


Co-Stimulatory Receptor Signaling in CAR-T Cells


Abstract
:1. Introduction
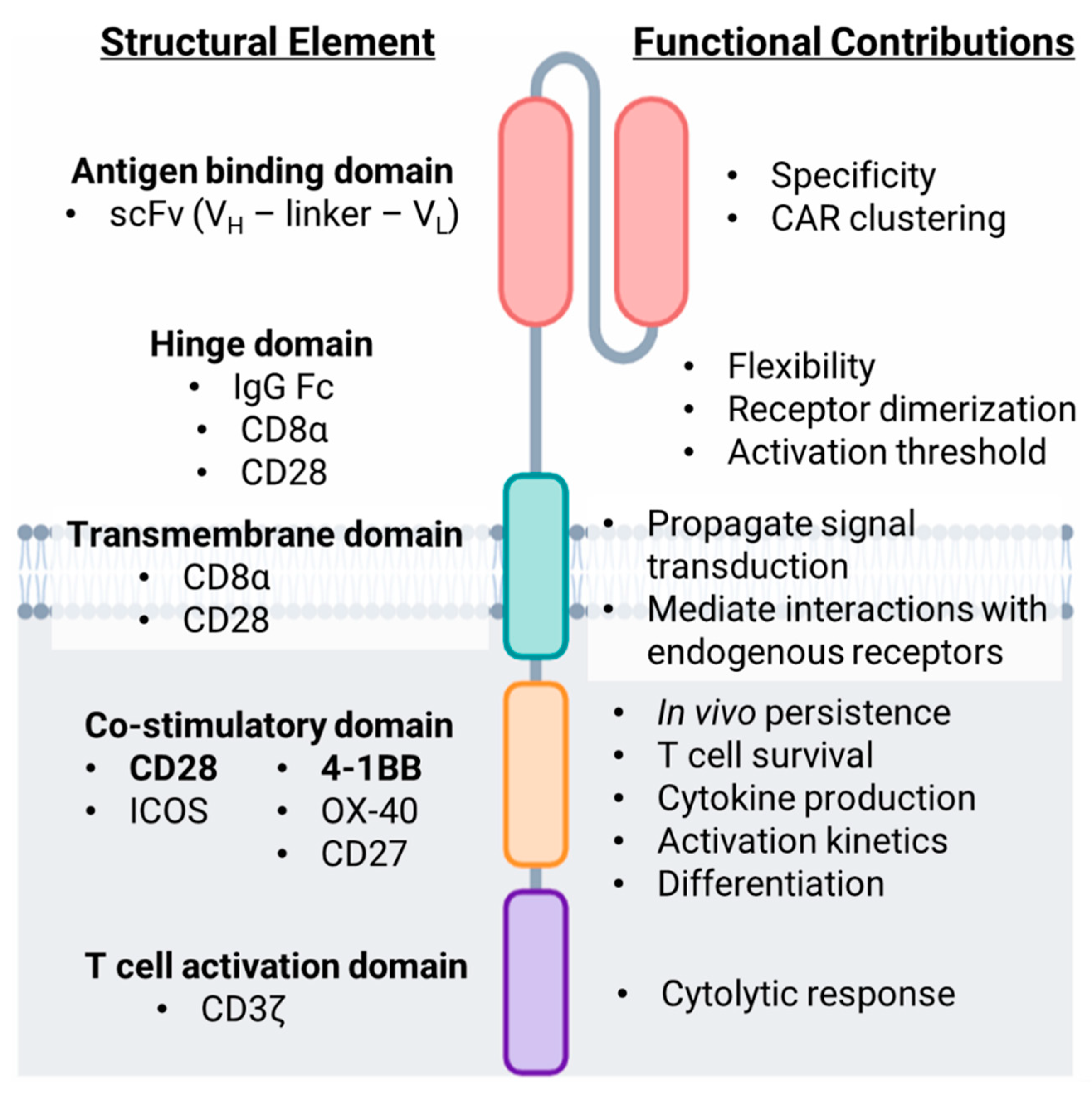
2. Structure and Design of the Chimeric Antigen Receptor (CAR)
2.1. Extracellular Domains
2.1.1. Antigen Targeting Domain
2.1.2. Hinge Domain
2.2. Intracellular Domains
2.2.1. Transmembrane Domain
2.2.2. T Cell Activation Domain
2.2.3. Co-Stimulatory Domain
3. Co-stimulatory Receptor Signaling Pathways
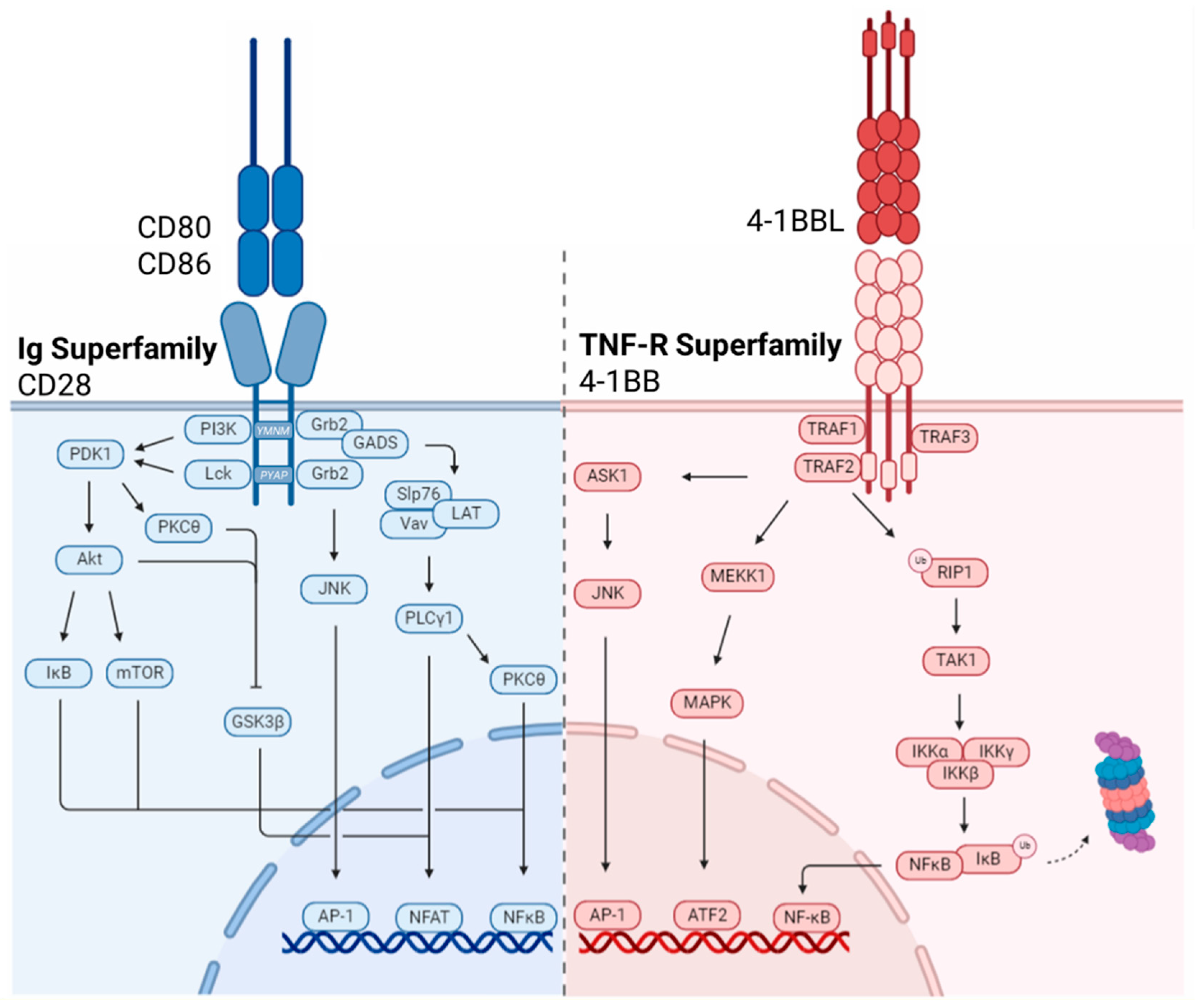
3.1. Immunoglobulin Superfamily
3.1.1. CD28
3.1.2. Inducible T Cell Co-Stimulator (ICOS)
3.2. TNF-R Superfamily
3.2.1. 4-1BB
3.2.2. OX40
3.2.3. CD27
4. Functional Implications of Co-Stimulation
4.1. Phenotype
4.1.1. T Cell Differentiation
4.1.2. Metabolic Profile
4.2. Response Kinetics
4.3. Persistence and Durability
4.4. Clinical Efficacy and Associated Toxicities
5. Future Perspectives: Harnessing Co-Stimulation to Enhance CAR-T Cell Efficacy
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eshhar, Z.; Waks, T.; Gross, G.; Schindler, D.G. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc. Natl. Acad. Sci. USA 1993, 90, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Vesely, M.D.; Schreiber, R.D. Cancer immunoediting: Antigens, mechanisms, and implications to cancer immunotherapy. Ann. N. Y. Acad. Sci. 2013, 1284, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Maher, J.; Brentjens, R.J.; Gunset, G.; Rivière, I.; Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRζ /CD28 receptor. Nat. Biotechnol. 2002, 20, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Eshhar, Z.; Waks, T.; Gross, G. The emergence of T-bodies/CAR T cells. Cancer J. 2014, 20, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.H.; Liu, J.; Wang, B.Y.; Chen, Y.X.; Cao, X.M.; Yang, Y.; Zhang, Y.L.; Wang, F.X.; Zhang, P.Y.; Lei, B.; et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J. Hematol. Oncol. 2018, 11, 141. [Google Scholar] [CrossRef]
- Munshi, N.C.; Larry, D.; Anderson, J.; Shah, N.; Jagannath, S.; Berdeja, J.G.; Lonial, S.; Raje, N.S.; Siegel, D.S.D.; Lin, Y.; et al. Idecabtagene vicleucel (ide-cel; bb2121), a BCMA-targeted CAR T-cell therapy, in patients with relapsed and refractory multiple myeloma (RRMM): Initial KarMMa results. J. Clin. Oncol. 2020, 38, 8503. [Google Scholar] [CrossRef]
- Wang, B.-Y.; Zhao, W.-H.; Liu, J.; Chen, Y.-X.; Cao, X.-M.; Yang, Y.; Zhang, Y.-L.; Wang, F.-X.; Zhang, P.-Y.; Lei, B.; et al. Long-Term Follow-up of a Phase 1, First-in-Human Open-Label Study of LCAR-B38M, a Structurally Differentiated Chimeric Antigen Receptor T (CAR-T) Cell Therapy Targeting B-Cell Maturation Antigen (BCMA), in Patients (pts) with Relapsed/Refractory Multiple Myeloma (RRMM). Blood 2019, 134, 579. [Google Scholar] [CrossRef]
- Roex, G.; Timmers, M.; Wouters, K.; Campillo-Davo, D.; Flumens, D.; Schroyens, W.; Chu, Y.; Berneman, Z.N.; Lion, E.; Luo, F.; et al. Safety and clinical efficacy of BCMA CAR-T-cell therapy in multiple myeloma. J. Hematol. Oncol. 2020, 13, 164. [Google Scholar] [CrossRef]
- Sadelain, M.; Rivière, I.; Brentjens, R. Targeting tumours with genetically enhanced T lymphocytes. Nat. Rev. Cancer 2003, 3, 35–45. [Google Scholar] [CrossRef]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T cells in solid tumors: Challenges and opportunities. Stem Cell Res. Ther. 2021, 12, 81. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef] [PubMed]
- Townsend, M.H.; Shrestha, G.; Robison, R.A.; O’Neill, K.L. The expansion of targetable biomarkers for CAR T cell therapy. J. Exp. Clin. Cancer Res. 2018, 37, 163. [Google Scholar] [CrossRef] [PubMed]
- Bailey, S.R.; Maus, M.V. Gene editing for immune cell therapies. Nat. Biotechnol. 2019, 37, 1425–1434. [Google Scholar] [CrossRef]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef]
- Singh, N.; Frey, N.V.; Engels, B.; Barrett, D.M.; Shestova, O.; Ravikumar, P.; Cummins, K.D.; Lee, Y.G.; Pajarillo, R.; Chun, I.; et al. Antigen-independent activation enhances the efficacy of 4-1BB-costimulated CD22 CAR T cells. Nat. Med. 2021, 27, 842–850. [Google Scholar] [CrossRef]
- Alabanza, L.; Pegues, M.; Geldres, C.; Shi, V.; Wiltzius, J.J.W.; Sievers, S.A.; Yang, S.; Kochenderfer, J.N. Function of Novel Anti-CD19 Chimeric Antigen Receptors with Human Variable Regions Is Affected by Hinge and Transmembrane Domains. Mol. Ther. 2017, 25, 2452–2465. [Google Scholar] [CrossRef]
- Geldres, C.; Savoldo, B.; Dotti, G. Chimeric antigen receptor-redirected T cells return to the bench. Semin. Immunol. 2016, 28, 3–9. [Google Scholar] [CrossRef]
- Guest, R.D.; Hawkins, R.E.; Kirillova, N.; Cheadle, E.J.; Arnold, J.; O’Neill, A.; Irlam, J.; Chester, K.A.; Kemshead, J.T.; Shaw, D.M.; et al. The Role of Extracellular Spacer Regions in the Optimal Design of Chimeric Immune Receptors: Evaluation of Four Different scFvs and Antigens. J. Immunother. 2005, 28. [Google Scholar] [CrossRef]
- Hudecek, M.; Sommermeyer, D.; Kosasih, P.L.; Silva-Benedict, A.; Liu, L.; Rader, C.; Jensen, M.C.; Riddell, S.R. The Nonsignaling Extracellular Spacer Domain of Chimeric Antigen Receptors Is Decisive for In Vivo Antitumor Activity. Cancer Immunol. Res. 2015, 3, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Majzner, R.G.; Rietberg, S.P.; Sotillo, E.; Dong, R.; Vachharajani, V.T.; Labanieh, L.; Myklebust, J.H.; Kadapakkam, M.; Weber, E.W.; Tousley, A.M.; et al. Tuning the Antigen Density Requirement for CAR T-cell Activity. Cancer Discov. 2020, 10, 702–723. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Calderon, H.; Posey, A.D.; Maus, M.V. Engineering and Design of Chimeric Antigen Receptors. Mol. Ther.-Methods Clin. Dev. 2019, 12, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Muller, Y.D.; Nguyen, D.P.; Ferreira, L.M.R.; Ho, P.; Raffin, C.; Valencia, R.V.B.; Congrave-Wilson, Z.; Roth, T.L.; Eyquem, J.; Van Gool, F.; et al. The CD28-Transmembrane Domain Mediates Chimeric Antigen Receptor Heterodimerization With CD28. Front. Immunol. 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Frigault, M.J.; Lee, J.; Basil, M.C.; Carpenito, C.; Motohashi, S.; Scholler, J.; Kawalekar, O.U.; Guedan, S.; McGettigan, S.E.; Posey, A.D., Jr.; et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol. Res. 2015, 3, 356–367. [Google Scholar] [CrossRef]
- Love, P.E.; Hayes, S.M. ITAM-mediated Signaling by the T-Cell Antigen Receptor. Cold Spring Harb. Perspect. Biol. 2010, 2, a002485. [Google Scholar] [CrossRef]
- June, C.H.; Fletcher, M.C.; Ledbetter, J.A.; Schieven, G.L.; Siegel, J.N.; Phillips, A.F.; Samelson, L.E. Inhibition of tyrosine phosphorylation prevents T-cell receptor-mediated signal transduction. Proc. Natl. Acad. Sci. USA 1990, 87, 7722–7726. [Google Scholar] [CrossRef]
- Samelson, L.E.; Patel, M.D.; Weissman, A.M.; Harford, J.B.; Klausner, R.D. Antigen activation of murine T cells induces tyrosine phosphorylation of a polypeptide associated with the T cell antigen receptor. Cell 1986, 46, 1083–1090. [Google Scholar] [CrossRef]
- Hartl, F.A.; Beck-Garcìa, E.; Woessner, N.M.; Flachsmann, L.J.; Cárdenas, R.M.H.V.; Brandl, S.M.; Taromi, S.; Fiala, G.J.; Morath, A.; Mishra, P.; et al. Noncanonical binding of Lck to CD3ε promotes TCR signaling and CAR function. Nat. Immunol. 2020, 21, 902–913. [Google Scholar] [CrossRef]
- Lindner, S.E.; Johnson, S.M.; Brown, C.E.; Wang, L.D. Chimeric antigen receptor signaling: Functional consequences and design implications. Sci. Adv. 2020, 6, eaaz3223. [Google Scholar] [CrossRef]
- Sun, C.; Shou, P.; Du, H.; Hirabayashi, K.; Chen, Y.; Herring, L.E.; Ahn, S.; Xu, Y.; Suzuki, K.; Li, G.; et al. THEMIS-SHP1 Recruitment by 4-1BB Tunes LCK-Mediated Priming of Chimeric Antigen Receptor-Redirected T Cells. Cancer Cell 2020, 37, 216–225.e216. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R.; et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.K.; Wang, L.-C.; Dolfi, D.V.; Wilson, C.B.; Ranganathan, R.; Sun, J.; Kapoor, V.; Scholler, J.; Puré, E.; Milone, M.C.; et al. Multifactorial T-cell Hypofunction That Is Reversible Can Limit the Efficacy of Chimeric Antigen Receptor–Transduced Human T cells in Solid Tumors. Clin. Cancer Res. 2014, 20, 4262–4273. [Google Scholar] [CrossRef]
- Feucht, J.; Sun, J.; Eyquem, J.; Ho, Y.-J.; Zhao, Z.; Leibold, J.; Dobrin, A.; Cabriolu, A.; Hamieh, M.; Sadelain, M. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat. Med. 2019, 25, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, M.H.; Westwood, J.A.; Hwu, P. Dual-specific T cells combine proliferation and antitumor activity. Nat. Biotechnol. 2002, 20, 1221–1227. [Google Scholar] [CrossRef]
- Cooper, L.J.N.; Topp, M.S.; Serrano, L.M.; Gonzalez, S.; Chang, W.-C.; Naranjo, A.; Wright, C.; Popplewell, L.; Raubitschek, A.; Forman, S.J.; et al. T-cell clones can be rendered specific for CD19: Toward the selective augmentation of the graft-versus-B–lineage leukemia effect. Blood 2003, 101, 1637–1644. [Google Scholar] [CrossRef]
- Hwu, P.; Shafer, G.E.; Treisman, J.; Schindler, D.G.; Gross, G.; Cowherd, R.; Rosenberg, S.A.; Eshhar, Z. Lysis of ovarian cancer cells by human lymphocytes redirected with a chimeric gene composed of an antibody variable region and the Fc receptor gamma chain. J. Exp. Med. 1993, 178, 361–366. [Google Scholar] [CrossRef]
- Gong, M.C.; Latouche, J.B.; Krause, A.; Heston, W.D.; Bander, N.H.; Sadelain, M. Cancer patient T cells genetically targeted to prostate-specific membrane antigen specifically lyse prostate cancer cells and release cytokines in response to prostate-specific membrane antigen. Neoplasia 1999, 1, 123–127. [Google Scholar] [CrossRef]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A Phase I Study on Adoptive Immunotherapy Using Gene-Modified T Cells for Ovarian Cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef]
- Jensen, M.C.; Popplewell, L.; Cooper, L.J.; Digiusto, D.; Kalos, M.; Ostberg, J.R.; Forman, S.J. Antitransgene Rejection Responses Contribute to Attenuated Persistence of Adoptively Transferred CD20/CD19-Specific Chimeric Antigen Receptor Redirected T Cells in Humans. Biol. Blood Marrow Transplant. 2010, 16, 1245–1256. [Google Scholar] [CrossRef] [Green Version]
- Krause, A.; Guo, H.-F.; Latouche, J.-B.; Tan, C.; Cheung, N.-K.V.; Sadelain, M. Antigen-dependent CD28 Signaling Selectively Enhances Survival and Proliferation in Genetically Modified Activated Human Primary T Lymphocytes. J. Exp. Med. 1998, 188, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Kowolik, C.M.; Topp, M.S.; Gonzalez, S.; Pfeiffer, T.; Olivares, S.; Gonzalez, N.; Smith, D.D.; Forman, S.J.; Jensen, M.C.; Cooper, L.J.N. CD28 Costimulation Provided through a CD19-Specific Chimeric Antigen Receptor Enhances In vivo Persistence and Antitumor Efficacy of Adoptively Transferred T Cells. Cancer Res. 2006, 66, 10995–11004. [Google Scholar] [CrossRef] [PubMed]
- Hombach, A.; Wieczarkowiecz, A.; Marquardt, T.; Heuser, C.; Usai, L.; Pohl, C.; Seliger, B.; Abken, H. Tumor-Specific T Cell Activation by Recombinant Immunoreceptors: CD3ζ Signaling and CD28 Costimulation Are Simultaneously Required for Efficient IL-2 Secretion and Can Be Integrated Into One Combined CD28/CD3ζ Signaling Receptor Molecule. J. Immunol. 2001, 167, 6123–6131. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.J.; Latouche, J.-B.; Santos, E.; Marti, F.; Gong, M.C.; Lyddane, C.; King, P.D.; Larson, S.; Weiss, M.; Rivière, I.; et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat. Med. 2003, 9, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, M.K.; Taylor, P.S.; Norton, S.D.; Urdahl, K.B. CD28 delivers a costimulatory signal involved in antigen-specific IL-2 production by human T cells. J. Immunol. 1991, 147, 2461–2466. [Google Scholar]
- Schwartz, R.H. Costimulation of T lymphocytes: The role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell 1992, 71, 1065–1068. [Google Scholar] [CrossRef]
- Harding, F.A.; McArthur, J.G.; Gross, J.A.; Raulet, D.H.; Allison, J.P. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature 1992, 356, 607–609. [Google Scholar] [CrossRef]
- Boise, L.H.; Minn, A.J.; Noel, P.J.; June, C.H.; Accavitti, M.A.; Lindsten, T.; Thompson, C.B. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity 1995, 3, 87–98. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): A phase 1b/2 open-label study. Lancet 2021, 398, 314–324. [Google Scholar] [CrossRef]
- Munshi, N.C.; Anderson, L.D.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.; Usmani, S.Z.; Berdeja, J.G.; Agha, M.; Cohen, A.D.; Hari, P.; Avigan, D.; Deol, A.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene Autoleucel, an Anti–B-cell Maturation Antigen Chimeric Antigen Receptor T-Cell Therapy, for Relapsed/Refractory Multiple Myeloma: CARTITUDE-1 2-Year Follow-Up. J. Clin. Oncol. 2022. [Google Scholar] [CrossRef]
- Shah, B.D.; Ghobadi, A.; Oluwole, O.O.; Logan, A.C.; Boissel, N.; Cassaday, R.D.; Leguay, T.; Bishop, M.R.; Topp, M.S.; Tzachanis, D.; et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: Phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet 2021, 398, 491–502. [Google Scholar] [CrossRef]
- Shah, B.D.; Bishop, M.R.; Oluwole, O.O.; Logan, A.C.; Baer, M.R.; Donnellan, W.B.; O’Dwyer, K.M.; Holmes, H.; Arellano, M.L.; Ghobadi, A.; et al. KTE-X19 anti-CD19 CAR T-cell therapy in adult relapsed/refractory acute lymphoblastic leukemia: ZUMA-3 phase 1 results. Blood 2021, 138, 11–22. [Google Scholar] [CrossRef]
- Melenhorst, J.J.; Chen, G.M.; Wang, M.; Porter, D.L.; Chen, C.; Collins, M.A.; Gao, P.; Bandyopadhyay, S.; Sun, H.; Zhao, Z.; et al. Decade-long leukaemia remissions with persistence of CD4+ CAR T cells. Nature 2022, 602, 503–509. [Google Scholar] [CrossRef]
- Roselli, E.; Boucher, J.C.; Li, G.; Kotani, H.; Spitler, K.; Reid, K.; Cervantes, E.V.; Bulliard, Y.; Tu, N.; Lee, S.B.; et al. 4-1BB and optimized CD28 co-stimulation enhances function of human mono-specific and bi-specific third-generation CAR T cells. J. ImmunoTherapy Cancer 2021, 9, e003354. [Google Scholar] [CrossRef]
- Ramos, C.A.; Rouce, R.; Robertson, C.S.; Reyna, A.; Narala, N.; Vyas, G.; Mehta, B.; Zhang, H.; Dakhova, O.; Carrum, G.; et al. In Vivo Fate and Activity of Second- versus Third-Generation CD19-Specific CAR-T Cells in B Cell Non-Hodgkin’s Lymphomas. Mol. Ther. 2018, 26, 2727–2737. [Google Scholar] [CrossRef]
- Zhong, X.S.; Matsushita, M.; Plotkin, J.; Riviere, I.; Sadelain, M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol. Ther. 2010, 18, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Abate-Daga, D.; Lagisetty, K.H.; Tran, E.; Zheng, Z.; Gattinoni, L.; Yu, Z.; Burns, W.R.; Miermont, A.M.; Teper, Y.; Rudloff, U.; et al. A novel chimeric antigen receptor against prostate stem cell antigen mediates tumor destruction in a humanized mouse model of pancreatic cancer. Hum. Gene Ther. 2014, 25, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Till, B.G.; Jensen, M.C.; Wang, J.; Qian, X.; Gopal, A.K.; Maloney, D.G.; Lindgren, C.G.; Lin, Y.; Pagel, J.M.; Budde, L.E.; et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: Pilot clinical trial results. Blood 2012, 119, 3940–3950. [Google Scholar] [CrossRef] [PubMed]
- Weinkove, R.; George, P.; Dasyam, N.; McLellan, A.D. Selecting costimulatory domains for chimeric antigen receptors: Functional and clinical considerations. Clin. Transl. Immunol. 2019, 8, e1049. [Google Scholar] [CrossRef] [PubMed]
- Carpenito, C.; Milone, M.C.; Hassan, R.; Simonet, J.C.; Lakhal, M.; Suhoski, M.M.; Varela-Rohena, A.; Haines, K.M.; Heitjan, D.F.; Albelda, S.M.; et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc. Natl. Acad. Sci. USA 2009, 106, 3360–3365. [Google Scholar] [CrossRef] [PubMed]
- George, P.; Dasyam, N.; Giunti, G.; Mester, B.; Bauer, E.; Andrews, B.; Perera, T.; Ostapowicz, T.; Frampton, C.; Li, P.; et al. Third-generation anti-CD19 chimeric antigen receptor T-cells incorporating a TLR2 domain for relapsed or refractory B-cell lymphoma: A phase I clinical trial protocol (ENABLE). BMJ Open 2020, 10, e034629. [Google Scholar] [CrossRef]
- Enblad, G.; Karlsson, H.; Gammelgård, G.; Wenthe, J.; Lövgren, T.; Amini, R.M.; Wikstrom, K.I.; Essand, M.; Savoldo, B.; Hallböök, H.; et al. A Phase I/IIa Trial Using CD19-Targeted Third-Generation CAR T Cells for Lymphoma and Leukemia. Clin Cancer Res. 2018, 24, 6185–6194. [Google Scholar] [CrossRef]
- Karlsson, H.; Svensson, E.; Gigg, C.; Jarvius, M.; Olsson-Strömberg, U.; Savoldo, B.; Dotti, G.; Loskog, A. Evaluation of Intracellular Signaling Downstream Chimeric Antigen Receptors. PLoS ONE 2015, 10, e0144787. [Google Scholar] [CrossRef]
- Guedan, S.; Posey, A.D., Jr.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3, e96976. [Google Scholar] [CrossRef]
- Milone, M.C.; Fish, J.D.; Carpenito, C.; Carroll, R.G.; Binder, G.K.; Teachey, D.; Samanta, M.; Lakhal, M.; Gloss, B.; Danet-Desnoyers, G.; et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther. 2009, 17, 1453–1464. [Google Scholar] [CrossRef]
- Salter, A.I.; Ivey, R.G.; Kennedy, J.J.; Voillet, V.; Rajan, A.; Alderman, E.J.; Voytovich, U.J.; Lin, C.; Sommermeyer, D.; Liu, L.; et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci. Signal. 2018, 11, eaat6753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijewarnasuriya, D.; Bebernitz, C.; Lopez, A.V.; Rafiq, S.; Brentjens, R.J. Excessive Costimulation Leads to Dysfunction of Adoptively Transferred T Cells. Cancer Immunol. Res. 2020, 8, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Madar, A.; Casado-Medrano, V.; Shaw, C.; Wing, A.; Liu, F.; Young, R.M.; June, C.H.; Posey, A.D., Jr. Single residue in CD28-costimulated CAR-T cells limits long-term persistence and antitumor durability. J. Clin. Investig. 2020, 130, 3087–3097. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.C.; Li, G.; Kotani, H.; Cabral, M.L.; Morrissey, D.; Lee, S.B.; Spitler, K.; Beatty, N.J.; Cervantes, E.V.; Shrestha, B.; et al. CD28 Costimulatory Domain–Targeted Mutations Enhance Chimeric Antigen Receptor T-cell Function. Cancer Immunol. Res. 2021, 9, 62–74. [Google Scholar] [CrossRef]
- Leddon, S.A.; Fettis, M.M.; Abramo, K.; Kelly, R.; Oleksyn, D.; Miller, J. The CD28 Transmembrane Domain Contains an Essential Dimerization Motif. Front. Immunol. 2020, 11, 1519. [Google Scholar] [CrossRef] [PubMed]
- Ramello, M.C.; Benzaïd, I.; Kuenzi, B.M.; Lienlaf-Moreno, M.; Kandell, W.M.; Santiago, D.N.; Pabón-Saldaña, M.; Darville, L.; Fang, B.; Rix, U.; et al. An immunoproteomic approach to characterize the CAR interactome and signalosome. Sci. Signal. 2019, 12, eaap9777. [Google Scholar] [CrossRef]
- June, C.H.; Ledbetter, J.A.; Linsley, P.S.; Thompson, C.B. Role of the CD28 receptor in T-cell activation. Immunol. Today 1990, 11, 211–216. [Google Scholar] [CrossRef]
- Burr, J.S.; Savage, N.D.; Messah, G.E.; Kimzey, S.L.; Shaw, A.S.; Arch, R.H.; Green, J.M. Cutting edge: Distinct motifs within CD28 regulate T cell proliferation and induction of Bcl-XL. J. Immunol. 2001, 166, 5331–5335. [Google Scholar] [CrossRef]
- Boomer, J.S.; Deppong, C.M.; Shah, D.D.; Bricker, T.L.; Green, J.M. Cutting edge: A double-mutant knockin of the CD28 YMNM and PYAP motifs reveals a critical role for the YMNM motif in regulation of T cell proliferation and Bcl-xL expression. J. Immunol. 2014, 192, 3465–3469. [Google Scholar] [CrossRef]
- Frauwirth, K.A.; Riley, J.L.; Harris, M.H.; Parry, R.V.; Rathmell, J.C.; Plas, D.R.; Elstrom, R.L.; June, C.H.; Thompson, C.B. The CD28 signaling pathway regulates glucose metabolism. Immunity 2002, 16, 769–777. [Google Scholar] [CrossRef]
- DuPage, M.; Chopra, G.; Quiros, J.; Rosenthal, W.L.; Morar, M.M.; Holohan, D.; Zhang, R.; Turka, L.; Marson, A.; Bluestone, J.A. The chromatin-modifying enzyme Ezh2 is critical for the maintenance of regulatory T cell identity after activation. Immunity 2015, 42, 227–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attema, J.L.; Reeves, R.; Murray, V.; Levichkin, I.; Temple, M.D.; Tremethick, D.J.; Shannon, M.F. The human IL-2 gene promoter can assemble a positioned nucleosome that becomes remodeled upon T cell activation. J. Immunol. 2002, 169, 2466–2476. [Google Scholar] [CrossRef]
- Thomas, R.M.; Gao, L.; Wells, A.D. Signals from CD28 induce stable epigenetic modification of the IL-2 promoter. J. Immunol. 2005, 174, 4639–4646. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; Ledbetter, J.A.; Gillespie, M.M.; Lindsten, T.; Thompson, C.B. T-cell proliferation involving the CD28 pathway is associated with cyclosporine-resistant interleukin 2 gene expression. Mol. Cell Biol. 1987, 7, 4472–4481. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, V.S.; Truitt, K.E.; Imboden, J.B.; Weiss, A. CD28 mediates transcriptional upregulation of the interleukin-2 (IL-2) promoter through a composite element containing the CD28RE and NF-IL-2B AP-1 sites. Mol. Cell Biol. 1997, 17, 4051–4058. [Google Scholar] [CrossRef]
- Fraser, J.D.; Irving, B.A.; Crabtree, G.R.; Weiss, A. Regulation of interleukin-2 gene enhancer activity by the T cell accessory molecule CD28. Science 1991, 251, 313–316. [Google Scholar] [CrossRef]
- Thompson, C.B.; Lindsten, T.; Ledbetter, J.A.; Kunkel, S.L.; Young, H.A.; Emerson, S.G.; Leiden, J.M.; June, C.H. CD28 activation pathway regulates the production of multiple T-cell-derived lymphokines/cytokines. Proc. Natl. Acad. Sci. USA 1989, 86, 1333–1337. [Google Scholar] [CrossRef]
- Lindstein, T.; June, C.H.; Ledbetter, J.A.; Stella, G.; Thompson, C.B. Regulation of lymphokine messenger RNA stability by a surface-mediated T cell activation pathway. Science 1989, 244, 339–343. [Google Scholar] [CrossRef]
- Salazar-Fontana, L.I.; Barr, V.; Samelson, L.E.; Bierer, B.E. CD28 engagement promotes actin polymerization through the activation of the small Rho GTPase Cdc42 in human T cells. J. Immunol. 2003, 171, 2225–2232. [Google Scholar] [CrossRef]
- Muscolini, M.; Camperio, C.; Porciello, N.; Caristi, S.; Capuano, C.; Viola, A.; Galandrini, R.; Tuosto, L. Phosphatidylinositol 4-phosphate 5-kinase α and Vav1 mutual cooperation in CD28-mediated actin remodeling and signaling functions. J. Immunol. 2015, 194, 1323–1333. [Google Scholar] [CrossRef]
- Butte, M.J.; Lee, S.J.; Jesneck, J.; Keir, M.E.; Haining, W.N.; Sharpe, A.H. CD28 costimulation regulates genome-wide effects on alternative splicing. PLoS ONE 2012, 7, e40032. [Google Scholar] [CrossRef] [PubMed]
- Holling, G.A.; Sharda, A.P.; Honikel, M.M.; James, C.M.; Lightman, S.M.; Qiao, G.; Singel, K.L.; Emmons, T.R.; Giridharan, T.; Hou, S.; et al. ARS2-directed alternative splicing mediates CD28 driven T cell glycolysis and effector function. bioRxiv 2021. [Google Scholar] [CrossRef]
- Esensten, J.H.; Helou, Y.A.; Chopra, G.; Weiss, A.; Bluestone, J.A. CD28 Costimulation: From Mechanism to Therapy. Immunity 2016, 44, 973–988. [Google Scholar] [CrossRef] [PubMed]
- Boomer, J.S.; Green, J.M. An Enigmatic Tail of CD28 Signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002436. [Google Scholar] [CrossRef] [PubMed]
- Frauwirth, K.A.; Thompson, C.B. Activation and inhibition of lymphocytes by costimulation. J. Clin. Investig. 2002, 109, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Harada, Y.; Watanabe, R.; Inutake, Y.; Ogawa, S.; Onuki, K.; Kagaya, S.; Tanabe, K.; Kishimoto, H.; Abe, R. CD28 stimulation triggers NF-kappaB activation through the CARMA1-PKCtheta-Grb2/Gads axis. Int. Immunol. 2008, 20, 1507–1515. [Google Scholar] [CrossRef]
- Watanabe, R.; Harada, Y.; Takeda, K.; Takahashi, J.; Ohnuki, K.; Ogawa, S.; Ohgai, D.; Kaibara, N.; Koiwai, O.; Tanabe, K.; et al. Grb2 and Gads exhibit different interactions with CD28 and play distinct roles in CD28-mediated costimulation. J. Immunol. 2006, 177, 1085–1091. [Google Scholar] [CrossRef]
- Coudronniere, N.; Villalba, M.; Englund, N.; Altman, A. NF-kappa B activation induced by T cell receptor/CD28 costimulation is mediated by protein kinase C-theta. Proc. Natl. Acad. Sci. USA 2000, 97, 3394–3399. [Google Scholar] [CrossRef]
- Dodson, L.F.; Boomer, J.S.; Deppong, C.M.; Shah, D.D.; Sim, J.; Bricker, T.L.; Russell, J.H.; Green, J.M. Targeted knock-in mice expressing mutations of CD28 reveal an essential pathway for costimulation. Mol. Cell Biol. 2009, 29, 3710–3721. [Google Scholar] [CrossRef]
- Andreotti, A.H.; Schwartzberg, P.L.; Joseph, R.E.; Berg, L.J. T-cell signaling regulated by the Tec family kinase, Itk. Cold Spring Harb. Perspect Biol. 2010, 2, a002287. [Google Scholar] [CrossRef]
- Gomez-Rodriguez, J.; Kraus, Z.J.; Schwartzberg, P.L. Tec family kinases Itk and Rlk/Txk in T lymphocytes: Cross-regulation of cytokine production and T-cell fates. Febs. J. 2011, 278, 1980–1989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, Y.; Tokushima, M.; Matsumoto, Y.; Ogawa, S.; Otsuka, M.; Hayashi, K.; Weiss, B.D.; June, C.H.; Abe, R. Critical requirement for the membrane-proximal cytosolic tyrosine residue for CD28-mediated costimulation in vivo. J. Immunol. 2001, 166, 3797–3803. [Google Scholar] [CrossRef] [PubMed]
- Okkenhaug, K.; Wu, L.; Garza, K.M.; La Rose, J.; Khoo, W.; Odermatt, B.; Mak, T.W.; Ohashi, P.S.; Rottapel, R. A point mutation in CD28 distinguishes proliferative signals from survival signals. Nat. Immunol. 2001, 2, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Friend, L.D.; Shah, D.D.; Deppong, C.; Lin, J.; Bricker, T.L.; Juehne, T.I.; Rose, C.M.; Green, J.M. A dose-dependent requirement for the proline motif of CD28 in cellular and humoral immunity revealed by a targeted knockin mutant. J. Exp. Med. 2006, 203, 2121–2133. [Google Scholar] [CrossRef]
- McAdam, A.J.; Chang, T.T.; Lumelsky, A.E.; Greenfield, E.A.; Boussiotis, V.A.; Duke-Cohan, J.S.; Chernova, T.; Malenkovich, N.; Jabs, C.; Kuchroo, V.K.; et al. Mouse inducible costimulatory molecule (ICOS) expression is enhanced by CD28 costimulation and regulates differentiation of CD4+ T cells. J. Immunol. 2000, 165, 5035–5040. [Google Scholar] [CrossRef]
- Hutloff, A.; Dittrich, A.M.; Beier, K.C.; Eljaschewitsch, B.; Kraft, R.; Anagnostopoulos, I.; Kroczek, R.A. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature 1999, 397, 263–266. [Google Scholar] [CrossRef]
- Yoshinaga, S.K.; Whoriskey, J.S.; Khare, S.D.; Sarmiento, U.; Guo, J.; Horan, T.; Shih, G.; Zhang, M.; Coccia, M.A.; Kohno, T.; et al. T-cell co-stimulation through B7RP-1 and ICOS. Nature 1999, 402, 827–832. [Google Scholar] [CrossRef]
- Fos, C.; Salles, A.; Lang, V.; Carrette, F.; Audebert, S.; Pastor, S.; Ghiotto, M.; Olive, D.; Bismuth, G.; Nunès, J.A. ICOS ligation recruits the p50alpha PI3K regulatory subunit to the immunological synapse. J. Immunol. 2008, 181, 1969–1977. [Google Scholar] [CrossRef]
- Coyle, A.J.; Lehar, S.; Lloyd, C.; Tian, J.; Delaney, T.; Manning, S.; Nguyen, T.; Burwell, T.; Schneider, H.; Gonzalo, J.A.; et al. The CD28-related molecule ICOS is required for effective T cell-dependent immune responses. Immunity 2000, 13, 95–105. [Google Scholar] [CrossRef]
- Gigoux, M.; Shang, J.; Pak, Y.; Xu, M.; Choe, J.; Mak, T.W.; Suh, W.K. Inducible costimulator promotes helper T-cell differentiation through phosphoinositide 3-kinase. Proc. Natl. Acad. Sci. USA 2009, 106, 20371–20376. [Google Scholar] [CrossRef]
- Wikenheiser, D.J.; Stumhofer, J.S. ICOS Co-Stimulation: Friend or Foe? Front. Immunol. 2016, 7, 304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, Y.; Ohgai, D.; Watanabe, R.; Okano, K.; Koiwai, O.; Tanabe, K.; Toma, H.; Altman, A.; Abe, R. A Single Amino Acid Alteration in Cytoplasmic Domain Determines IL-2 Promoter Activation by Ligation of CD28 but Not Inducible Costimulator (ICOS). J. Exp. Med. 2003, 197, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Heinrichs, J.; Leconte, J.; Haarberg, K.; Semple, K.; Liu, C.; Gigoux, M.; Kornete, M.; Piccirillo, C.A.; Suh, W.K.; et al. Phosphatidylinositol 3-kinase-independent signaling pathways contribute to ICOS-mediated T cell costimulation in acute graft-versus-host disease in mice. J. Immunol. 2013, 191, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Pedros, C.; Zhang, Y.; Hu, J.K.; Choi, Y.S.; Canonigo-Balancio, A.J.; Yates, J.R.; Altman, A.; Crotty, S.; Kong, K.-F. A TRAF-like motif of the inducible costimulator ICOS controls development of germinal center TFH cells via the kinase TBK1. Nat. Immunol. 2016, 17, 825–833. [Google Scholar] [CrossRef]
- Leconte, J.; Bagherzadeh Yazdchi, S.; Panneton, V.; Suh, W.-K. Inducible costimulator (ICOS) potentiates TCR-induced calcium flux by augmenting PLCγ1 activation and actin remodeling. Mol. Immunol. 2016, 79, 38–46. [Google Scholar] [CrossRef]
- Wen, T.; Bukczynski, J.; Watts, T.H. 4-1BB Ligand-Mediated Costimulation of Human T Cells Induces CD4 and CD8 T Cell Expansion, Cytokine Production, and the Development of Cytolytic Effector Function. J. Immunol. 2002, 168, 4897–4906. [Google Scholar] [CrossRef]
- Croft, M. Costimulation of T cells by OX40, 4-1BB, and CD27. Cytokine Growth Factor Rev. 2003, 14, 265–273. [Google Scholar] [CrossRef]
- Pollok, K.E.; Kim, Y.J.; Zhou, Z.; Hurtado, J.; Kim, K.K.; Pickard, R.T.; Kwon, B.S. Inducible T cell antigen 4-1BB. Analysis of expression and function. J. Immunol. 1993, 150, 771–781. [Google Scholar]
- Cannons, J.L.; Hoeflich, K.P.; Woodgett, J.R.; Watts, T.H. Role of the Stress Kinase Pathway in Signaling Via the T Cell Costimulatory Receptor 4-1BB. J. Immunol. 1999, 163, 2990–2998. [Google Scholar]
- Croft, M. The role of TNF superfamily members in T-cell function and diseases. Nat. Rev. Immunol. 2009, 9, 271–285. [Google Scholar] [CrossRef]
- Arch, R.H.; Gedrich, R.W.; Thompson, C.B. Tumor necrosis factor receptor-associated factors (TRAFs)—a family of adapter proteins that regulates life and death. Genes Amp. Dev. 1998, 12, 2821–2830. [Google Scholar] [CrossRef] [PubMed]
- Watts, T.H. TNF/TNFR family members in costimulation of T cell responses. Annu. Rev. Immunol. 2005, 23, 23–68. [Google Scholar] [CrossRef]
- Jang, I.K.; Lee, Z.H.; Kim, Y.J.; Kim, S.H.; Kwon, B.S. Human 4-1BB (CD137) signals are mediated by TRAF2 and activate nuclear factor-kappa B. Biochem. Biophys. Res. Commun. 1998, 242, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Zapata, J.M.; Perez-Chacon, G.; Carr-Baena, P.; Martinez-Forero, I.; Azpilikueta, A.; Otano, I.; Melero, I. CD137 (4-1BB) Signalosome: Complexity Is a Matter of TRAFs. Front. Immunol. 2018, 9, 2618. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, L.; Pulle, G.; Liu, Y.; Tsitsikov, E.N.; Watts, T.H. ERK-dependent Bim modulation downstream of the 4-1BB-TRAF1 signaling axis is a critical mediator of CD8 T cell survival in vivo. J. Immunol. 2008, 180, 8093–8101. [Google Scholar] [CrossRef] [PubMed]
- Arch, R.H.; Thompson, C.B. 4-1BB and Ox40 are members of a tumor necrosis factor (TNF)-nerve growth factor receptor subfamily that bind TNF receptor-associated factors and activate nuclear factor kappaB. Mol. Cell Biol. 1998, 18, 558–565. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.; Matsuzawa, A.; Zhang, W.; Tseng, P.H.; Keats, J.J.; Wang, H.; Vignali, D.A.; Bergsagel, P.L.; Karin, M. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. Nat. Immunol. 2008, 9, 1364–1370. [Google Scholar] [CrossRef]
- Zarnegar, B.J.; Wang, Y.; Mahoney, D.J.; Dempsey, P.W.; Cheung, H.H.; He, J.; Shiba, T.; Yang, X.; Yeh, W.C.; Mak, T.W.; et al. Noncanonical NF-kappaB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat. Immunol. 2008, 9, 1371–1378. [Google Scholar] [CrossRef]
- Ichijo, H.; Nishida, E.; Irie, K.; ten Dijke, P.; Saitoh, M.; Moriguchi, T.; Takagi, M.; Matsumoto, K.; Miyazono, K.; Gotoh, Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 1997, 275, 90–94. [Google Scholar] [CrossRef]
- Nishitoh, H.; Saitoh, M.; Mochida, Y.; Takeda, K.; Nakano, H.; Rothe, M.; Miyazono, K.; Ichijo, H. ASK1 Is Essential for JNK/SAPK Activation by TRAF2. Mol. Cell 1998, 2, 389–395. [Google Scholar] [CrossRef]
- Menk, A.V.; Scharping, N.E.; Rivadeneira, D.B.; Calderon, M.J.; Watson, M.J.; Dunstane, D.; Watkins, S.C.; Delgoffe, G.M. 4-1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J. Exp. Med. 2018, 215, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Cannons, J.L.; Choi, Y.; Watts, T.H. Role of TNF Receptor-Associated Factor 2 and p38 Mitogen-Activated Protein Kinase Activation During 4-1BB-Dependent Immune Response. J. Immunol. 2000, 165, 6193–6204. [Google Scholar] [CrossRef] [PubMed]
- Saoulli, K.; Lee, S.Y.; Cannons, J.L.; Yeh, W.C.; Santana, A.; Goldstein, M.D.; Bangia, N.; DeBenedette, M.A.; Mak, T.W.; Choi, Y.; et al. CD28-independent, TRAF2-dependent costimulation of resting T cells by 4-1BB ligand. J. Exp. Med. 1998, 187, 1849–1862. [Google Scholar] [CrossRef] [PubMed]
- Croft, M. Co-stimulatory members of the TNFR family: Keys to effective T-cell immunity? Nat. Rev. Immunol. 2003, 3, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Paterson, D.J.; Jefferies, W.A.; Green, J.R.; Brandon, M.R.; Corthesy, P.; Puklavec, M.; Williams, A.F. Antigens of activated rat T lymphocytes including a molecule of 50,000 Mr detected only on CD4 positive T blasts. Mol. Immunol. 1987, 24, 1281–1290. [Google Scholar] [CrossRef]
- Godfrey, W.R.; Fagnoni, F.F.; Harara, M.A.; Buck, D.; Engleman, E.G. Identification of a human OX-40 ligand, a costimulator of CD4+ T cells with homology to tumor necrosis factor. J. Exp. Med. 1994, 180, 757–762. [Google Scholar] [CrossRef]
- Redmond, W.L.; Ruby, C.E.; Weinberg, A.D. The Role of OX40-Mediated Co-stimulation in T-Cell Activation and Survival. Crit. Rev. ™ Immunol. 2009, 29, 187–201. [Google Scholar] [CrossRef]
- Rogers, P.R.; Song, J.; Gramaglia, I.; Killeen, N.; Croft, M. OX40 promotes Bcl-xL and Bcl-2 expression and is essential for long-term survival of CD4 T cells. Immunity 2001, 15, 445–455. [Google Scholar] [CrossRef]
- Gramaglia, I.; Jember, A.; Pippig, S.D.; Weinberg, A.D.; Killeen, N.; Croft, M. The OX40 costimulatory receptor determines the development of CD4 memory by regulating primary clonal expansion. J. Immunol. 2000, 165, 3043–3050. [Google Scholar] [CrossRef]
- Weinberg, A.D.; Bourdette, D.N.; Sullivan, T.J.; Lemon, M.; Wallin, J.J.; Maziarz, R.; Davey, M.; Palida, F.; Godfrey, W.; Engleman, E.; et al. Selective depletion of myelin-reactive T cells with the anti-OX-40 antibody ameliorates autoimmune encephalomyelitis. Nat. Med. 1996, 2, 183–189. [Google Scholar] [CrossRef]
- Croft, M. Control of immunity by the TNFR-related molecule OX40 (CD134). Annu. Rev. Immunol. 2010, 28, 57–78. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, S.; Hori, T.; Imura, A.; Takaori-Kondo, A.; Uchiyama, T. Activation of OX40 Signal Transduction Pathways Leads to Tumor Necrosis Factor Receptor-associated Factor (TRAF) 2- and TRAF5-mediated NF-κB Activation. J. Biol. Chem. 1998, 273, 5808–5814. [Google Scholar] [CrossRef] [PubMed]
- So, T.; Choi, H.; Croft, M. OX40 complexes with phosphoinositide 3-kinase and protein kinase B (PKB) to augment TCR-dependent PKB signaling. J. Immunol. 2011, 186, 3547–3555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- So, T.; Song, J.; Sugie, K.; Altman, A.; Croft, M. Signals from OX40 regulate nuclear factor of activated T cells c1 and T cell helper 2 lineage commitment. Proc. Natl. Acad. Sci. USA 2006, 103, 3740–3745. [Google Scholar] [CrossRef]
- Redmond, W.L.; Gough, M.J.; Charbonneau, B.; Ratliff, T.L.; Weinberg, A.D. Defects in the Acquisition of CD8 T Cell Effector Function after Priming with Tumor or Soluble Antigen Can Be Overcome by the Addition of an OX40 Agonist. J. Immunol. 2007, 179, 7244–7253. [Google Scholar] [CrossRef]
- Ruby, C.E.; Montler, R.; Zheng, R.; Shu, S.; Weinberg, A.D. IL-12 Is Required for Anti-OX40-Mediated CD4 T Cell Survival. J. Immunol. 2008, 180, 2140–2148. [Google Scholar] [CrossRef]
- Prell, R.A.; Evans, D.E.; Thalhofer, C.; Shi, T.; Funatake, C.; Weinberg, A.D. OX40-mediated memory T cell generation is TNF receptor-associated factor 2 dependent. J. Immunol. 2003, 171, 5997–6005. [Google Scholar] [CrossRef]
- Krause, P.; Bruckner, M.; Uermösi, C.; Singer, E.; Groettrup, M.; Legler, D.F. Prostaglandin E2 enhances T-cell proliferation by inducing the costimulatory molecules OX40L, CD70, and 4-1BBL on dendritic cells. Blood J. Am. Soc. Hematol. 2009, 113, 2451–2460. [Google Scholar] [CrossRef]
- Iwamoto, S.; Iwai, S.-i.; Tsujiyama, K.; Kurahashi, C.; Takeshita, K.; Naoe, M.; Masunaga, A.; Ogawa, Y.; Oguchi, K.; Miyazaki, A. TNF-α drives human CD14+ monocytes to differentiate into CD70+ dendritic cells evoking Th1 and Th17 responses. J. Immunol. 2007, 179, 1449–1457. [Google Scholar] [CrossRef]
- Bullock, T.N.; Yagita, H. Induction of CD70 on dendritic cells through CD40 or TLR stimulation contributes to the development of CD8+ T cell responses in the absence of CD4+ T cells. J. Immunol. 2005, 174, 710–717. [Google Scholar] [CrossRef]
- Yamamoto, H.; Kishimoto, T.; Minamoto, S. NF-κB Activation in CD27 Signaling: Involvement of TNF Receptor-Associated Factors in Its Signaling and Identification of Functional Region of CD27. J. Immunol. 1998, 161, 4753–4759. [Google Scholar] [PubMed]
- Akiba, H.; Nakano, H.; Nishinaka, S.; Shindo, M.; Kobata, T.; Atsuta, M.; Morimoto, C.; Ware, C.F.; Malinin, N.L.; Wallach, D.; et al. CD27, a Member of the Tumor Necrosis Factor Receptor Superfamily, Activates NF-κB and Stress-activated Protein Kinase/c-Jun N-terminal Kinase via TRAF2, TRAF5, and NF-κB-inducing Kinase*. J. Biol. Chem. 1998, 273, 13353–13358. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, J.; Gravestein, L.A.; Tesselaar, K.; van Lier, R.A.; Schumacher, T.N.; Borst, J. CD27 is required for generation and long-term maintenance of T cell immunity. Nat. Immunol. 2000, 1, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Kobata, T.; Agematsu, K.; Kameoka, J.; Schlossman, S.F.; Morimoto, C. CD27 is a signal-transducing molecule involved in CD45RA+ naive T cell costimulation. J. Immunol. 1994, 153, 5422–5432. [Google Scholar]
- Keller, A.M.; Xiao, Y.; Peperzak, V.; Naik, S.H.; Borst, J. Costimulatory ligand CD70 allows induction of CD8+ T-cell immunity by immature dendritic cells in a vaccination setting. Blood 2009, 113, 5167–5175. [Google Scholar] [CrossRef]
- Dolfi, D.V.; Boesteanu, A.C.; Petrovas, C.; Xia, D.; Butz, E.A.; Katsikis, P.D. Late Signals from CD27 Prevent Fas-Dependent Apoptosis of Primary CD8+T Cells. J. Immunol. 2008, 180, 2912–2921. [Google Scholar] [CrossRef]
- Hendriks, J.; Xiao, Y.; Borst, J. CD27 Promotes Survival of Activated T Cells and Complements CD28 in Generation and Establishment of the Effector T Cell Pool. J. Exp. Med. 2003, 198, 1369–1380. [Google Scholar] [CrossRef]
- Loenen, W.A.; de Vries, E.; Gravestein, L.A.; Hintzen, R.Q.; Van Lier, R.A.; Borst, J. The CD27 membrane receptor, a lymphocyte-specific member of the nerve growth factor receptor family, gives rise to a soluble form by protein processing that does not involve receptor endocytosis. Eur. J. Immunol. 1992, 22, 447–455. [Google Scholar] [CrossRef]
- Hendriks, J.; Xiao, Y.; Rossen, J.W.; Van Der Sluijs, K.F.; Sugamura, K.; Ishii, N.; Borst, J. During Viral Infection of the Respiratory Tract, CD27, 4-1BB, and OX40 Collectively Determine Formation of CD8+ Memory T Cells and Their Capacity for Secondary Expansion. J. Immunol. 2005, 175, 1665–1676. [Google Scholar] [CrossRef]
- Xiao, Y.; Peperzak, V.; Keller, A.M.; Borst, J. CD27 instructs CD4+ T cells to provide help for the memory CD8+ T cell response after protein immunization. J. Immunol. 2008, 181, 1071–1082. [Google Scholar] [CrossRef]
- Schildknecht, A.; Miescher, I.; Yagita, H.; van den Broek, M. Priming of CD8+ T cell responses by pathogens typically depends on CD70-mediated interactions with dendritic cells. Eur. J. Immunol. 2007, 37, 716–728. [Google Scholar] [CrossRef] [PubMed]
- Laouar, A.; Haridas, V.; Vargas, D.; Zhinan, X.; Chaplin, D.; van Lier, R.A.; Manjunath, N. CD70+ antigen-presenting cells control the proliferation and differentiation of T cells in the intestinal mucosa. Nat. Immunol. 2005, 6, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Sommermeyer, D.; Hudecek, M.; Kosasih, P.L.; Gogishvili, T.; Maloney, D.G.; Turtle, C.J.; Riddell, S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 2016, 30, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Hinrichs, C.S.; Borman, Z.A.; Cassard, L.; Gattinoni, L.; Spolski, R.; Yu, Z.; Sanchez-Perez, L.; Muranski, P.; Kern, S.J.; Logun, C.; et al. Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc. Natl. Acad. Sci. USA 2009, 106, 17469–17474. [Google Scholar] [CrossRef] [Green Version]
- Gattinoni, L.; Klebanoff, C.A.; Palmer, D.C.; Wrzesinski, C.; Kerstann, K.; Yu, Z.; Finkelstein, S.E.; Theoret, M.R.; Rosenberg, S.A.; Restifo, N.P. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J. Clin. Investig. 2005, 115, 1616–1626. [Google Scholar] [CrossRef]
- Klebanoff, C.A.; Gattinoni, L.; Torabi-Parizi, P.; Kerstann, K.; Cardones, A.R.; Finkelstein, S.E.; Palmer, D.C.; Antony, P.A.; Hwang, S.T.; Rosenberg, S.A.; et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc. Natl. Acad. Sci. USA 2005, 102, 9571–9576. [Google Scholar] [CrossRef]
- Gattinoni, L.; Speiser, D.E.; Lichterfeld, M.; Bonini, C. T memory stem cells in health and disease. Nat. Med. 2017, 23, 18–27. [Google Scholar] [CrossRef]
- Biasco, L.; Izotova, N.; Rivat, C.; Ghorashian, S.; Richardson, R.; Guvenel, A.; Hough, R.; Wynn, R.; Popova, B.; Lopes, A.; et al. Clonal expansion of T memory stem cells determines early anti-leukemic responses and long-term CAR T cell persistence in patients. Nat. Cancer 2021, 2, 629–642. [Google Scholar] [CrossRef]
- Gattinoni, L.; Klebanoff, C.A.; Restifo, N.P. Paths to stemness: Building the ultimate antitumour T cell. Nat. Rev. Cancer 2012, 12, 671–684. [Google Scholar] [CrossRef]
- Klebanoff, C.A.; Crompton, J.G.; Leonardi, A.J.; Yamamoto, T.N.; Chandran, S.S.; Eil, R.L.; Sukumar, M.; Vodnala, S.K.; Hu, J.; Ji, Y.; et al. Inhibition of AKT signaling uncouples T cell differentiation from expansion for receptor-engineered adoptive immunotherapy. JCI Insight 2017, 2, e95103. [Google Scholar] [CrossRef]
- Drent, E.; Poels, R.; Ruiter, R.; van de Donk, N.; Zweegman, S.; Yuan, H.; de Bruijn, J.; Sadelain, M.; Lokhorst, H.M.; Groen, R.W.J.; et al. Combined CD28 and 4-1BB Costimulation Potentiates Affinity-tuned Chimeric Antigen Receptor-engineered T Cells. Clin. Cancer Res. 2019, 25, 4014–4025. [Google Scholar] [CrossRef] [PubMed]
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D., Jr.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Chen, X.; Madar, A.; Carpenito, C.; McGettigan, S.E.; Frigault, M.J.; Lee, J.; Posey, A.D., Jr.; Scholler, J.; Scholler, N.; et al. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood 2014, 124, 1070–1080. [Google Scholar] [CrossRef]
- Song, D.-G.; Ye, Q.; Poussin, M.; Harms, G.M.; Figini, M.; Powell, D.J. CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood 2012, 119, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Carr, J.M.; Carrasco, M.J.; Thaventhiran, J.E.; Bambrough, P.J.; Kraman, M.; Edwards, A.D.; Al-Shamkhani, A.; Fearon, D.T. CD27 mediates interleukin-2-independent clonal expansion of the CD8+ T cell without effector differentiation. Proc. Natl. Acad. Sci. USA 2006, 103, 19454–19459. [Google Scholar] [CrossRef]
- Finney, H.M.; Akbar, A.N.; Lawson, A.D. Activation of resting human primary T cells with chimeric receptors: Costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J. Immunol. 2004, 172, 104–113. [Google Scholar] [CrossRef]
- Hombach, A.A.; Chmielewski, M.; Rappl, G.; Abken, H. Adoptive immunotherapy with redirected T cells produces CCR7- cells that are trapped in the periphery and benefit from combined CD28-OX40 costimulation. Hum Gene Ther. 2013, 24, 259–269. [Google Scholar] [CrossRef]
- Hombach, A.A.; Heiders, J.; Foppe, M.; Chmielewski, M.; Abken, H. OX40 costimulation by a chimeric antigen receptor abrogates CD28 and IL-2 induced IL-10 secretion by redirected CD4(+) T cells. Oncoimmunology 2012, 1, 458–466. [Google Scholar] [CrossRef]
- Pulè, M.A.; Straathof, K.C.; Dotti, G.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol. Ther. 2005, 12, 933–941. [Google Scholar] [CrossRef]
- Lynn, R.C.; Weber, E.W.; Sotillo, E.; Gennert, D.; Xu, P.; Good, Z.; Anbunathan, H.; Lattin, J.; Jones, R.; Tieu, V.; et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature 2019, 576, 293–300. [Google Scholar] [CrossRef]
- Gomes-Silva, D.; Mukherjee, M.; Srinivasan, M.; Krenciute, G.; Dakhova, O.; Zheng, Y.; Cabral, J.M.S.; Rooney, C.M.; Orange, J.S.; Brenner, M.K.; et al. Tonic 4-1BB Costimulation in Chimeric Antigen Receptors Impedes T Cell Survival and Is Vector-Dependent. Cell Rep. 2017, 21, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Buck, M.D.; O’Sullivan, D.; Pearce, E.L. T cell metabolism drives immunity. J. Exp. Med. 2015, 212, 1345–1360. [Google Scholar] [CrossRef]
- van der Windt, G.J.; Pearce, E.L. Metabolic switching and fuel choice during T-cell differentiation and memory development. Immunol. Rev. 2012, 249, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Buck, M.D.; O’Sullivan, D.; Geltink, R.I.K.; Curtis, J.D.; Chang, C.-H.; Sanin, D.E.; Qiu, J.; Kretz, O.; Braas, D.; van der Windt, G.J.W.; et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 2016, 166, 63–76. [Google Scholar] [CrossRef]
- Corrado, M.; Pearce, E.L. Targeting memory T cell metabolism to improve immunity. J. Clin. Investig. 2022, 132. [Google Scholar] [CrossRef]
- van der Windt, G.J.; Everts, B.; Chang, C.H.; Curtis, J.D.; Freitas, T.C.; Amiel, E.; Pearce, E.J.; Pearce, E.L. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 2012, 36, 68–78. [Google Scholar] [CrossRef]
- Jacobs, S.R.; Herman, C.E.; Maciver, N.J.; Wofford, J.A.; Wieman, H.L.; Hammen, J.J.; Rathmell, J.C. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J. Immunol. 2008, 180, 4476–4486. [Google Scholar] [CrossRef]
- Menk, A.V.; Scharping, N.E.; Moreci, R.S.; Zeng, X.; Guy, C.; Salvatore, S.; Bae, H.; Xie, J.; Young, H.A.; Wendell, S.G.; et al. Early TCR Signaling Induces Rapid Aerobic Glycolysis Enabling Distinct Acute T Cell Effector Functions. Cell Rep. 2018, 22, 1509–1521. [Google Scholar] [CrossRef]
- Klein Geltink, R.I.; O’Sullivan, D.; Corrado, M.; Bremser, A.; Buck, M.D.; Buescher, J.M.; Firat, E.; Zhu, X.; Niedermann, G.; Caputa, G.; et al. Mitochondrial Priming by CD28. Cell 2017, 171, 385–397.e311. [Google Scholar] [CrossRef]
- Zeng, H.; Cohen, S.; Guy, C.; Shrestha, S.; Neale, G.; Brown, S.A.; Cloer, C.; Kishton, R.J.; Gao, X.; Youngblood, B.; et al. mTORC1 and mTORC2 Kinase Signaling and Glucose Metabolism Drive Follicular Helper T Cell Differentiation. Immunity 2016, 45, 540–554. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Jia, Y.; Zhou, M.; Fu, C.; Tuhin, I.J.; Ye, J.; Monty, M.A.; Xu, N.; Kang, L.; Li, M.; et al. Chimeric antigen receptors containing the OX40 signalling domain enhance the persistence of T cells even under repeated stimulation with multiple myeloma target cells. J. Hematol. Oncol. 2022, 15, 39. [Google Scholar] [CrossRef] [PubMed]
- van der Stegen, S.J.; Hamieh, M.; Sadelain, M. The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov. 2015, 14, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Brentjens, R.J.; Santos, E.; Nikhamin, Y.; Yeh, R.; Matsushita, M.; La Perle, K.; Quintás-Cardama, A.; Larson, S.M.; Sadelain, M. Genetically targeted T cells eradicate systemic acute lymphoblastic leukemia xenografts. Clin. Cancer Res. 2007, 13, 5426–5435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Condomines, M.; van der Stegen, S.J.C.; Perna, F.; Kloss, C.C.; Gunset, G.; Plotkin, J.; Sadelain, M. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell 2015, 28, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Dudley, M.E.; Feldman, S.A.; Wilson, W.H.; Spaner, D.E.; Maric, I.; Stetler-Stevenson, M.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 2012, 119, 2709–2720. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Wilson, W.H.; Janik, J.E.; Dudley, M.E.; Stetler-Stevenson, M.; Feldman, S.A.; Maric, I.; Raffeld, M.; Nathan, D.A.; Lanier, B.J.; et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010, 116, 4099–4102. [Google Scholar] [CrossRef]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and Toxicity Management of 19-28z CAR T Cell Therapy in B Cell Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2014, 6, 224ra225. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef]
- Lam, N.; Trinklein, N.D.; Buelow, B.; Patterson, G.H.; Ojha, N.; Kochenderfer, J.N. Anti-BCMA chimeric antigen receptors with fully human heavy-chain-only antigen recognition domains. Nat. Commun. 2020, 11, 283. [Google Scholar] [CrossRef] [PubMed]
- Cappell, K.M.; Kochenderfer, J.N. A comparison of chimeric antigen receptors containing CD28 versus 4-1BB costimulatory domains. Nat. Rev. Clin. Oncol. 2021, 18, 715–727. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Dudley, M.E.; Kassim, S.H.; Somerville, R.P.; Carpenter, R.O.; Stetler-Stevenson, M.; Yang, J.C.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J. Clin. Oncol. 2015, 33, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Somerville, R.P.T.; Lu, T.; Shi, V.; Bot, A.; Rossi, J.; Xue, A.; Goff, S.L.; Yang, J.C.; Sherry, R.M.; et al. Lymphoma Remissions Caused by Anti-CD19 Chimeric Antigen Receptor T Cells Are Associated With High Serum Interleukin-15 Levels. J. Clin. Oncol. 2017, 35, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Lam, N.; Vanasse, D.; Shen, Y.W.; Rose, J.J.; Rossi, J.; Xue, A.; Bot, A.; Scholler, N.; Mikkilineni, L.; et al. Safety and feasibility of anti-CD19 CAR T cells with fully human binding domains in patients with B-cell lymphoma. Nat. Med. 2020, 26, 270–280. [Google Scholar] [CrossRef]
- Ying, Z.; Yang, H.; Guo, Y.; Li, W.; Zou, D.; Zhou, D.; Wang, Z.; Zhang, M.; Wu, J.; Liu, H.; et al. Relmacabtagene autoleucel (relma-cel) CD19 CAR-T therapy for adults with heavily pretreated relapsed/refractory large B-cell lymphoma in China. Cancer Med. 2021, 10, 999–1011. [Google Scholar] [CrossRef]
- Hirayama, A.V.; Gauthier, J.; Hay, K.A.; Voutsinas, J.M.; Wu, Q.; Pender, B.S.; Hawkins, R.M.; Vakil, A.; Steinmetz, R.N.; Riddell, S.R.; et al. High rate of durable complete remission in follicular lymphoma after CD19 CAR-T cell immunotherapy. Blood 2019, 134, 636–640. [Google Scholar] [CrossRef]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Curran, K.J.; Margossian, S.P.; Kernan, N.A.; Silverman, L.B.; Williams, D.A.; Shukla, N.; Kobos, R.; Forlenza, C.J.; Steinherz, P.; Prockop, S.; et al. Toxicity and response after CD19-specific CAR T-cell therapy in pediatric/young adult relapsed/refractory B-ALL. Blood 2019, 134, 2361–2368. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Yang, J.F.; Deng, B.P.; Zhao, X.J.; Zhang, X.; Lin, Y.H.; Wu, Y.N.; Deng, Z.L.; Zhang, Y.L.; Liu, S.H.; et al. High efficacy and safety of low-dose CD19-directed CAR-T cell therapy in 51 refractory or relapsed B acute lymphoblastic leukemia patients. Leukemia 2017, 31, 2587–2593. [Google Scholar] [CrossRef] [PubMed]
- Hay, K.A.; Gauthier, J.; Hirayama, A.V.; Voutsinas, J.M.; Wu, Q.; Li, D.; Gooley, T.A.; Cherian, S.; Chen, X.; Pender, B.S.; et al. Factors associated with durable EFS in adult B-cell ALL patients achieving MRD-negative CR after CD19 CAR T-cell therapy. Blood 2019, 133, 1652–1663. [Google Scholar] [CrossRef] [PubMed]
- Ghorashian, S.; Kramer, A.M.; Onuoha, S.; Wright, G.; Bartram, J.; Richardson, R.; Albon, S.J.; Casanovas-Company, J.; Castro, F.; Popova, B.; et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat. Med. 2019, 25, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Frey, N.V.; Shaw, P.A.; Hexner, E.O.; Pequignot, E.; Gill, S.; Luger, S.M.; Mangan, J.K.; Loren, A.W.; Perl, A.E.; Maude, S.L.; et al. Optimizing Chimeric Antigen Receptor T-Cell Therapy for Adults With Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2020, 38, 415–422. [Google Scholar] [CrossRef]
- Anagnostou, T.; Riaz, I.B.; Hashmi, S.K.; Murad, M.H.; Kenderian, S.S. Anti-CD19 chimeric antigen receptor T-cell therapy in acute lymphocytic leukaemia: A systematic review and meta-analysis. Lancet Haematol. 2020, 7, e816–e826. [Google Scholar] [CrossRef]
- Sesques, P.; Ferrant, E.; Safar, V.; Wallet, F.; Tordo, J.; Dhomps, A.; Karlin, L.; Brisou, G.; Vercasson, M.; Hospital-Gustem, C.; et al. Commercial anti-CD19 CAR T cell therapy for patients with relapsed/refractory aggressive B cell lymphoma in a European center. Am. J. Hematol. 2020, 95, 1324–1333. [Google Scholar] [CrossRef]
- Cappell, K.M.; Sherry, R.M.; Yang, J.C.; Goff, S.L.; Vanasse, D.A.; McIntyre, L.; Rosenberg, S.A.; Kochenderfer, J.N. Long-Term Follow-Up of Anti-CD19 Chimeric Antigen Receptor T-Cell Therapy. J. Clin. Oncol. 2020, 38, 3805–3815. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hanafi, L.A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef]
- Brudno, J.N.; Maric, I.; Hartman, S.D.; Rose, J.J.; Wang, M.; Lam, N.; Stetler-Stevenson, M.; Salem, D.; Yuan, C.; Pavletic, S.; et al. T Cells Genetically Modified to Express an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Poor-Prognosis Relapsed Multiple Myeloma. J. Clin. Oncol. 2018, 36, 2267–2280. [Google Scholar] [CrossRef]
- Ali, S.A.; Shi, V.; Maric, I.; Wang, M.; Stroncek, D.F.; Rose, J.J.; Brudno, J.N.; Stetler-Stevenson, M.; Feldman, S.A.; Hansen, B.G.; et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood 2016, 128, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.-H.; Wang, B.-Y.; Chen, L.-J.; Fu, W.-J.; Xu, J.; Liu, J.; Jin, S.-W.; Chen, Y.-X.; Cao, X.-M.; Yang, Y.; et al. Four-year follow-up of LCAR-B38M in relapsed or refractory multiple myeloma: A phase 1, single-arm, open-label, multicenter study in China (LEGEND-2). J. Hematol. Oncol. 2022, 15, 86. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, J.; Hu, G.; Wang, W.; Xiao, Y.; Cai, H.; Jiang, L.; Meng, L.; Yang, Y.; Zhou, X.; et al. A phase 1 study of a novel fully human BCMA-targeting CAR (CT103A) in patients with relapsed/refractory multiple myeloma. Blood 2021, 137, 2890–2901. [Google Scholar] [CrossRef]
- Chen, W.; Fu, C.; Cai, Z.; Li, Z.; Wang, H.; Yan, L.; Wu, Y.; Shi, X.; Gao, W.; Yan, S.; et al. Sustainable Efficacy and Safety Results from Lummicar Study 1: A Phase 1/2 Study of Fully Human B-Cell Maturation Antigen-Specific CAR T Cells (CT053) in Chinese Subjects with Relapsed and/or Refractory Multiple Myeloma. Blood 2021, 138, 2821. [Google Scholar] [CrossRef]
- Brudno, J.N.; Kochenderfer, J.N. Toxicities of chimeric antigen receptor T cells: Recognition and management. Blood 2016, 127, 3321–3330. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef]
- Gust, J.; Ponce, R.; Liles, W.C.; Garden, G.A.; Turtle, C.J. Cytokines in CAR T Cell-Associated Neurotoxicity. Front. Immunol. 2020, 11, 577027. [Google Scholar] [CrossRef]
- Locke, F.L.; Neelapu, S.S.; Bartlett, N.L.; Siddiqi, T.; Chavez, J.C.; Hosing, C.M.; Ghobadi, A.; Budde, L.E.; Bot, A.; Rossi, J.M.; et al. Phase 1 Results of ZUMA-1: A Multicenter Study of KTE-C19 Anti-CD19 CAR T Cell Therapy in Refractory Aggressive Lymphoma. Mol. Ther. 2017, 25, 285–295. [Google Scholar] [CrossRef]
- Teachey, D.T.; Lacey, S.F.; Shaw, P.A.; Melenhorst, J.J.; Maude, S.L.; Frey, N.; Pequignot, E.; Gonzalez, V.E.; Chen, F.; Finklestein, J.; et al. Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discov. 2016, 6, 664–679. [Google Scholar] [CrossRef]
- Porter, D.L.; Hwang, W.T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013, 5, 177ra138. [Google Scholar] [CrossRef] [PubMed]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Sun, J.; Wu, Z.; Yu, J.; Cui, Q.; Pu, C.; Liang, B.; Luo, Y.; Shi, J.; Jin, A.; et al. Predominant cerebral cytokine release syndrome in CD19-directed chimeric antigen receptor-modified T cell therapy. J. Hematol. Oncol. 2016, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Kalos, M.; Levine, B.L.; Porter, D.L.; Katz, S.; Grupp, S.A.; Bagg, A.; June, C.H. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 2011, 3, 95ra73. [Google Scholar] [CrossRef] [Green Version]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]


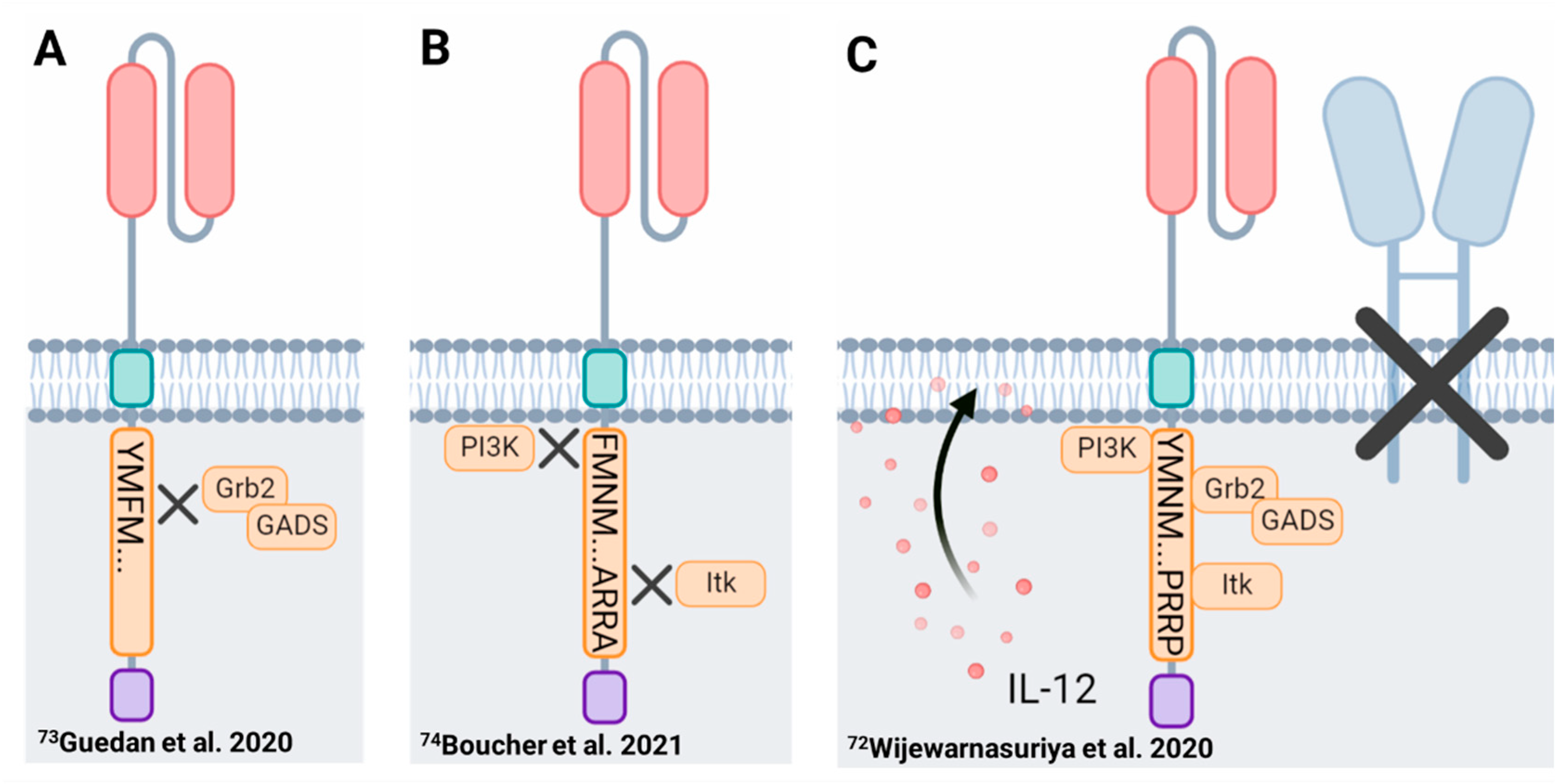

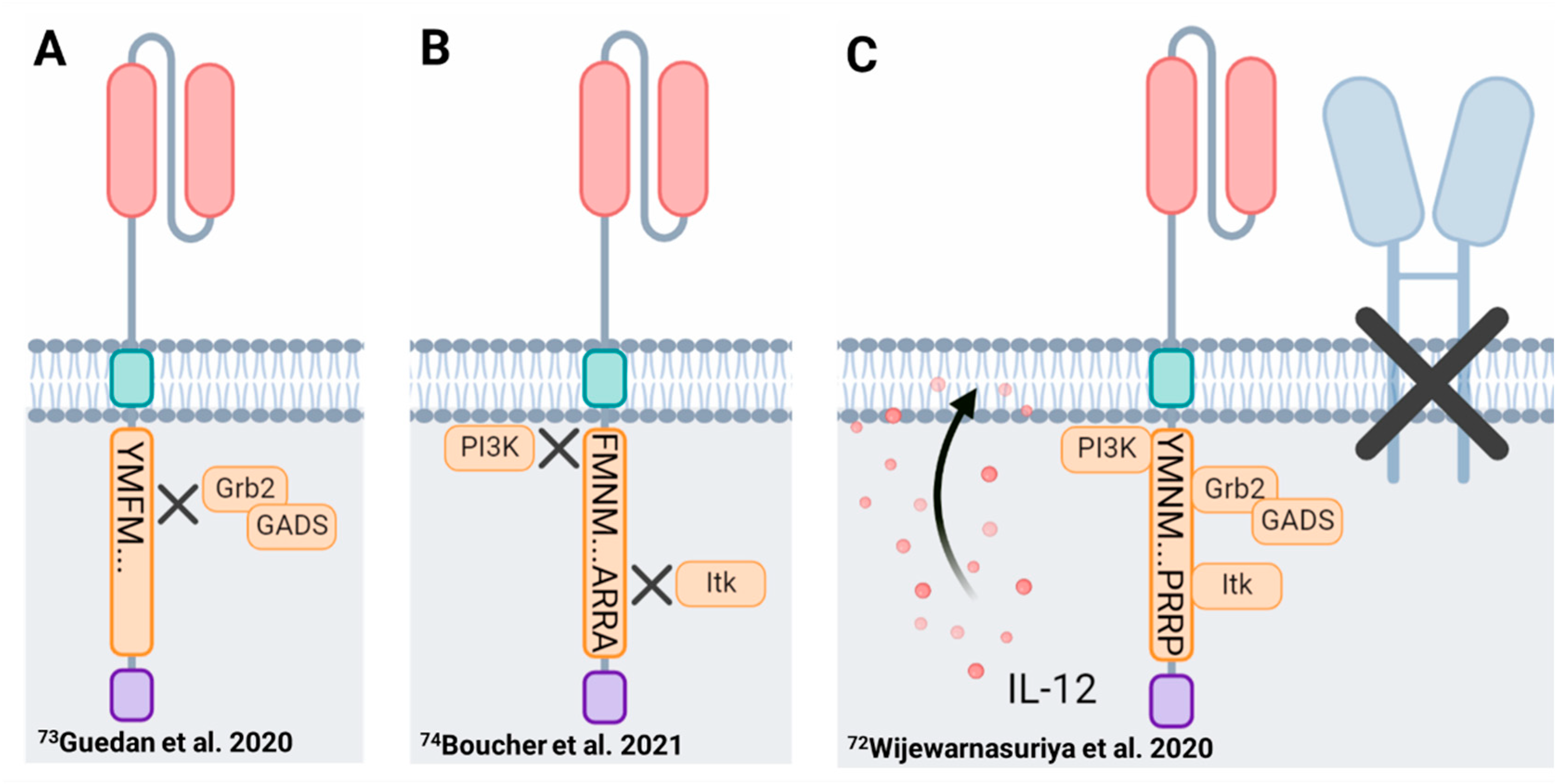
{kind=link}
{kind=link}
{kind=link}
| Co-Stimulatory Domain | Receptor Family | Differentiation | Exhaustion | Metabolic Landscape | Kinetics | Persistence | Toxicity |
|---|---|---|---|---|---|---|---|
| CD28 | Ig Superfamily | Effector memory | Prone to exhaustion | Aerobic glycolysis | Rapid signaling kinetics, greater phosphorylation intensity, greater cytokine release, rapid tumor regression | Short-lived | Rapid symptom onset (within 48 h. of infusion), greater frequency and severity of CRS, greater frequency of patients require intervention |
| ICOS | Ig Superfamily | TH17 polarization (self renewal and stem-like properties) | Less susceptible to exhaustion | Aerobic glycolysis | Rapid signaling kinetics, greater phosphorylation intensity, greater cytokine release, rapid tumor regression | Long-lived | Pending clinical investigation |
| 4-1BB | TNF-R Superfamily | Central memory | Less susceptible to exhaustion | Oxidative phosphorylation and fatty acid oxidation | Slower, less intense signaling, reduced cytokine release, gradual tumor regression | Long-lived | Delayed symptom onset (3–5 days of infusion), lower frequency and severity of CRS, lower frequency of patients require intervention |
| OX40 | TNF-R Superfamily | Central memory | Less susceptible to exhaustion | Oxidative phosphorylation (transcriptomic level analysis) | Reduced cytokine release, gradual tumor regression | Long-lived | Pending clinical investigation |
| CD27 | TNF-R Superfamily | Central memory | Less susceptible to exhaustion | --- | Rapid tumor regression | Long-lived | Pending clinical investigation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Honikel, M.M.; Olejniczak, S.H. Co-Stimulatory Receptor Signaling in CAR-T Cells. Biomolecules 2022, 12, 1303. https://doi.org/10.3390/biom12091303
Honikel MM, Olejniczak SH. Co-Stimulatory Receptor Signaling in CAR-T Cells. Biomolecules. 2022; 12(9):1303. https://doi.org/10.3390/biom12091303
Chicago/Turabian StyleHonikel, Mackenzie M., and Scott H. Olejniczak. 2022. "Co-Stimulatory Receptor Signaling in CAR-T Cells" Biomolecules 12, no. 9: 1303. https://doi.org/10.3390/biom12091303
APA StyleHonikel, M. M., & Olejniczak, S. H. (2022). Co-Stimulatory Receptor Signaling in CAR-T Cells. Biomolecules, 12(9), 1303. https://doi.org/10.3390/biom12091303