1. Introduction
Colorectal cancer (CRC) represents the third-most-commonly diagnosed cancer and the second leading cause of cancer death worldwide [
1]. Although most (80%) of CRCs are diagnosed as surgically resectable, a significant fraction of them (25–30%) will relapse in 3–5 years of surgery, notwithstanding the resort to adjuvant chemotherapy [
2], in a fashion that is difficult to predict based on current biomolecular and clinical knowledge. A better understanding of the biological causes and molecular interactions leading to the onset and clinical progression of this life-threatening disease is, therefore, imperative.
While somatic DNA mutations fuel cancer initiation and evolution in CRC and other malignancies, additional environmental factors, including chronic inflammation and intestinal microbes, are increasingly recognized as causative or complicit in colorectal carcinogenesis [
3]. In particular, converging lines of investigation have identified bacterial pathogens that are not part of the normal intestinal microbiota, such as
Fusobacterium nucleatum (
Fn) [
4] and
Streptococcus gallolyticus (
Sg) [
5], as potential etiologic factors in CRC based on the over-representation of DNA sequences or even cultivable microorganisms in malignant tissues compared with normal colon specimens. Notably,
Fn also promotes the chemoresistance of colon cancer cells, and
Fn presence predicts lower patient survival [
6]. CRC-associated bacteria promote cancer by perturbing the equilibrium among different microbiota components and between bacterial populations and inflammatory and immune cells so as to trigger a vicious dysbiosis–inflammation circle [
7]. Additionally, specific pathogens directly attack and invade epithelial cells, hijacking signaling components and cascades that control cell proliferation and normal mucosal repair. More specifically,
Fn modulates E-cadherin/β-catenin signaling via its FadA (Fusobacterium adhesin A) so as to promote oncogenic and proinflammatory responses in CRC cells [
8]; the same pathogen uses the lectin Fap2 to bind the tumor-cell-expressed disaccharide Gal-GalNac [
9], and host cell binding and invasion leads to the secretion of the chemokines IL-8 and CXCL1 and enhanced CRC cell migration [
10]. Moreover, on the stromal tumor side,
Fn binding to the T lymphocyte inhibitory receptors TIGIT and CEACAM-1 (via Fap-2 and the trimeric autotransporter adhesin CbpF, respectively [
11]) suppresses antitumor immunity, thereby indirectly promoting malignant growth [
12,
13]. Importantly,
Fn has been detected and cultivated from distant CRC metastatic lesions [
4], suggesting that either this microbe localizes to disseminated cancer colonies through the hematogenous route, or can stably persist within colonic metastasis-initiating cells.
Over the last few years, intense research has highlighted the presence, in several solid malignancies, including CRC, of a unique subset of “stem-like” cells endowed with tumor-initiating capacity that are arguably responsible for cancer metastatic spreading and local recurrence after surgery. These colorectal cancer stem cells (CR-CSCs) may truly descend from intestinal stem cells undergoing oncogenic transformation, or rather reflect the de-differentiation of more mature or even fully differentiated enterocytes; either way, they can be identified by the expression of molecular markers (CD133, EpCAM, CD44v6, LGR5, ALDH1, and DCLK1) and the activation of intracellular signaling pathways (Wnt-APC/β-catenin, Notch, TGF-β/BMP) (see [
14] for a comprehensive literature review) functionally related to self-renewal activity and pluripotency.
While several studies have investigated the molecular determinants and downstream functional consequences of the interaction between oncomicrobes and colon cancer epithelial and stromal/immune cells, little information is available on whether such interactions also or even preferentially involve CSC. It is, however, known that colorectal-tumor-initiating cells exploit autocrine cytokine-triggered circuitries to resist chemotherapy-induced cell death [
15], and that normal intestinal stem cells express innate immune receptors that mediate protection from oxidative damage and ROS cytotoxicity in response to bacterial components [
16]. Additionally,
Fn induces CSC-like traits in cultured CRC lines via epithelial–mesenchymal transition (EMT) [
17], and the
Fn-derived metabolite formate enhances the stemness and self-renewal capacity of patient-derived colorectal tumor organoids [
18].
In the present work, we set out to address the microbe–CSC interaction by exposing CSC-enriched primary spheroidal cultures of colorectal tumors to Fn in vitro. Our results demonstrate that Fn avidly binds to colonsphere-derived cells and triggers intracellular proinflammatory and oncogenic cascades superimposable to those previously described in mature CRC cells. Additionally, we highlight the role of the cellular CEACAM-1 and its associated tyrosine phosphatase SHP-2/PTPN11 in mediating early phosphorylation responses downstream of cell–pathogen interactions. These findings provide original information on the role of Fn in CRC and suggest a novel paradigm of bacterial carcinogenesis centered on the direct bacterial targeting of cancer-initiating cells.
2. Materials and Methods
Cell lines. The primary spheroidal colon cancer cultures CSC-P and SA-22 used in the present study were initially derived at Istituto Superiore di Sanità, Rome, Italy, and made available to GBP under a Material Transfer Agreement. The procedure of isolation and characterization is described in detail in ref. [
19]. Briefly, surgical specimens of primary CRCs were cut into small pieces, digested in Collagenase II + DNAse for 1 h at 37 °C, and resuspended in a serum-free defined growth medium containing 10 ng/mL human bFGF and 20 ng/mL human EGF. CSC-enriched spheroids developing within 2–4 weeks from the primary seeding were expanded and further characterized for their mutational profile and in vivo tumorigenicity. CSCs were routinely maintained in CSC medium (Neurobasal-A or Advanced DMEM/F12, supplemented with Vitamin A-free B27 (Life Technologies, Carlsbad, CA, USA), 10 mM of nicotinamide (Sigma, St. Louis, MO, USA), 1 μM of RhoK inhibitor Y-27632 (Tocris Biosciences, Bristol, UK; cat. #1254), 6 g/L of glucose, 2 μg/mL of Heparin (StemCell Technology, Vancouver, BC, Canada; cat. #07980), 10 ng/mL of bFGF, and 20 ng/mL of hEGF (Peprotech, Thermo Fisher Scientific, Waltham, MA, USA) and passaged weekly. Spheroid aggregates were dissociated by gentle trypsinization; cell suspensions were passed through a 70 μm-pore-size strainer (FlowMi
®, SP Bel-Art, Wayne, NJ, USA; cat. #136800070) and re-seeded at 1.5 × 10
5 cells/mL in 25 cm
2 ultralow-attachment tissue culture flasks. In order to induce intestinal differentiation, the cells were dissociated and cultivated for 7–10 days in regular (attachment-permissive) tissue culture plates in CSC medium supplemented with 2% FBS and 10 mM sodium butyrate (Sigma Aldrich).
The colorectal carcinoma cell lines CaCo-2 (cat. HTB-37™) and HT-29 (cat. HTB-38™) were obtained from the American Type Culture Collection (ATCC) and routinely grown in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% FBS (v/v) and antibiotics. Both lines were periodically checked for mycoplasma infection.
Bacterial strains.
Fusobacterium nucleatum subsp. nucleatum Knorr (25586™) was obtained from ATCC and maintained in anaerobiosis according to the accompanying instructions. The
Streptococcus gallolyticus (
Sg) used in the present study was a clinical strain isolated from a patient hospitalized at IRCCS Fondazione Policlinico A. Gemelli—Università Cattolica del Sacro Cuore (Rome).
Sg was grown at 37 °C in brain–heart infusion (BHI) broth with shaking or on BHI agar (Difco Laboratories, Sparks, MD, USA, under aerobic conditions) [
5]. The
Escherichia coli strain C43 expressing the fusobacterial adhesin CbpF (variant 1) and the relative control strain are described in ref. [
20]. Recombinant
E. coli strains were grown in LB broth (Difco) or on LB agar plates (Difco) containing 100 μg/mL of ampicillin at 37 °C under aerobic conditions. CbpF1 expression was induced with 0.4 mM of isopropyl-b-d-thio-galactoside (IPTG, Sigma) at 22 °C overnight.
Antibodies. The mouse monoclonal antibodies anti-CEACAM-1 (E1, cat. #sc-166453), anti-GFP (B2, cat. #sc-9996), and anti-p-(Ser 32) IkB-α (clone B9, cat. #sc-8404), as well as the polyclonal anti-SHP-2 and anti-SHP-1 rabbit antibodies (C-18, cat. #sc-280 and C19, cat. #sc-287, respectively), were obtained from Santa Cruz Biotechnology. The mouse/rat monoclonal antibodies anti-β-actin (8H10D10, cat. #3700), anti-E-Cadherin (24E10, cat. #3195), anti-GSK3β (27C10, cat. #9315), and anti-p(Ser 9) GSK3β (5B3, cat. #9323), and the polyclonal rabbit antibodies anti-p(Thr202-Tyr204) p44/42 MAPK (cat. #9101) and anti-p(Tyr 542) SHP-2 (cat- #3751) were purchased from Cell Signaling Technology. The rabbit polyclonal antiserum anti-p42/44 MAP Kinase ½ (Erk1/2) was obtained from Millipore/Merck (cat. #06-182). The PE-conjugated anti-hCD133/1 (AC133, cat. #130-113-670) and anti-hCEACAM1/CD66a (282640, cat. #FAB2244P), used for flow cytometry, were from Miltenyi Biotec and R&D Systems, respectively.
Plasmids. The double-color lentiviral Wnt-reporter TOP-GFPmC was a gift from Ramesh Shivdasani (Addgene plasmid #35491;
http://n2t.net/addgene:35491 (accessed on 11 July 2022); RRID:Addgene_35491) [
21]. This third-generation lentiviral construct encodes eGFP under the transcriptional control of 7× TCF/LEF promoter elements, while constitutive PGK-driven mCherry fused to H2B marks successfully transduces cells.
The pre-designed shRNA lentiviral construct directed against human CEACAM-1 (cat. #SHCLNG clone TRCN0000377692) and the MISSION
® pLKO.1-puro non-target shRNA control vector (cat. #SHC016) were purchased from Sigma/Merck. The NF-kB-responsive
Firefly luciferase reporter construct and the CMV-driven
Renilla internal control (Cignal Reporter Assay, cat. CCS-013L) were from QIAGEN. The IPTG-inducible plasmid encoding the two SHP-2 SH2 domains (N- and C-terminal) fused with GST (pGEX SHP-2(NC)-SH2) was a gift from Bruce Mayer (Addgene plasmid #46499;
http://n2t.net/addgene:46499 (accessed on 11 July 2022); RRID:Addgene_46499) [
22]. The plasmid originally provided in the DH5α
E. coli strain was amplified, purified, and transformed into the protease-deficient BL21 strain to maximize GST-fusion protein recovery.
CSC infection and Luciferase reporter assays. Lentiviral supernatants were produced according to Tiscornia et al. [
23], with minor changes; the plasmid mixture containing the transfer vector, pMDL, pRev, and pVSVG, was introduced to HEK-293T cells by calcium phosphate precipitation. Supernatants from the second and third days post-transfection were pooled and concentrated 100 times by ultracentrifugation (72,000 g for 2 h at 20 °C). Pooled supernatants from one or two 10 cm Petri dishes (4–8 × 10
6 packaging cells) were used to infect 2 × 10
5 CSC cells in 2 mL of complete CSC medium. To increase the infection efficiency, cells were co-centrifugated with lentiviral particles at RT for 2.5 h at 2500 g (“spinoculation”) in presence of 8 μg/mL of polybrene (Sigma-Aldrich/Merck, Darmstadt, Germany).
For the transfection of luc reporter plasmids, 2.5 × 105 cells from freshly dissociated spheroids were seeded in 0.5 mL of complete CSC medium without Heparin (found to interfere with transfection) and left to recover for 8 h. Plasmid DNA (1 μg of Firefly reporter and 25 ng of CMV-Renilla internal control) was transfected using the Lipofectamine 3000 reagent (Life Technologies, Carlsbad, CA, USA) in 24-well ultralow-attachment cell culture clusters. After overnight incubation, stimuli were applied as needed. Following an additional 24 h, the normalized reporter activity (Firefly/Renilla) was measured by luminometry using a dual luciferase assay kit (Promega, cat. E1910) according to the manufacturer’s instructions.
Flow Cytometry. For FC analysis, spheroid cultures were dissociated with trypsin, and cell suspensions were filtered through a 70 μm cell strainer. For surface staining, cells (5 × 105 in 75 μL) were incubated in PFA buffer (PBS + 1% FBS + 0.05% Azide) with 0.5–1 μg of primary antibody for 60 min on ice, followed by two washes in cold PFA. Labeling with a secondary reagent, if necessary, was performed sequentially following an identical procedure. For cell staining with FITC-labeled peanut lectin (PNA, Sigma/Merck, cat. #L7381), 2.5 × 105 cells were incubated in 100 μL PFA buffer with 2 μg/mL lectin for 30 min on ice.
Samples were analyzed with either a three-laser, 12-fluorescence Cytoflex cytometer (Beckman Coulter, Brea, CA, USA) or with a single-laser (488 nm), 3-fluorescence MCL-XL Epics (Coulter) instrument. Dead cells were excluded by staining with propidium iodide or based on their position in the forward-scatter/side-scatter plot.
Detection of Nitric Oxide by DAF-FM. Dissociated CSC-P cells (2.5 × 105) were seeded in ultralow-attachment 24-well clusters in 500 μL of complete CSC medium without antibiotics and incubated for 24 h with 200 or 500 MOI Fn. DAF-FM diacetate (Thermo-Fisher, cat. #D23844) was added at 10 μM for the last 60 min of incubation. Cells were then washed in cold PBS, re-dissociated with trypsin, and immediately analyzed by flow cytometry.
Bacterial pull-down assay (Bactoprecipitation). In order to isolate CSC proteins potentially involved in cell interaction with bacteria, 0.5–1 × 106 cells were lysed in 100 μL of PBS containing 1% Triton X-100 and protease inhibitors, incubated on ice for 10 min, and spun down at 14,000 rpm, 4 °C to remove unlysed cells, nuclei, and cell debris. Next, the supernatant was mixed with 900 μL of PBS containing 109 bacterial cells (final Triton X-100 concentration 0.1%) and incubated at 4 °C for 45 min on a rotating wheel. Bacteria were then centrifuged at 3000× g for 10 min, washed twice in cold PBS, and finally resuspended in 60 μL of 1× SDS Laemmli sample buffer (50 mM Tris-HCl pH 6.8, 5% β-mercaptoethanol, 10% glycerol, 1% SDS, and 1.5 mM bromo-phenol blue), briefly sonicated, and boiled for 5 min to elute bacteria-adsorbed cellular proteins. After a brief centrifugation step, the supernatants were used for regular Western blot analysis.
Fn fluorescent labeling and CSC–bacteria binding assay. Fn and other bacterial strains were fluorescently labeled using the Bac-Light™ Red (Thermo Fisher, Waltham, MA, USA; cat. #B35001) or the BactoView™ Live Green (Biotium, Fremont, CA, USA; cat. #40102) bacterial stains according to the manufacturer’s indications. After two washes in PBS to remove the unbound dye, bacteria were mixed with CSC in a 100:1 ratio and incubated at 37 °C for 30 min in 500 μL of RPMI, with occasional agitation. Samples were then briefly centrifuged (14,000 rpm for 15 s) to separate cells from unbound bacteria, washed once in RPMI, and analyzed by flow cytometry. Labeled bacteria were also separately analyzed to identify and gate out the corresponding population on the FS-SS plot. Comparable labeling intensities among different strains (i.e., Fn vs. Sg) were also verified.
Confocal microscopy imaging. The binding of Red-
Fn after 30 min of CSC co-incubation with fluorescent bacteria was qualitatively assessed by confocal microscopy imaging [
24,
25]. Images were acquired with a Nikon A1-MP inverted confocal microscope equipped with an on-stage incubator (OKOLAB), which kept a constant temperature of 37 °C and 5% CO
2. Fluorescence emission, excited with a single-photon laser (excitation wavelength: 561 nm), was collected in the wavelength range of 570–620 nm using a 60× oil-immersion objective (1.4 NA). Brightfield images, collected along with fluorescent images, were used to highlight the distribution of red-stained bacteria.
Cell stimulation. CSCs from dissociated spheroids were counted with a hemocytometer, spun down at 14,000 rpm for 20 s, and resuspended in RPMI without additives at 0.5–1 × 10
7 cells/mL in 100 μL (short-term stimulation) or 1 × 10
6/mL in 500 μL (16–24 h stimulation). Bacterial liquid cultures were spun down at 3000 g (3900 rpm) for 10 min, washed once in PBS, and quantified by spectrophotometry at 660 nm (1 OD = 10
9 cells/mL) [
26]. The desired bacterial MOI were resuspended in 100 μL of RPMI, briefly (5 s) sonicated to dissolve gross aggregates, and mixed with CSCs (final stimulation volumes of 200 μL and 600 μL, respectively). At the end of the incubation, the cells were quickly pelleted in a benchtop centrifuge (14,000 rpm × 20 s) and flash-frozen for further processing or directly lysed in 80 μL of 1× SDS Laemmli sample buffer. For overnight incubation, Gentamycin (100 μg/mL) was added to the culture after 3 h of stimulation to prevent bacterial overgrowth in the medium.
Western blotting. Protein samples in Laemmli buffer were heated at 95 °C for 5 min, subjected to SDS-PAGE, and electroblotted onto a nitrocellulose membrane. Immunocomplexes were visualized by enhanced chemiluminescence (Westernbright™ ECL, Advansta, San Jose, CA, USA, cat. #K-12045) with the Alliance Q9® advanced chemiluminescence imager (Uvitec, Cambridge, UK). Digital images were quantified using the Image J software (Analize→Gels).
Cell lysis and immunoprecipitation. For co-immunoprecipitation studies, cell pellets (3–5 × 106 cells) were lysed in 1 mL of cold lysis buffer (50 mM Tris-HCl pH 8.0; 150 mM NaCl; 5 mM EDTA; and 0.05% Na+ Azide) containing 1% (v/v) Triton-X100, and protease and phosphatase inhibitors. After 15 min of incubation on ice, the tubes were spun down (14,000 rpm for 10 min at 4 °C) to remove cell debris and unlysed nuclei, and the supernatants were precleared with empty Protein A/G sepharose beads (100 μL of a 10% v/v slurry) for 1 h at 4 °C on a rocking plate. After centrifugation, 1/20 of the supernatant was kept for Western blot analysis (input) and 19/20 was incubated with 1 μg of antibody (anti-CEACAM-1 or anti-SHP-2) and 100 μL of protein A/G slurry for 16 h in rotation at 4 °C. Sepharose-bound immunocomplexes were collected by centrifugation (14,000 rpm × 30 s), washed 4–5 times in lysis buffer with inhibitors, eluted in Laemmli buffer and analyzed by Western blotting.
GST-pull down assay. To obtain the immobilized GST-2SH2-SHP2 fusion protein, the encoding pGEX plasmid (Addgene #46499) was transformed into the low-protease E. coli strain BL21; overnight bacterial cultures were diluted 1:10, incubated until OD > 0.6, and induced for 3 h with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). Pellets were lysed in PBS/Triton-X100 1%/PMSF, and the GST fusion protein was affinity-purified with Glutathione-Sepharose (10 µL packed resin/mL lysate, 30 min at RT) in batch, extensively washed, aliquoted, and frozen. For the pull-down assay, Sepharose-bound GST-2(SH2)-SHP2 (the equivalent of 1 mL of bacterial culture) was incubated with CSC lysates, obtained as for a conventional immunoprecipitation, for 2.5 h at 4 °C in rotation. The following washing and elution steps were conducted as for immunoprecipitation. After protein blotting, equal amounts of the GST fusion proteins throughout the samples were verified by membrane-reversible Ponceau S staining.
Multiplex cytokine screening of CSC supernatants. Cells (2 × 106 in 2 mL CSC medium) were stimulated for 24 h with 100 MOI Fn or left untreated. Supernatants were overlaid on twin membranes of a semi-quantitative, sandwich-based, antibody array kit (Cytokine Array C3, Ray Biotech, Peachtree Corners, GA, USA, cat. #AAH-CYT-3) and incubated for 16 h at 4 °C on a rocking plate. Subsequent steps for chemiluminescent immunocomplex detection were conducted according to the manufacturer’s instructions.
Quantitative Real-Time PCR. For RNA extraction, undissociated CSC spheroids or plastic-adherent (differentiated CSCs or HT-29 and CaCo2 cultures) cell clusters were washed once in PBS and processed using a Direct-zol RNA Miniprep kit (Zymo Research, Irvine, CA, USA, R2052) according to the manufacturer’s instructions. The amount and purity of RNA were determined by NanoDrop™ (Thermo Fisher Scientific). SensiFAST™ cDNA Synthesis Kit (Meridian Bioscience, Cincinnati, OH, USA) cDNA was used for RNA retro-transcription. Real-time PCR was performed using a SensiFAST™ SYBR® No-ROX Kit (Meridian Bioscience) with a CFX96 qPCR Instrument (Bio-Rad, Hercules, CA, USA). Primer sets for human NANOG (amplicon size 129 bp), human OCT4/POU5F1 (amplicon size 154 bp), and the human housekeeping gene LDHA (a.s. 130 bp) were from the Human Stem Cell Pluripotency Detection qPCR Kit (ScienCell, Carlsbad, CA, USA, Catalog #0853). The reaction conditions were as per the manufacturer’s recommendations.
To detect Fn DNA in CSC-P cultures, genomic DNA was obtained from cell pellets (106 cells) following a standard procedure. A 244 bp fragment from the Fn (subspecies nucleatum, MT482608.1) 16S ribosomal RNA gene was amplified using the following primer pair: F: AAAGCGCGTCTAGGTGGTTA and R: GTTTACGGCGTGGACTACCA.
Cell viability assay. CSC-P cells were seeded in complete medium at 2.5 × 104/100μL in 96 flat-bottom well plates and treated for 4 days with the alkylating agent Oxaliplatin (Selleckchem, Houston, TX, USA, cat. #S1224) at 250–7.5 μM or left untreated. A total of 250 heath-killed MOI (H-K, 60 °C for 40 min.) were added immediately after seeding where needed. Cell survival was measured by the CellTiter-Glo® Luminescent Cell Viability Assay (PROMEGA, Madison, WI, USA; cat. #G7571) according to the manufacturer’s instructions. In each sample group (control and H-K Fn), the average RLU reading (background-subtracted) of the untreated wells (No Oxa) was assumed as 100% survival, and cell viability throughout the treatments was calculated as (RLU well/average RLU of untreated wells) × 100. RLU readings from wells containing H-K Fn without CSC-P cells were not different from the background.
Statistical analysis. Differences between two sample means were evaluated by a two-tailed Student’s
t-test for independent or correlated samples, where appropriate. One- or two-way ANOVA followed by Tukey Honest Significant Difference post hoc analysis were used for multiple comparisons. Experimental measurements from independent experiments (i.e., control vs.
Fn) were grouped and analyzed pairwise by the Wilcoxon signed rank test, or by ANOVA for correlated samples if k > 2. Where data were reported as the fold induction (compared with untreated control), relevant statistical tests were performed for row values or stimulation indexes (treated/untreated). The single-sample Student’s
t-test (two-tailed) was used to compare the mean fold induction value with the null hypothesis = 1 (no effect). The threshold for statistical significance was set at
p < 0.05 (two-tailed). Calculations were performed on the online Vassar Stats platform (
http://vassarstats.net/index.html, accessed between 1 March–1 May 2022).
4. Discussion
Intestinal bacteria have recently drawn unprecedented attention as a determinant of human health and disease; this is especially true for inflammatory bowel diseases and colorectal cancer as their most severe and worrisome complications. The proposed model whereby altered bacterial–host communication and the ensuing inflammatory response leads to un-resolving epithelial damage–regeneration cycles, eventually conducive to malignant transformation and carcinogenesis, implies a primary involvement of intestinal stem cells in the process; however, our understanding of whether and how stem cells, and particularly CSCs, communicate with the microbiota is still incomplete [
16]. The present work preliminarily addresses this critical knowledge gap by investigating the interaction between CRC stem-like cells and
Fn, a pathobiont recently recognized as a potential etiologic factor in colon cancer development and malignant progression. Our results provide evidence for a direct interaction of bacteria with immature cancer cells, investigate the downstream cellular responses, and highlight a novel signaling circuitry whereby
Fn triggers a growth-factor-like signaling cascade in CSC by impinging on the complex between the bacterial receptor CEACAM-1 and its associated cytosolic tyrosine phosphatase SHP-2.
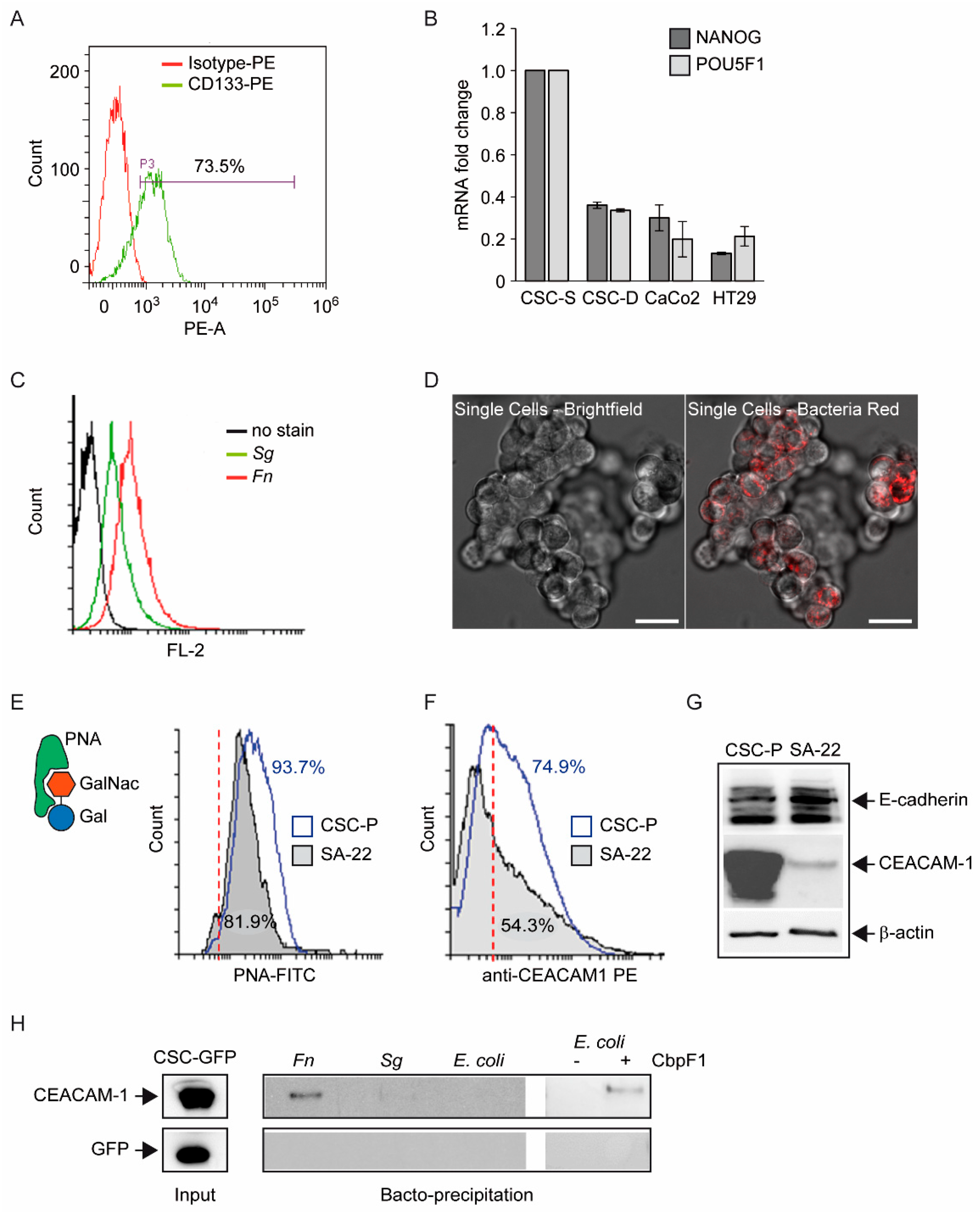
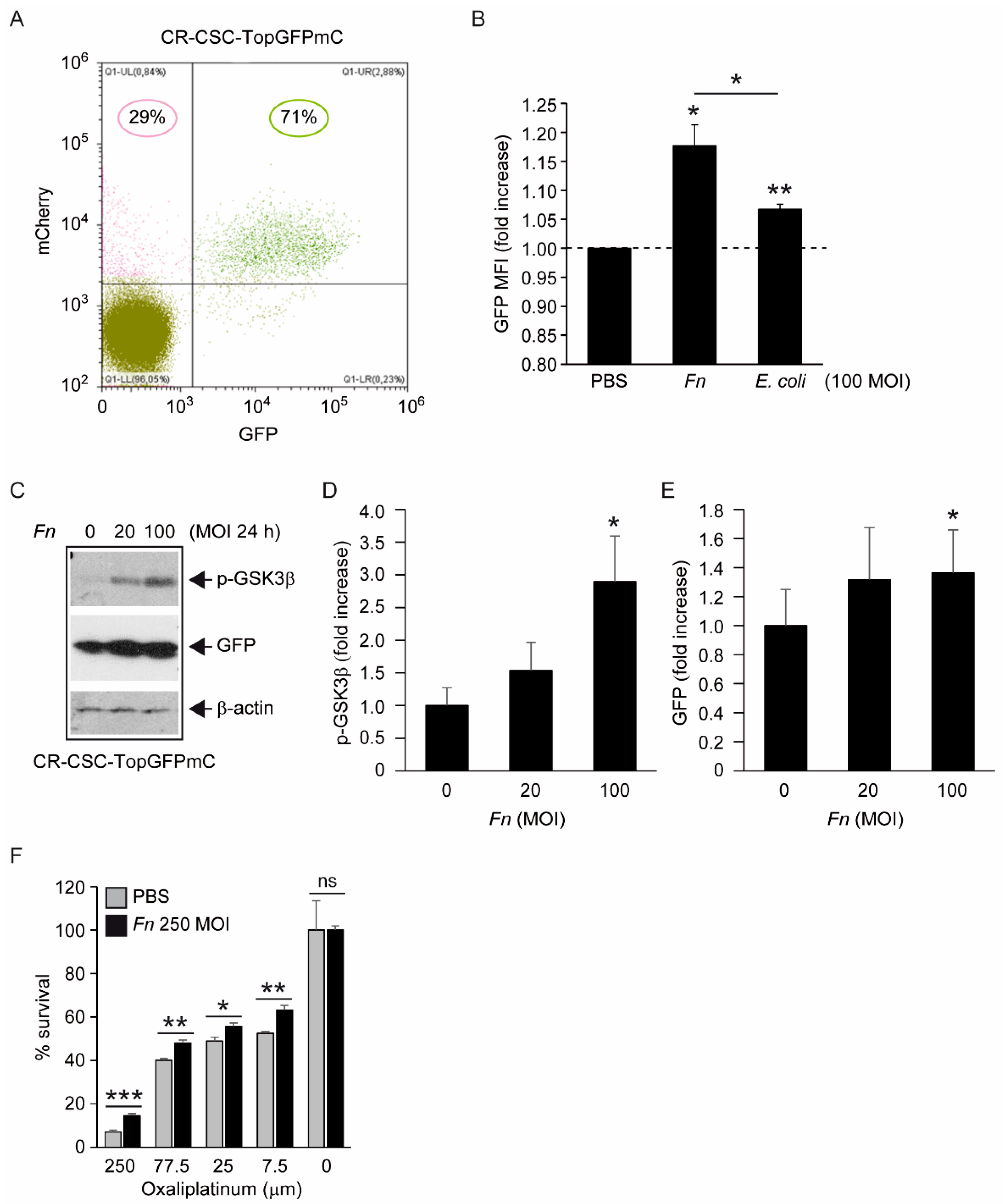
One first element of novelty in the research presented here is the focus on a stem-like cancer cell population. Although spheroidal cultures of colon cancer cells can still be heterogeneous in terms of clonogenicity and malignant potential, evidence for the (a) expression of CD133 in the majority of CSC-P cells, (b) downregulation of the pluripotency factors Nanog and Oct4 in differentiative culture conditions, and (c) constitutive activation of the Wnt pathway as revealed by the fluorescent reporter TopGFP clearly argues in favor of this cell model being representative of the CRC stem cell subset. Additionally, experiments of cell–bacteria interactions with fluorescent Fn confirm that nearly 100% of spheroid-derived cells are physically targeted by the pathogen. Therefore, it is unlikely that the cell responses to Fn infection described here are accounted for by a minority of more differentiated cells phenotypically similar to cancer cell lines employed in previous studies.
The finding of the direct and stable binding of
Fn to CSC, presented in
Figure 1, is not trivial. The reported enrichment of
Fn, not only at primary CRC sites, but also in distant metastatic colonies [
4], may, in fact, reflect the capacity of the pathogen to hitchhike a specific subpopulation of tumor-initiating and tumor-disseminating cells. On the other hand, while CSCs appear to express multiple potential
Fn docking molecules (Gal-GalNac, E-cadherin, CEACAM-1), they may not be easily accessible for microbial contact when confined to their hypoxic niche in vivo. Nevertheless, spontaneous or therapy-induced tumor necrosis may occasionally expose CSCs so as to infringe their isolation. Additionally, Gal-GalNac expression (
Figure 1E) renders CSCs targetable by systemically spread
Fn, and even more so in hypoxic regions, which is more favorable for this obligate anaerobic pathogen. Notably, the spheroid 3D culture model creates a unique microenvironment permissive to the growth of anaerobic bacteria [
37], which adds to the relevance of this experimental setting for studying the interaction between
Fn and CSCs.
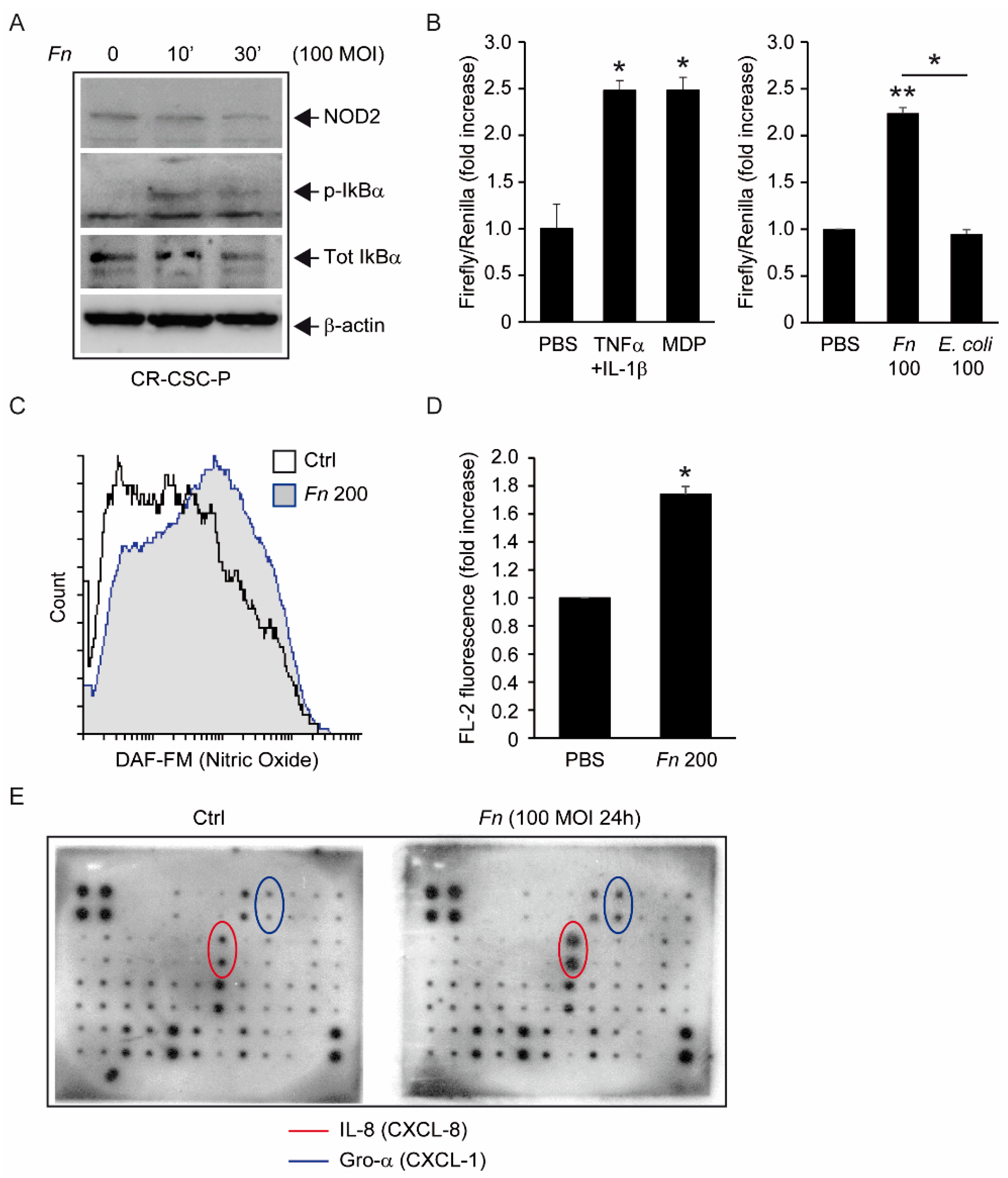
The molecular analyses displayed in
Figure 2 clearly show that
Fn can elicit proinflammatory and innate immune responses in intestinal cells, including the activation of NF-kB and secretion of the cancer-associated chemokines CXCL1 (Gro-α) and CXCL-8 (IL-8) [
10]; while in part confirmative, these observations gain particular relevance in revealing the potential of CSC to actively participate in the establishment of a tumor-promoting and immunosuppressive microenvironment. From this perspective, of special interest is the
Fn-elicited generation of nitric oxide, a gaseous mediator involved in microbicidal responses in macrophages, but also previously linked to colorectal CSCs’ malignant capacity [
29]. Along a parallel line of speculation, while our data show that spheroid-derived cells are competent for bacterial sensing, it is still possible that the antibacterial responses triggered in these progenitors are somewhat less efficient than those in mature epithelial cells, making them a preferential infection target for
Fn or other pathogens. This possibility, which entails far-reaching implications for microbe-driven colorectal carcinogenesis, deserves further investigation by systematically comparing innate immune responses in stem versus differentiated cancer cells.
Fn reportedly activates Wnt signaling [
8] and produces CSC characteristics and a mesenchymal phenotype in CRC cells [
17,
18]. The results depicted in
Figure 3 complement this information, showing that, although constitutively active, the Wnt cascade can be further stimulated by
Fn in CSC, as revealed by the increased inhibitory phosphorylation of the β-catenin destruction factor GSK3β [
31] and by the induction of TCF/LEF-driven recombinant GFP. Additionally, the increased chemoresistance of CSCs to oxaliplatin when co-cultured with
Fn, although not directly demonstrated in the present work, is consistent with enhanced stem-like features downstream of Wnt activation. Of note, Vermeulen et al. elegantly showed that Wnt signaling in CSCs can be triggered by extrinsic cues, such as HGF secreted by stromal myofibroblasts [
30]. Thus, by adding
Fn to the list of “environmental” stemness-promoting factors, our observations align perfectly with this idea. Nevertheless, changes in cells transduced with TCF-driven GFP were relatively modest, with a slight fluorescence increase in GFP+ cells and no detectable cell shift from the GFP- to the GFP+ population (
Figure 3A,B, and data not shown). Whether this reflects an exceptionally high baseline Wnt activity in our CSC line or is the result of a technical limitation (i.e., the use of non-cloned Top-GFP cells bearing varying copy numbers of the probe) is currently being evaluated.
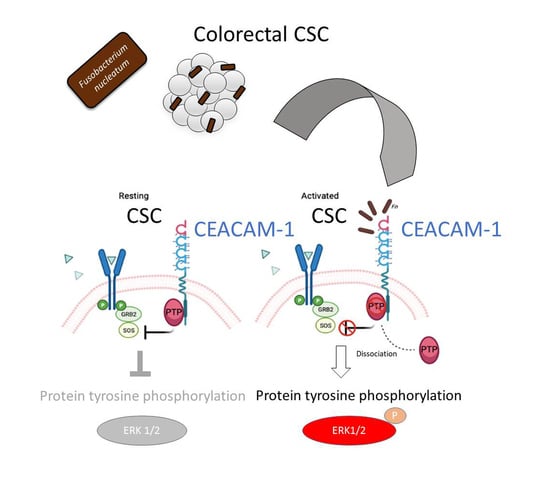
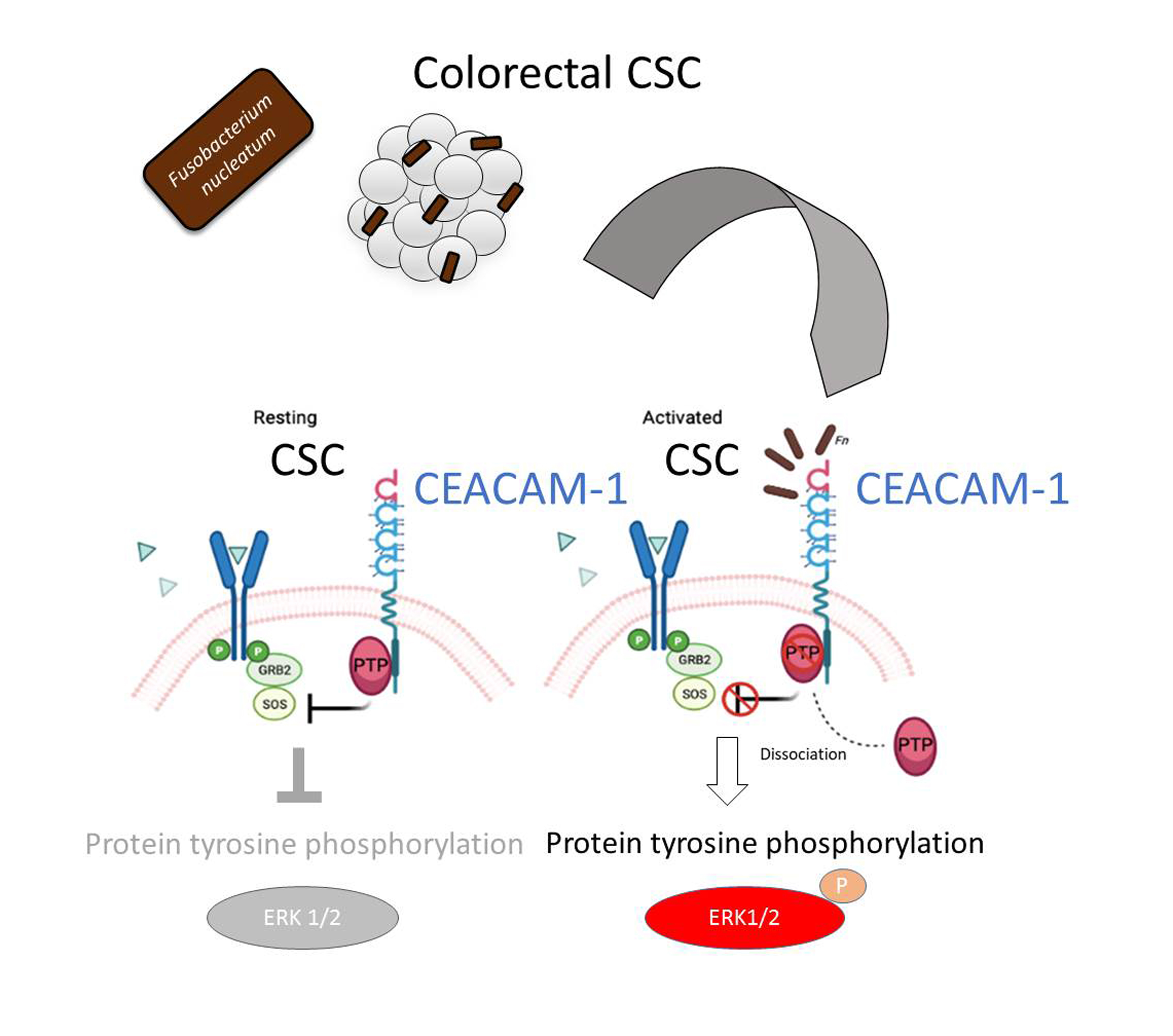
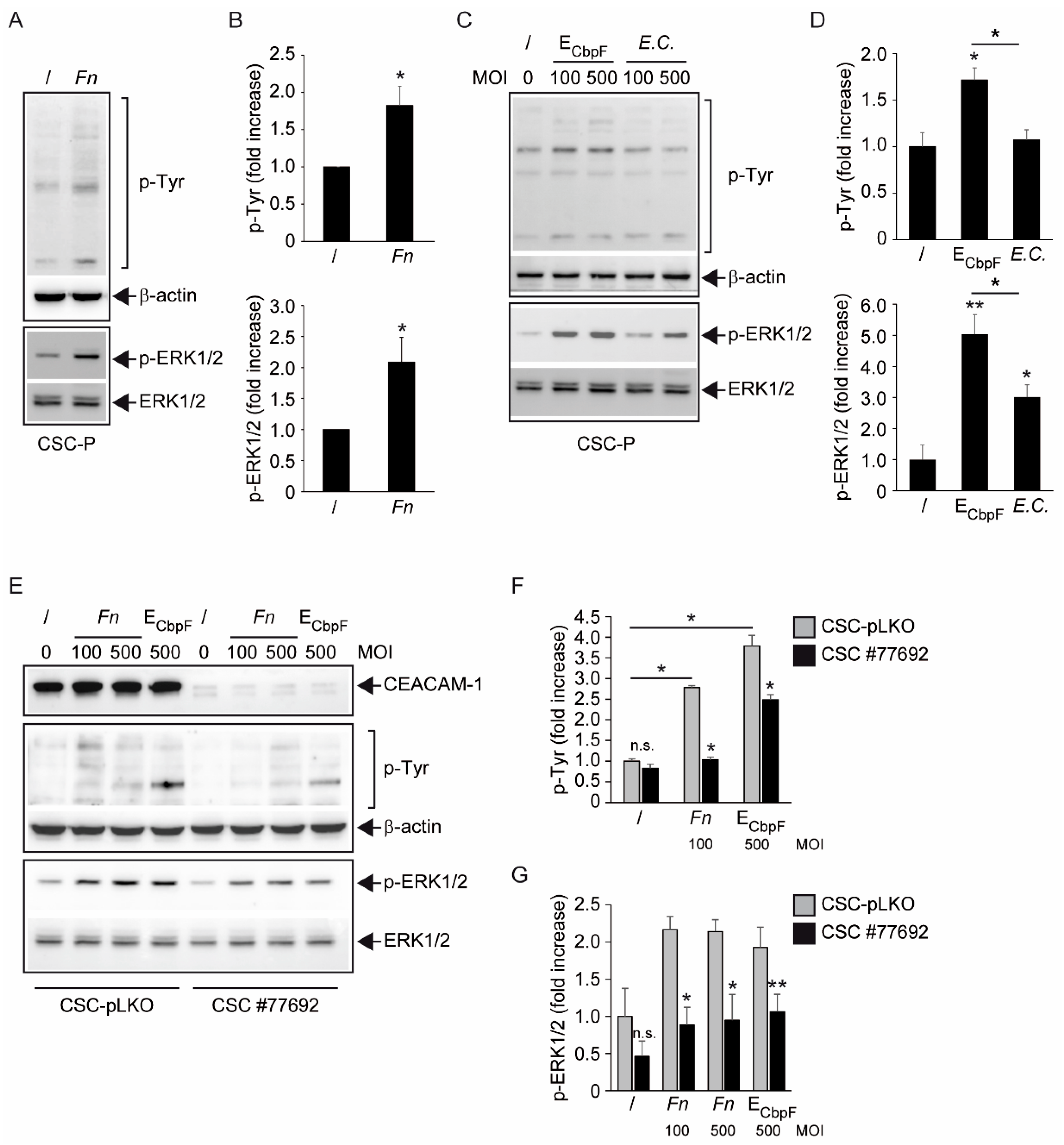
The analysis of protein tyrosine phosphorylation events elicited by
Fn binding to CSCs represents the most innovative contribution of the present work. This growth factor-like cascade, culminating in the phosphorylation of ERK1/2, is largely mediated by CEACAM-1, as indicated by the blunted response observed in cells depleted of this adhesion molecule (
Figure 4). Accordingly, an
E. coli strain recombinant for the fusobacterial autotransporter adhesin and CEACAM-1 ligand CbpF [
11,
20] elicited a similar set of cellular responses in CSCs more efficiently compared with the parental, non-CEACAM-1 binding parental EC (
Figure 1H and
Figure 4C,D). However, the mechanistic interactions whereby
Fn activates an RTK-like cascade in CSCs remain largely undefined. The signaling capacity of CEACAM-1 resides in the ability of the “long” (71 aa) cytosolic tail, harbored by the L isoforms, to recruit tyrosine phosphatases (via the ITIM domain) and Src-like kinases [
38]. Notably, the relative affinity toward these different transducers is regulated by homophilic interactions and receptor cis-dimerization dictated by the extracellular domains. Rigorous FRET-based studies by Muller et al. suggested that transmembrane signaling by CEACAM-1 operates by altering the monomer/dimer equilibrium, which leads to changes in the SHP-2/c-Src–binding ratio [
36]. Thus,
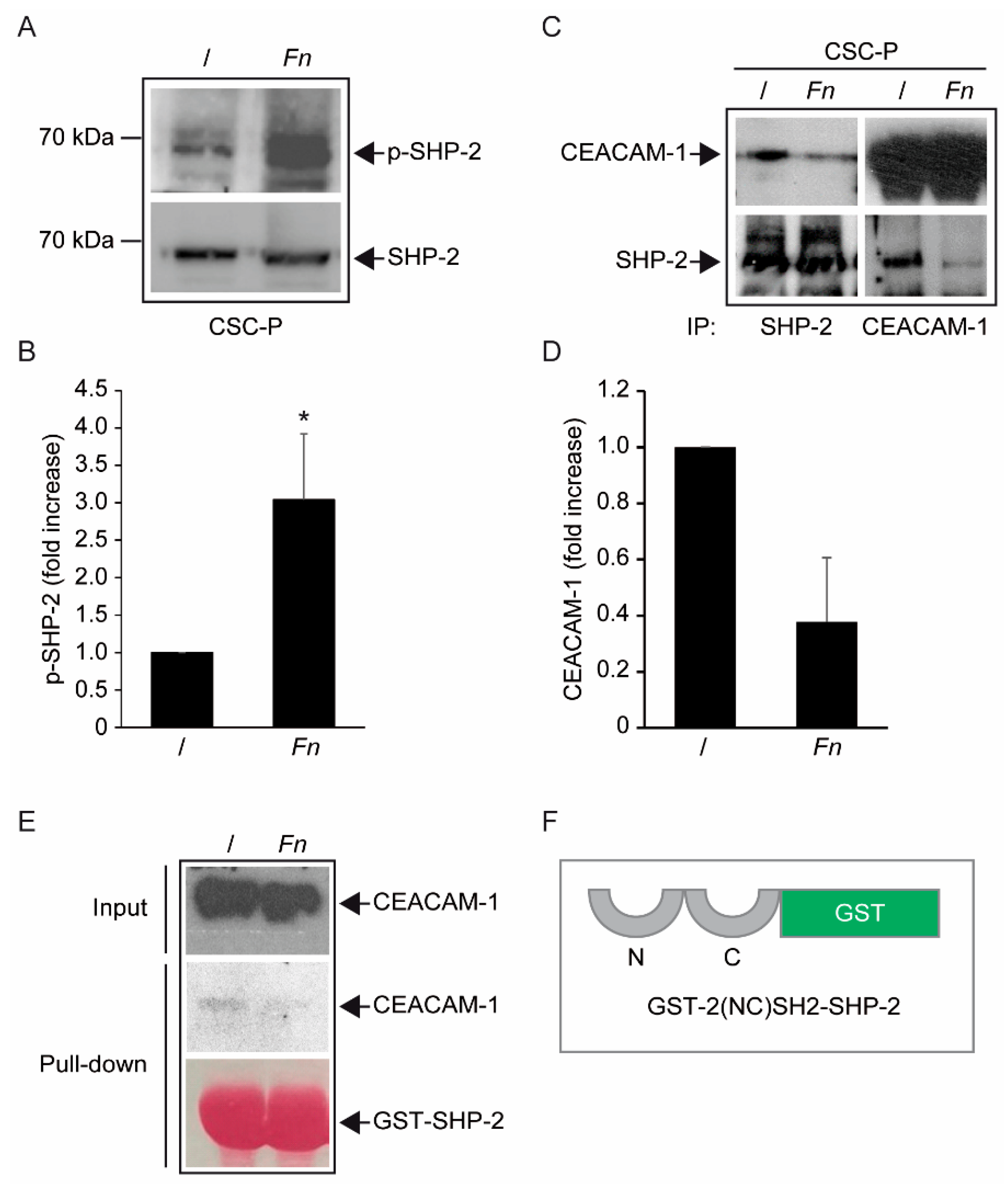
Fn, by engaging CEACAM-1 through CbpF, could impact the supramolecular organization of CEACAM-1-L aggregates so as to tilt the phosphorylation–dephosphorylation balance in favor of the former. In keeping with this view, the co-immunoprecipitation studies presented in
Figure 5 are consistent with the dissociation of the CEACAM-1–SHP2 complex in response to bacterial stimulation, although the possibility of a simultaneous recruitment and the activation of Src-like kinases by CEACAM-1 was not directly investigated. Notably, the above model diverges from the one proposed by Bachrach and colleagues for the immunosuppressive action of
Fn-CEACAM-1 signaling in T lymphocytes and NK cells, whereby bacterial engagement leads to CEACAM-1 dimerization/activation, and possibly the phosphatase-dependent downregulation of antigen-triggered activation signals [
20,
33].
The mechanism leading to SHP-2 dissociation from CEACAM-1 upon
Fn binding remains elusive, although the dephosphorylation of the CEACAM-1 ITIM at positions Y493 and Y520, the putative SHP-2 docking sites, represents a plausible explanation. This idea is consistent with the reduced CEACAM-1 recovery from
Fn-stimulated CSC lysates in pull-down experiments, where the two SHP-2 SH2 phosphotyrosine binding domains were used as bait (
Figure 5E). Unfortunately, we could not detect constitutive CEACAM-1 tyrosine phosphorylation in unstimulated CSCs, nor dephosphorylation upon
Fn challenge (
Supplementary Figure S8); future experiments with CSCs expressing CEACAM-1 mutant forms lacking the ITIM region will hopefully help to clarify this point.
In a broader perspective, our results carry significant implications for CRC biology. While recent works have focused on the immunomodulatory and immunosuppressive consequences of CEACAM-1 engagement by
Fn in T and NK cells, the data presented here underscore the relevance of this pathogenic interaction on the cancer cell side. Relevant to this aspect, CEACAM-1 has been recognized as playing a dual role in colorectal cancerogenesis, possibly tumor-suppressive in the early phases, and supportive for malignant progression and metastasis in advanced disease [
39,
40]. The evidence presented for
Fn dissociating CEACAM-1 from its inhibitory effector SHP-2 is consistent with the well-established paradigm whereby microbial carcinogenesis targets tumor suppressor mechanisms [
41]. On the other hand, by operating in CSCs, the
Fn–CEACAM-1–SHP2 axis qualifies well to link bacterial infection with cancer progression and dissemination. In keeping with this attractive hypothesis, CEACAM-1 is highly expressed in EpCAM+ liver CSCs [
42], and its overexpression induces stem cell markers and EMT, a stem-cell-related phenomenon, in HT29 and HCT16 CRC cell lines [
43]. Additionally, CD66c (a.k.a. CEACAM-6), another member of the CEACAM family, has been reported to mark a population of CD133+ CR-CSC, and its downregulation arrests tumor growth in vivo [
44]. While the preliminary studies presented in
Supplementary Figure S9 suggest that CEACAM-1 protein expression is not affected by CSC differentiation, we are currently evaluating whether the CEACAM-1 signaling properties, and by extension biological responses to
Fn, vary across spheroidal cell subsets in a fashion that correlates with stemness and Wnt signaling capacity. Likewise, further studies aimed at better dissecting the role of the CEACAM-1–SHP axis in the proinflammatory and Wnt-related responses elicited by
Fn in CSCs (
Figure 2 and
Figure 3) are warranted [
45,
46].



 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




