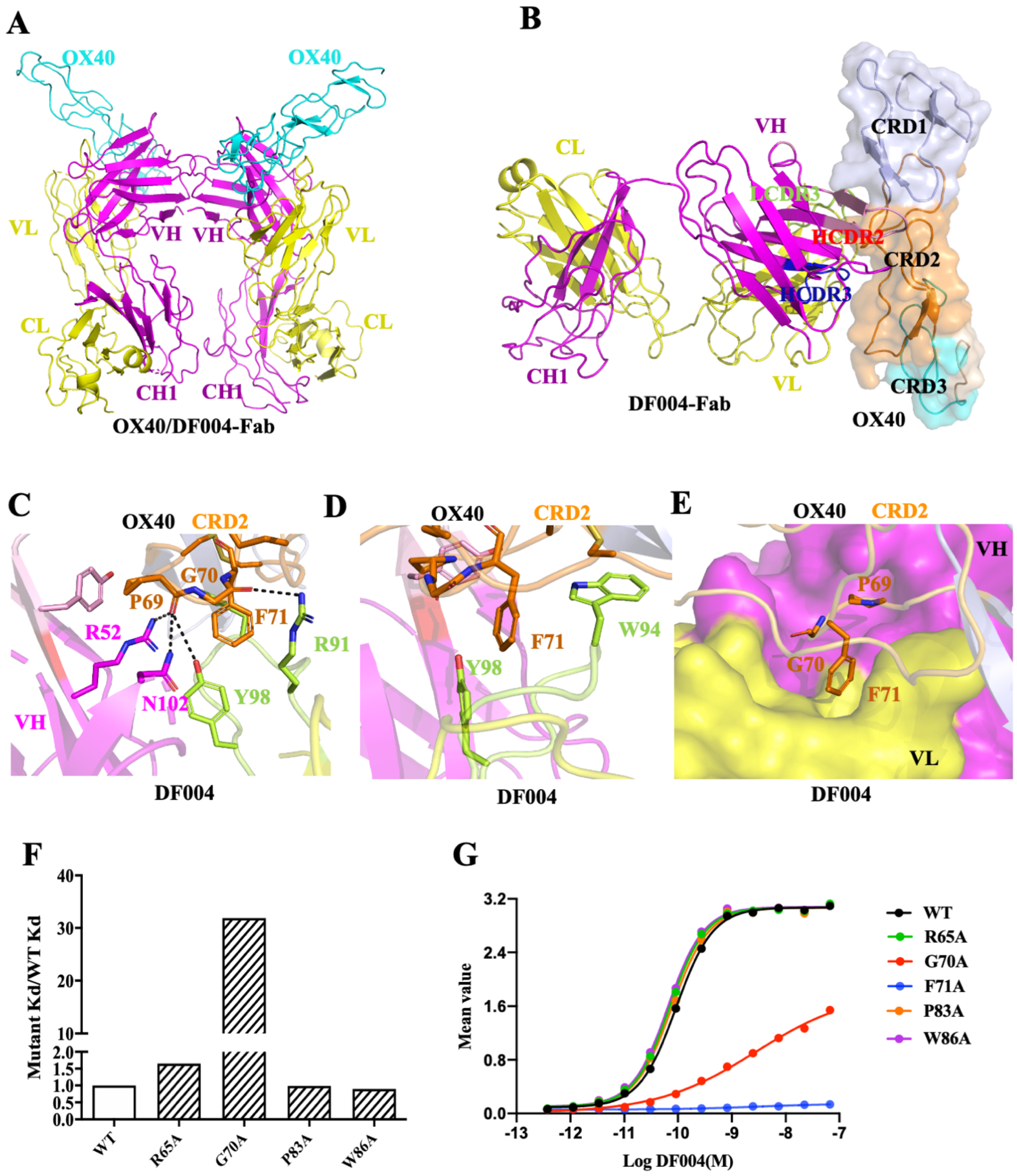
3.2. Structure of OX40/DF004-Fab Complex
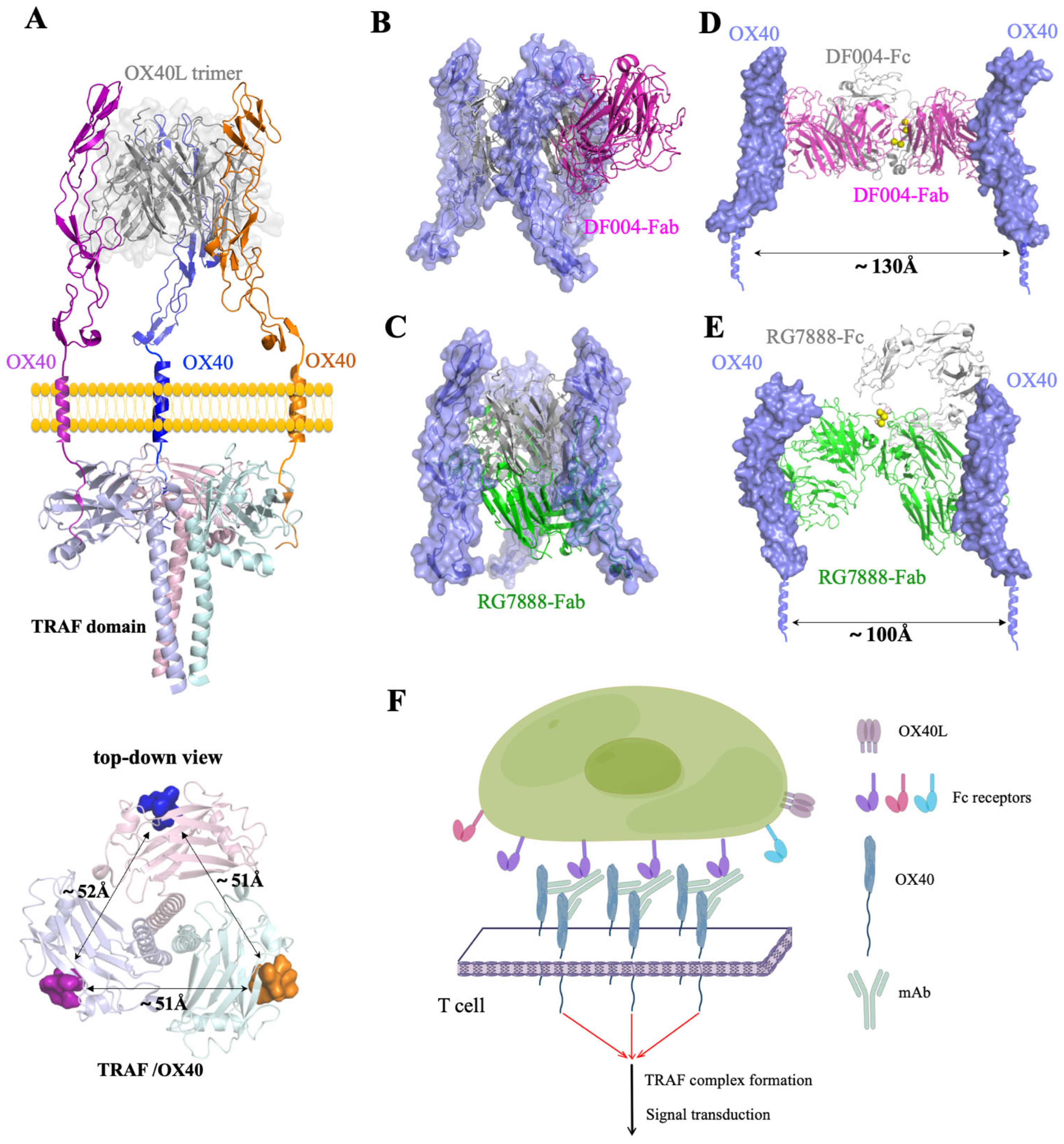
3.3. Comparison of OX40-Antibody Complex Structures
3.4. Mechanism of the Agonistic Activities
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Morad, G.; Helmink, B.A.; Sharma, P.; Wargo, J.A. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell 2021, 184, 5309–5337. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Swart, M.; Verbrugge, I.; Beltman, J.B. Combination Approaches with Immune-Checkpoint Blockade in Cancer Therapy. Front. Oncol. 2016, 6, 233. [Google Scholar] [CrossRef]
- Waight, J.D.; Gombos, R.B.; Wilson, N.S. Harnessing co-stimulatory TNF receptors for cancer immunotherapy: Current approaches and future opportunities. Hum. Antibodies 2017, 25, 87–109. [Google Scholar] [CrossRef]
- Croft, M. Co-stimulatory members of the TNFR family: Keys to effective T-cell immunity? Nat. Rev. Immunol. 2003, 3, 609–620. [Google Scholar] [CrossRef]
- Buchan, S.L.; Rogel, A.; Al-Shamkhani, A. The immunobiology of CD27 and OX40 and their potential as targets for cancer immunotherapy. Blood 2018, 131, 39–48. [Google Scholar] [CrossRef] [PubMed]
- So, T.; Song, J.; Sugie, K.; Altman, A.; Croft, M. Signals from OX40 regulate nuclear factor of activated T cells c1 and T cell helper 2 lineage commitment. Proc. Natl. Acad. Sci. USA 2006, 103, 3740–3745. [Google Scholar] [CrossRef] [PubMed]
- Croft, M. Costimulation of T cells by OX40, 4-1BB, and CD27. Cytokine Growth Factor Rev. 2003, 14, 265–273. [Google Scholar] [CrossRef]
- Croft, M. Control of immunity by the TNFR-related molecule OX40 (CD134). Annu. Rev. Immunol. 2010, 28, 57–78. [Google Scholar] [CrossRef]
- Aspeslagh, S.; Postel-Vinay, S.; Rusakiewicz, S.; Soria, J.C.; Zitvogel, L.; Marabelle, A. Rationale for anti-OX40 cancer immunotherapy. Eur. J. Cancer 2016, 52, 50–66. [Google Scholar] [CrossRef]
- Massarelli, E.; Lam, V.K.; Parra, E.R.; Rodriguez-Canales, J.; Behrens, C.; Diao, L.; Wang, J.; Blando, J.; Byers, L.A.; Yanamandra, N.; et al. High OX-40 expression in the tumor immune infiltrate is a favorable prognostic factor of overall survival in non-small cell lung cancer. J. Immunother. Cancer 2019, 7, 351. [Google Scholar] [CrossRef]
- Oberst, M.D.; Auge, C.; Morris, C.; Kentner, S.; Mulgrew, K.; McGlinchey, K.; Hair, J.; Hanabuchi, S.; Du, Q.; Damschroder, M.; et al. Potent Immune Modulation by MEDI6383, an Engineered Human OX40 Ligand IgG4P Fc Fusion Protein. Mol. Cancer Ther. 2018, 17, 1024–1038. [Google Scholar] [CrossRef]
- Kuang, Z.; Jing, H.; Wu, Z.; Wang, J.; Li, Y.; Ni, H.; Zhang, P.; Wu, W.; Wu, M.; Zhou, S.; et al. Development and characterization of a novel anti-OX40 antibody for potent immune activation. Cancer Immunol. Immunother. 2020, 69, 939–950. [Google Scholar] [CrossRef]
- Marin-Acevedo, J.A.; Dholaria, B.; Soyano, A.E.; Knutson, K.L.; Chumsri, S.; Lou, Y. Next generation of immune checkpoint therapy in cancer: New developments and challenges. J. Hematol. Oncol. 2018, 11, 39. [Google Scholar] [CrossRef]
- Glisson, B.S.; Leidner, R.S.; Ferris, R.L.; Powderly, J.; Rizvi, N.A.; Keam, B.; Schneider, R.; Goel, S.; Ohr, J.P.; Burton, J.; et al. Safety and Clinical Activity of MEDI0562, a Humanized OX40 Agonist Monoclonal Antibody, in Adult Patients with Advanced Solid Tumors. Clin. Cancer Res. 2020, 26, 5358–5367. [Google Scholar] [CrossRef]
- Mayes, P.A.; Hance, K.W.; Hoos, A. The promise and challenges of immune agonist antibody development in cancer. Nat. Rev. Drug Discov. 2018, 17, 509–527. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yeh, S.H.; Madireddi, S.; Matochko, W.L.; Gu, C.; Pacheco Sanchez, P.; Ultsch, M.; De Leon Boenig, G.; Harris, S.F.; Leonard, B.; et al. Tetravalent biepitopic targeting enables intrinsic antibody agonism of tumor necrosis factor receptor superfamily members. MAbs 2019, 11, 996–1011. [Google Scholar] [CrossRef] [PubMed]
- Cleary, K.L.S.; Chan, H.T.C.; James, S.; Glennie, M.J.; Cragg, M.S. Antibody Distance from the Cell Membrane Regulates Antibody Effector Mechanisms. J. Immunol. 2017, 198, 3999–4011. [Google Scholar] [CrossRef]
- Yu, X.; Chan, H.T.C.; Orr, C.M.; Dadas, O.; Booth, S.G.; Dahal, L.N.; Penfold, C.A.; O’Brien, L.; Mockridge, C.I.; French, R.R.; et al. Complex Interplay between Epitope Specificity and Isotype Dictates the Biological Activity of Anti-human CD40 Antibodies. Cancer Cell 2018, 33, 664–675.e4. [Google Scholar] [CrossRef]
- Zhang, P.; Tu, G.H.; Wei, J.; Santiago, P.; Larrabee, L.R.; Liao-Chan, S.; Mistry, T.; Chu, M.L.; Sai, T.; Lindquist, K.; et al. Ligand-Blocking and Membrane-Proximal Domain Targeting Anti-OX40 Antibodies Mediate Potent T Cell-Stimulatory and Anti-Tumor Activity. Cell Rep. 2019, 27, 3117–3123.e5. [Google Scholar] [CrossRef]
- Griffiths, J.; Hussain, K.; Smith, H.L.; Sanders, T.; Cox, K.L.; Semmrich, M.; Martensson, L.; Kim, J.; Inzhelevskaya, T.; Penfold, C.A.; et al. Domain binding and isotype dictate the activity of anti-human OX40 antibodies. J. Immunother. Cancer 2020, 8, e001557. [Google Scholar] [CrossRef]
- Boder, E.T.; Wittrup, K.D. Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 1997, 15, 553–557. [Google Scholar] [CrossRef]
- Gao, H.; Cai, H.; Liu, J.; Wang, X.; Zheng, P.; Devenport, M.; Xu, T.; Dou, F.; Liu, Y.; Zhou, A. Structure of CTLA-4 complexed with a pH-sensitive cancer immunotherapeutic antibody. Cell Discov. 2020, 6, 79. [Google Scholar] [CrossRef]
- Wang, Z.; Pan, Q.; Yang, L.; Zhou, H.; Xu, C.; Yu, F.; Wang, Q.; Huang, S.; He, J. Automatic crystal centring procedure at the SSRF macromolecular crystallography beamline. J. Synchrotron Radiat 2016, 23, 1323–1332. [Google Scholar] [CrossRef]
- Collaborative Computational Project, N. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994, 50, 760–763. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Storoni, L.C.; Read, R.J. Likelihood-enhanced fast translation functions. Acta Crystallogr. D Biol. Crystallogr. 2005, 61, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Compaan, D.M.; Hymowitz, S.G. The crystal structure of the costimulatory OX40-OX40L complex. Structure 2006, 14, 1321–1330. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Isupov, M.N.; Murshudov, G.N. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D Biol. Crystallogr. 2001, 57, 122–133. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, version 2.0; Schrödinger, LLC: New York, NY, USA, 2017.
- Bulliard, Y.; Jolicoeur, R.; Windman, M.; Rue, S.M.; Ettenberg, S.; Knee, D.A.; Wilson, N.S.; Dranoff, G.; Brogdon, J.L. Activating Fc gamma receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J. Exp. Med. 2013, 210, 1685–1693. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Ravetch, J.V. Inhibitory Fcgamma receptor engagement drives adjuvant and anti-tumor activities of agonistic CD40 antibodies. Science 2011, 333, 1030–1034. [Google Scholar] [CrossRef]
- Maxwell, J.R.; Weinberg, A.; Prell, R.A.; Vella, A.T. Danger and OX40 receptor signaling synergize to enhance memory T cell survival by inhibiting peripheral deletion. J. Immunol. 2000, 164, 107–112. [Google Scholar] [CrossRef]
- Naismith, J.H.; Sprang, S.R. Modularity in the TNF-receptor family. Trends Biochem. Sci. 1998, 23, 74–79. [Google Scholar] [CrossRef]
- Bodmer, J.L.; Schneider, P.; Tschopp, J. The molecular architecture of the TNF superfamily. Trends Biochem Sci 2002, 27, 19–26. [Google Scholar] [CrossRef]
- Jay, J.W.; Bray, B.; Qi, Y.; Igbinigie, E.; Wu, H.; Li, J.; Ren, G. IgG Antibody 3D Structures and Dynamics. Antibodies 2018, 7, 18. [Google Scholar] [CrossRef] [Green Version]
- Rayner, L.E.; Kadkhodayi-Kholghi, N.; Heenan, R.K.; Gor, J.; Dalby, P.A.; Perkins, S.J. The solution structure of rabbit IgG accounts for its interactions with the Fc receptor and complement C1q and its conformational stability. J. Mol. Biol. 2013, 425, 506–523. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Park, Y.C.; Kreishman, M.; Kieff, E.; Wu, H. The structural basis for the recognition of diverse receptor sequences by TRAF2. Mol. Cell 1999, 4, 321–330. [Google Scholar] [CrossRef]
- Saphire, E.O.; Parren, P.W.; Pantophlet, R.; Zwick, M.B.; Morris, G.M.; Rudd, P.M.; Dwek, R.A.; Stanfield, R.L.; Burton, D.R.; Wilson, I.A. Crystal structure of a neutralizing human IGG against HIV-1: A template for vaccine design. Science 2001, 293, 1155–1159. [Google Scholar] [CrossRef] [PubMed]
- Wajant, H. Principles of antibody-mediated TNF receptor activation. Cell Death Differ. 2015, 22, 1727–1741. [Google Scholar] [CrossRef]
- Vanamee, E.S.; Faustman, D.L. Structural principles of tumor necrosis factor superfamily signaling. Sci. Signal. 2018, 11, eaao4910. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Armstrong, A.A.; Tam, S.H.; McCarthy, S.G.; Luo, J.; Gilliland, G.L.; Chiu, M.L. Functional optimization of agonistic antibodies to OX40 receptor with novel Fc mutations to promote antibody multimerization. MAbs 2017, 9, 1129–1142. [Google Scholar] [CrossRef]
- Dahan, R.; Barnhart, B.C.; Li, F.; Yamniuk, A.P.; Korman, A.J.; Ravetch, J.V. Therapeutic Activity of Agonistic, Human Anti-CD40 Monoclonal Antibodies Requires Selective FcgammaR Engagement. Cancer Cell 2016, 29, 820–831. [Google Scholar] [CrossRef] [PubMed]
- Croft, M.; So, T.; Duan, W.; Soroosh, P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunol. Rev. 2009, 229, 173–191. [Google Scholar] [CrossRef]
- Kim, T.W.; Burris, H.A.; de Miguel Luken, M.J.; Pishvaian, M.J.; Bang, Y.J.; Gordon, M.; Awada, A.; Camidge, D.R.; Hodi, F.S.; McArthur, G.A.; et al. First-In-Human Phase I Study of the OX40 Agonist MOXR0916 in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2022, 8, 3452–3463. [Google Scholar] [CrossRef]
- Goulet, D.R.; Atkins, W.M. Considerations for the Design of Antibody-Based Therapeutics. J. Pharm. Sci. 2020, 109, 74–103. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Jiang, X.; Gao, H.; Zhang, F.; Zhang, X.; Zhou, A.; Xu, T.; Cai, H. Structural Basis of a Novel Agonistic Anti-OX40 Antibody. Biomolecules 2022, 12, 1209. https://doi.org/10.3390/biom12091209
Zhang J, Jiang X, Gao H, Zhang F, Zhang X, Zhou A, Xu T, Cai H. Structural Basis of a Novel Agonistic Anti-OX40 Antibody. Biomolecules. 2022; 12(9):1209. https://doi.org/10.3390/biom12091209
Chicago/Turabian StyleZhang, Jing, Xiaoyong Jiang, Han Gao, Fei Zhang, Xin Zhang, Aiwu Zhou, Ting Xu, and Haiyan Cai. 2022. "Structural Basis of a Novel Agonistic Anti-OX40 Antibody" Biomolecules 12, no. 9: 1209. https://doi.org/10.3390/biom12091209