
The Role of JAK/STAT Pathway in Fibrotic Diseases: Molecular and Cellular Mechanisms

Abstract
1. Introduction
2. Molecular Structure of JAK and STAT
3. Activation of the JAK/STAT Signaling Pathway
4. The Roles of JAK/STAT in Hepatic Fibrosis
4.1. STAT1
4.2. STAT2
4.3. STAT3
4.4. STAT4
4.5. STAT5
4.6. STAT6
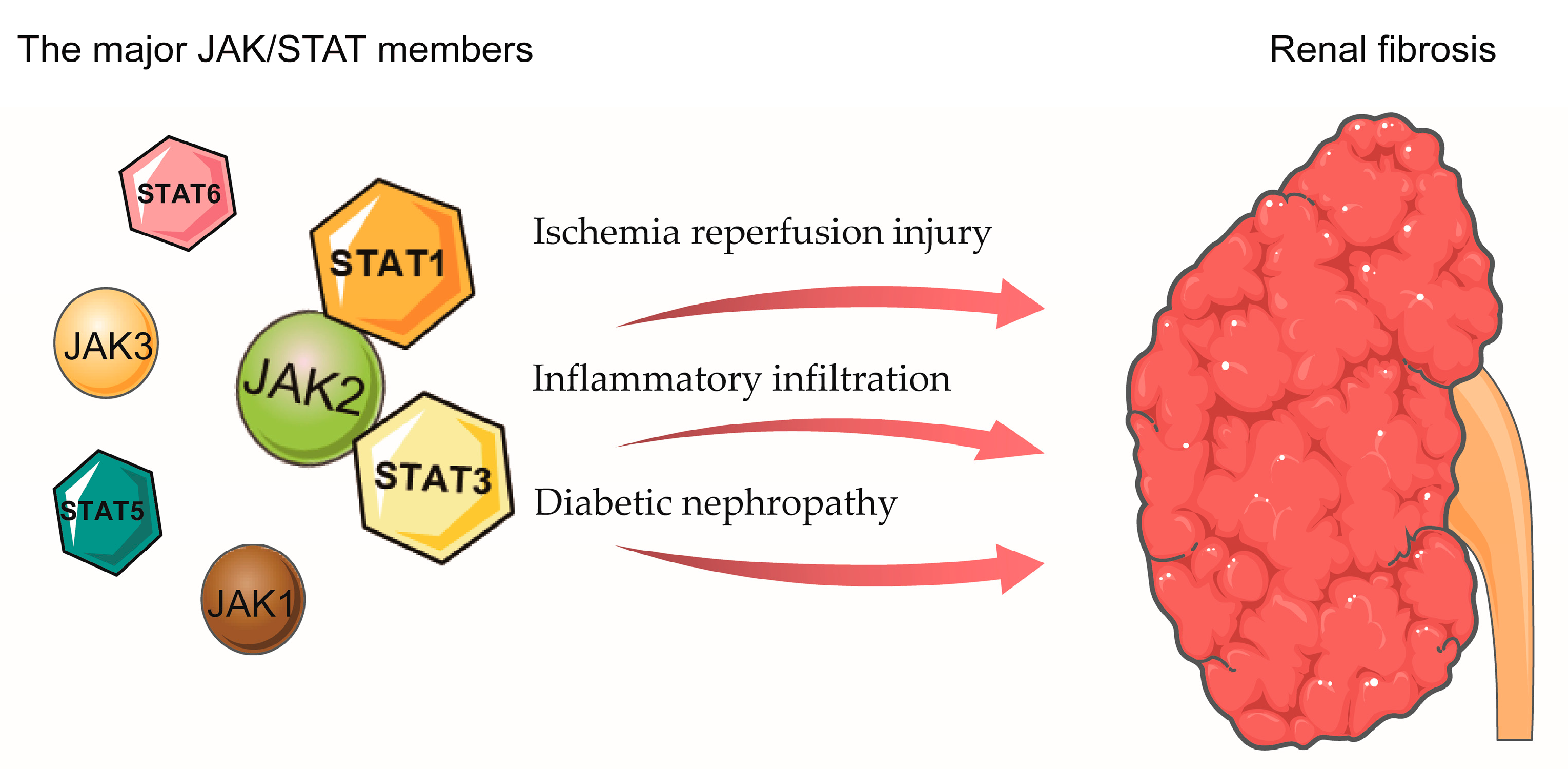
5. The Role of JAK/STAT in Renal Fibrosis
6. The Role of JAK/STAT in Cardiac Fibrosis
7. The Role of JAK/STAT in Myelofibrosis
8. The Role of JAK/STAT in Pulmonary Fibrosis
9. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From mechanisms to medicines. Nature 2020, 587, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Lv, W.; Booz, G.W.; Wang, Y.; Fan, F.; Roman, R.J. Inflammation and renal fibrosis: Recent developments on key signaling molecules as potential therapeutic targets. Eur. J. Pharmacol. 2018, 820, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, S.; Thadathil, N.; Selvarani, R.; Nicklas, E.H.; Wang, D.; Miller, B.F.; Richardson, A.; Deepa, S.S. Necroptosis contributes to chronic inflammation and fibrosis in aging liver. Aging Cell 2021, 20, e13512. [Google Scholar] [CrossRef]
- Sivakumar, P.; Das, A.M. Fibrosis, chronic inflammation and new pathways for drug discovery. Inflamm. Res. 2008, 57, 410–418. [Google Scholar] [CrossRef]
- Eming, S.A.; Wynn, T.A.; Martin, P. Inflammation and metabolism in tissue repair and regeneration. Science 2017, 356, 1026–1030. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E., Jr.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef]
- Jatiani, S.S.; Baker, S.J.; Silverman, L.R.; Reddy, E.P. Jak/STAT pathways in cytokine signaling and myeloproliferative disorders: Approaches for targeted therapies. Genes Cancer 2010, 1, 979–993. [Google Scholar] [CrossRef]
- Bromberg, J.; Darnell, J.E., Jr. The role of STATs in transcriptional control and their impact on cellular function. Oncogene 2000, 19, 2468–2473. [Google Scholar] [CrossRef]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E., Jr. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Kisseleva, T.; Bhattacharya, S.; Braunstein, J.; Schindler, C.W. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene 2002, 285, 1–24. [Google Scholar] [CrossRef]
- Murray, P.J. The JAK-STAT signaling pathway: Input and output integration. J. Immunol. 2007, 178, 2623–2629. [Google Scholar] [CrossRef]
- Bharadwaj, U.; Kasembeli, M.M.; Robinson, P.; Tweardy, D.J. Targeting Janus Kinases and Signal Transducer and Activator of Transcription 3 to Treat Inflammation, Fibrosis, and Cancer: Rationale, Progress, and Caution. Pharmacol. Rev. 2020, 72, 486–526. [Google Scholar] [CrossRef]
- Schindler, C.; Darnell, J.E., Jr. Transcriptional responses to polypeptide ligands: The JAK-STAT pathway. Annu. Rev. Biochem. 1995, 64, 621–651. [Google Scholar] [CrossRef]
- Gnanasambandan, K.; Sayeski, P.P. A structure-function perspective of Jak2 mutations and implications for alternate drug design strategies: The road not taken. Curr. Med. Chem. 2011, 18, 4659–4673. [Google Scholar] [CrossRef]
- Bousoik, E.; Montazeri Aliabadi, H. “Do We Know Jack” About JAK? A Closer Look at JAK/STAT Signaling Pathway. Front. Oncol. 2018, 8, 287. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef]
- LaFave, L.M.; Levine, R.L. JAK2 the future: Therapeutic strategies for JAK-dependent malignancies. Trends Pharm. Sci. 2012, 33, 574–582. [Google Scholar] [CrossRef]
- Schindler, C.; Strehlow, I. Cytokines and STAT signaling. Adv. Pharm. 2000, 47, 113–174. [Google Scholar]
- Hammaren, H.M.; Ungureanu, D.; Grisouard, J.; Skoda, R.C.; Hubbard, S.R.; Silvennoinen, O. ATP binding to the pseudokinase domain of JAK2 is critical for pathogenic activation. Proc. Natl. Acad. Sci. USA 2015, 112, 4642–4647. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. Publ. Protein Soc. 2018, 27, 1984–2009. [Google Scholar] [CrossRef] [PubMed]
- Bohmer, F.D.; Friedrich, K. Protein tyrosine phosphatases as wardens of STAT signaling. JAKSTAT 2014, 3, e28087. [Google Scholar] [CrossRef] [PubMed]
- Swiatek-Machado, K.; Kaminska, B. STAT Signaling in Glioma Cells. Adv. Exp. Med. Biol. 2020, 1202, 203–222. [Google Scholar]
- Yu, H.; Jove, R. The STATs of cancer-new molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Decker, T.; Kovarik, P. Serine phosphorylation of STATs. Oncogene 2000, 19, 2628–2637. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Darnell, J.E., Jr. Stats: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, L.A.; Liongue, C.; Lewis, R.S.; Stephenson, S.E.; Ward, A.C. Cytokine receptor signaling through the Jak-Stat-Socs pathway in disease. Mol. Immunol. 2007, 44, 2497–2506. [Google Scholar] [CrossRef]
- Yoshimura, A.; Naka, T.; Kubo, M. SOCS proteins, cytokine signalling and immune regulation. Nat. Rev. Immunol. 2007, 7, 454–465. [Google Scholar] [CrossRef]
- Aaronson, D.S.; Horvath, C.M. A road map for those who don’t know JAK-STAT. Science 2002, 296, 1653–1655. [Google Scholar] [CrossRef]
- Ferrao, R.; Wallweber, H.J.; Ho, H.; Tam, C.; Franke, Y.; Quinn, J.; Lupardus, P.J. The Structural Basis for Class II Cytokine Receptor Recognition by JAK1. Structure 2016, 24, 897–905. [Google Scholar] [CrossRef]
- Ozaki, K.; Leonard, W.J. Cytokine and cytokine receptor pleiotropy and redundancy. J. Biol. Chem. 2002, 277, 29355–29358. [Google Scholar] [CrossRef]
- Kotenko, S.V.; Langer, J.A. Full house: 12 receptors for 27 cytokines. Int. Immunopharmacol. 2004, 4, 593–608. [Google Scholar] [CrossRef]
- Abroun, S.; Saki, N.; Ahmadvand, M.; Asghari, F.; Salari, F.; Rahim, F. STATs: An Old Story, Yet Mesmerizing. Cell J. 2015, 17, 395–411. [Google Scholar]
- Clark, J.D.; Flanagan, M.E.; Telliez, J.B. Discovery and development of Janus kinase (JAK) inhibitors for inflammatory diseases. J. Med. Chem. 2014, 57, 5023–5038. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Loomba, R.; Anstee, Q.M.; Rinella, M.E.; Bugianesi, E.; Marchesini, G.; Neuschwander-Tetri, B.A.; Serfaty, L.; Negro, F.; Caldwell, S.H.; et al. Diagnostic modalities for nonalcoholic fatty liver disease, nonalcoholic steatohepatitis, and associated fibrosis. Hepatology 2018, 68, 349–360. [Google Scholar] [CrossRef]
- Gao, B. Cytokines, STATs and liver disease. Cell. Mol. Immunol. 2005, 2, 92–100. [Google Scholar]
- Ruff-Jamison, S.; Chen, K.; Cohen, S. Induction by EGF and interferon-gamma of tyrosine phosphorylated DNA binding proteins in mouse liver nuclei. Science 1993, 261, 1733–1736. [Google Scholar] [CrossRef]
- Machida, K.; Tsukamoto, H.; Liu, J.C.; Han, Y.P.; Govindarajan, S.; Lai, M.M.; Akira, S.; Ou, J.H. c-Jun mediates hepatitis C virus hepatocarcinogenesis through signal transducer and activator of transcription 3 and nitric oxide-dependent impairment of oxidative DNA repair. Hepatology 2010, 52, 480–492. [Google Scholar] [CrossRef]
- Jeong, W.I.; Park, O.; Radaeva, S.; Gao, B. STAT1 inhibits liver fibrosis in mice by inhibiting stellate cell proliferation and stimulating NK cell cytotoxicity. Hepatology 2006, 44, 1441–1451. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, F.; Fan, X.; Lin, C.; Hao, Y.; Wei, H.; Lin, W.; Jiang, Y.; He, F. Quantitative Proteomic analysis on Activated Hepatic Stellate Cells reversion Reveal STAT1 as a key regulator between Liver Fibrosis and recovery. Sci. Rep. 2017, 7, 44910. [Google Scholar] [CrossRef]
- Sun, R.; Gao, B. Negative regulation of liver regeneration by innate immunity (natural killer cells/interferon-gamma). Gastroenterology 2004, 127, 1525–1539. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Park, O.; Horiguchi, N.; Kulkarni, S.; Jeong, W.I.; Sun, H.Y.; Radaeva, S.; Gao, B. STAT1 contributes to dsRNA inhibition of liver regeneration after partial hepatectomy in mice. Hepatology 2006, 44, 955–966. [Google Scholar] [CrossRef]
- Hong, F.; Jaruga, B.; Kim, W.H.; Radaeva, S.; El-Assal, O.N.; Tian, Z.; Nguyen, V.A.; Gao, B. Opposing roles of STAT1 and STAT3 in T cell-mediated hepatitis: Regulation by SOCS. J. Clin. Investig. 2002, 110, 1503–1513. [Google Scholar] [CrossRef] [PubMed]
- Jaruga, B.; Hong, F.; Sun, R.; Radaeva, S.; Gao, B. Crucial role of IL-4/STAT6 in T cell-mediated hepatitis: Up-regulating eotaxins and IL-5 and recruiting leukocytes. J. Immunol. 2003, 171, 3233–3244. [Google Scholar] [CrossRef] [PubMed]
- Siebler, J.; Wirtz, S.; Klein, S.; Protschka, M.; Blessing, M.; Galle, P.R.; Neurath, M.F. A key pathogenic role for the STAT1/T-bet signaling pathway in T-cell-mediated liver inflammation. Hepatology 2003, 38, 1573–1580. [Google Scholar]
- Wen, J.; Zhou, Y.; Wang, J.; Chen, J.; Yan, W.; Wu, J.; Yan, J.; Zhou, K.; Xiao, Y.; Wang, Y.; et al. Retraction Note: Interactions between Th1 cells and Tregs affect regulation of hepatic fibrosis in biliary atresia through the IFN-gamma/STAT1 pathway. Cell Death Differ. 2020, 27, 2295. [Google Scholar] [CrossRef]
- Kim, W.H.; Hong, F.; Radaeva, S.; Jaruga, B.; Fan, S.; Gao, B. STAT1 plays an essential role in LPS/D-galactosamine-induced liver apoptosis and injury. Am. J. Physiol. Gastrointest Liver Physiol. 2003, 285, G761–G768. [Google Scholar] [CrossRef]
- Car, B.D.; Eng, V.M.; Schnyder, B.; Ozmen, L.; Huang, S.; Gallay, P.; Heumann, D.; Aguet, M.; Ryffel, B. Interferon gamma receptor deficient mice are resistant to endotoxic shock. J. Exp. Med. 1994, 179, 1437–1444. [Google Scholar] [CrossRef]
- Gao, B.; Wang, H.; Lafdil, F.; Feng, D. STAT proteins—Key regulators of anti-viral responses, inflammation, and tumorigenesis in the liver. J. Hepatol. 2012, 57, 430–441. [Google Scholar] [CrossRef]
- Torisu, T.; Nakaya, M.; Watanabe, S.; Hashimoto, M.; Yoshida, H.; Chinen, T.; Yoshida, R.; Okamoto, F.; Hanada, T.; Torisu, K.; et al. Suppressor of cytokine signaling 1 protects mice against concanavalin A-induced hepatitis by inhibiting apoptosis. Hepatology 2008, 47, 1644–1654. [Google Scholar] [CrossRef]
- Park, O.; Wang, H.; Weng, H.; Feigenbaum, L.; Li, H.; Yin, S.; Ki, S.H.; Yoo, S.H.; Dooley, S.; Wang, F.S.; et al. In vivo consequences of liver-specific interleukin-22 expression in mice: Implications for human liver disease progression. Hepatology 2011, 54, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Wustefeld, T.; Assmus, U.; Roskams, T.; Rose-John, S.; Muller, M.; Manns, M.P.; Ernst, M.; Trautwein, C. The IL-6-gp130-STAT3 pathway in hepatocytes triggers liver protection in T cell-mediated liver injury. J. Clin. Investig. 2005, 115, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Lafdil, F.; Wang, H.; Park, O.; Zhang, W.; Moritoki, Y.; Yin, S.; Fu, X.Y.; Gershwin, M.E.; Lian, Z.X.; Gao, B. Myeloid STAT3 inhibits T cell-mediated hepatitis by regulating T helper 1 cytokine and interleukin-17 production. Gastroenterology 2009, 137, 2125–2135. [Google Scholar] [CrossRef] [PubMed]
- Heim, M.H.; Thimme, R. Innate and adaptive immune responses in HCV infections. J. Hepatol. 2014, 61, S14–S25. [Google Scholar] [CrossRef] [PubMed]
- Blaszczyk, K.; Olejnik, A.; Nowicka, H.; Ozgyin, L.; Chen, Y.L.; Chmielewski, S.; Kostyrko, K.; Wesoly, J.; Balint, B.L.; Lee, C.K.; et al. STAT2/IRF9 directs a prolonged ISGF3-like transcriptional response and antiviral activity in the absence of STAT1. Biochem. J. 2015, 466, 511–524. [Google Scholar] [CrossRef]
- Shrivastava, S.; Meissner, E.G.; Funk, E.; Poonia, S.; Shokeen, V.; Thakur, A.; Poonia, B.; Sarin, S.K.; Trehanpati, N.; Kottilil, S. Elevated hepatic lipid and interferon stimulated gene expression in HCV GT3 patients relative to non-alcoholic steatohepatitis. Hepatol. Int. 2016, 10, 937–946. [Google Scholar] [CrossRef]
- Orlent, H.; Reynaert, H.; Bourgeois, S.; Dideberg, V.; Adler, M.; Colle, I.; De Maeght, S.; Laleman, W.; Michielsen, P.; Moreno, C.; et al. IL28B polymorphism and the control of hepatitis C virus infection: Ready for clinical use? Acta Gastroenterol. Belg. 2011, 74, 317–322. [Google Scholar]
- Kong, X.; Horiguchi, N.; Mori, M.; Gao, B. Cytokines and STATs in Liver Fibrosis. Front. Physiol. 2012, 3, 69. [Google Scholar] [CrossRef]
- Ibrahim, M.K.; Khedr, A.; Bader El Din, N.G.; Khairy, A.; El Awady, M.K. Increased incidence of cytomegalovirus coinfection in HCV-infected patients with late liver fibrosis is associated with dysregulation of JAK-STAT pathway. Sci. Rep. 2017, 7, 10364. [Google Scholar] [CrossRef]
- Bieche, I.; Asselah, T.; Laurendeau, I.; Vidaud, D.; Degot, C.; Paradis, V.; Bedossa, P.; Valla, D.C.; Marcellin, P.; Vidaud, M. Molecular profiling of early stage liver fibrosis in patients with chronic hepatitis C virus infection. Virology 2005, 332, 130–144. [Google Scholar] [CrossRef]
- Bender, H.; Wiesinger, M.Y.; Nordhoff, C.; Schoenherr, C.; Haan, C.; Ludwig, S.; Weiskirchen, R.; Kato, N.; Heinrich, P.C.; Haan, S. Interleukin-27 displays interferon-gamma-like functions in human hepatoma cells and hepatocytes. Hepatology 2009, 50, 585–591. [Google Scholar] [CrossRef]
- Wang, H.; Lafdil, F.; Wang, L.; Park, O.; Yin, S.; Niu, J.; Miller, A.M.; Sun, Z.; Gao, B. Hepatoprotective versus oncogenic functions of STAT3 in liver tumorigenesis. Am. J. Pathol. 2011, 179, 714–724. [Google Scholar] [CrossRef]
- Horiguchi, N.; Lafdil, F.; Miller, A.M.; Park, O.; Wang, H.; Rajesh, M.; Mukhopadhyay, P.; Fu, X.Y.; Pacher, P.; Gao, B. Dissociation between liver inflammation and hepatocellular damage induced by carbon tetrachloride in myeloid cell-specific signal transducer and activator of transcription 3 gene knockout mice. Hepatology 2010, 51, 1724–1734. [Google Scholar] [CrossRef]
- Ding, Y.F.; Wu, Z.H.; Wei, Y.J.; Shu, L.; Peng, Y.R. Hepatic inflammation-fibrosis-cancer axis in the rat hepatocellular carcinoma induced by diethylnitrosamine. J. Cancer Res. Clin. Oncol. 2017, 143, 821–834. [Google Scholar] [CrossRef]
- Ogata, H.; Chinen, T.; Yoshida, T.; Kinjyo, I.; Takaesu, G.; Shiraishi, H.; Iida, M.; Kobayashi, T.; Yoshimura, A. Loss of SOCS3 in the liver promotes fibrosis by enhancing STAT3-mediated TGF-beta1 production. Oncogene 2006, 25, 2520–2530. [Google Scholar] [CrossRef]
- Tang, L.Y.; Heller, M.; Meng, Z.; Yu, L.R.; Tang, Y.; Zhou, M.; Zhang, Y.E. Transforming Growth Factor-beta (TGF-beta) Directly Activates the JAK1-STAT3 Axis to Induce Hepatic Fibrosis in Coordination with the SMAD Pathway. J. Biol. Chem. 2017, 292, 4302–4312. [Google Scholar] [CrossRef]
- Gong, Z.; Ye, H.; Huo, Y.; Wang, L.; Huang, Y.; Huang, M.; Yuan, X. S-allyl-cysteine attenuates carbon tetrachloride-induced liver fibrosis in rats by targeting STAT3/SMAD3 pathway. Am. J. Transl. Res. 2018, 10, 1337–1346. [Google Scholar]
- Kagan, P.; Sultan, M.; Tachlytski, I.; Safran, M.; Ben-Ari, Z. Both MAPK and STAT3 signal transduction pathways are necessary for IL-6-dependent hepatic stellate cells activation. PLoS ONE 2017, 12, e0176173. [Google Scholar] [CrossRef]
- Sallam, A.M.; Esmat, A.; Abdel-Naim, A.B. Cucurbitacin-B attenuates CCl4 -induced hepatic fibrosis in mice through inhibition of STAT-3. Chem. Biol. Drug Des. 2018, 91, 933–941. [Google Scholar] [CrossRef]
- Khawar, M.B.; Azam, F.; Sheikh, N.; Abdul Mujeeb, K. How Does Interleukin-22 Mediate Liver Regeneration and Prevent Injury and Fibrosis? J. Immunol. Res. 2016, 2016, 2148129. [Google Scholar] [CrossRef]
- Kong, X.; Feng, D.; Wang, H.; Hong, F.; Bertola, A.; Wang, F.S.; Gao, B. Interleukin-22 induces hepatic stellate cell senescence and restricts liver fibrosis in mice. Hepatology 2012, 56, 1150–1159. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lafdil, F.; Wang, L.; Yin, S.; Feng, D.; Gao, B. Tissue inhibitor of metalloproteinase 1 (TIMP-1) deficiency exacerbates carbon tetrachloride-induced liver injury and fibrosis in mice: Involvement of hepatocyte STAT3 in TIMP-1 production. Cell Biosci. 2011, 1, 14. [Google Scholar] [CrossRef] [PubMed]
- Kasembeli, M.M.; Bharadwaj, U.; Robinson, P.; Tweardy, D.J. Contribution of STAT3 to Inflammatory and Fibrotic Diseases and Prospects for its Targeting for Treatment. Int. J. Mol. Sci. 2018, 19, 2299. [Google Scholar] [CrossRef] [PubMed]
- Edlich, B.; Ahlenstiel, G.; Zabaleta Azpiroz, A.; Stoltzfus, J.; Noureddin, M.; Serti, E.; Feld, J.J.; Liang, T.J.; Rotman, Y.; Rehermann, B. Early changes in interferon signaling define natural killer cell response and refractoriness to interferon-based therapy of hepatitis C patients. Hepatology 2012, 55, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qu, A.; Wang, H. Signal transducer and activator of transcription 4 in liver diseases. Int. J. Biol. Sci. 2015, 11, 448–455. [Google Scholar] [CrossRef]
- El Sharkawy, R.; Thabet, K.; Lampertico, P.; Petta, S.; Mangia, A.; Berg, T.; Metwally, M.; Bayoumi, A.; Boonstra, A.; Brouwer, W.P.; et al. A STAT4 variant increases liver fibrosis risk in Caucasian patients with chronic hepatitis B. Aliment. Pharm. 2018, 48, 564–573. [Google Scholar] [CrossRef]
- Jiang, D.K.; Ma, X.P.; Wu, X.; Peng, L.; Yin, J.; Dan, Y.; Huang, H.X.; Ding, D.L.; Zhang, L.Y.; Shi, Z.; et al. Genetic variations in STAT4,C2,HLA-DRB1 and HLA-DQ associated with risk of hepatitis B virus-related liver cirrhosis. Sci. Rep. 2015, 5, 16278. [Google Scholar] [CrossRef]
- Cheng, Y.L.; Song, W.J.; Liu, W.Q.; Lei, J.H.; Kong, Z.; Li, Y.L. The effects of interleukin (IL)-12 and IL-4 deficiency on worm development and granuloma formation in Schistosoma japonicum-infected mice. Parasitol. Res. 2012, 110, 287–293. [Google Scholar] [CrossRef]
- Harada, N.; Shimada, M.; Okano, S.; Suehiro, T.; Soejima, Y.; Tomita, Y.; Maehara, Y. IL-12 gene therapy is an effective therapeutic strategy for hepatocellular carcinoma in immunosuppressed mice. J. Immunol. 2004, 173, 6635–6644. [Google Scholar] [CrossRef]
- Chang, C.J.; Chen, Y.H.; Huang, K.W.; Cheng, H.W.; Chan, S.F.; Tai, K.F.; Hwang, L.H. Combined GM-CSF and IL-12 gene therapy synergistically suppresses the growth of orthotopic liver tumors. Hepatology 2007, 45, 746–754. [Google Scholar] [CrossRef]
- Mair, M.; Blaas, L.; Osterreicher, C.H.; Casanova, E.; Eferl, R. JAK-STAT signaling in hepatic fibrosis. Front. Biosci. 2011, 16, 2794–2811. [Google Scholar] [CrossRef]
- Hosui, A.; Kimura, A.; Yamaji, D.; Zhu, B.M.; Na, R.; Hennighausen, L. Loss of STAT5 causes liver fibrosis and cancer development through increased TGF-{beta} and STAT3 activation. J. Exp. Med. 2009, 206, 819–831. [Google Scholar] [CrossRef]
- Cui, Y.; Hosui, A.; Sun, R.; Shen, K.; Gavrilova, O.; Chen, W.; Cam, M.C.; Gao, B.; Robinson, G.W.; Hennighausen, L. Loss of signal transducer and activator of transcription 5 leads to hepatosteatosis and impaired liver regeneration. Hepatology 2007, 46, 504–513. [Google Scholar] [CrossRef]
- Kaplan, M.H.; Schindler, U.; Smiley, S.T.; Grusby, M.J. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity 1996, 4, 313–319. [Google Scholar] [CrossRef]
- Farah, I.O.; Mola, P.W.; Kariuki, T.M.; Nyindo, M.; Blanton, R.E.; King, C.L. Repeated exposure induces periportal fibrosis in Schistosoma mansoni-infected baboons: Role of TGF-beta and IL-4. J. Immunol. 2000, 164, 5337–5343. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Epithelial to mesenchymal transition in renal fibrogenesis: Pathologic significance, molecular mechanism, and therapeutic intervention. J. Am. Soc. Nephrol. 2004, 15, 1–12. [Google Scholar] [CrossRef]
- Humphreys, B.D. Mechanisms of Renal Fibrosis. Annu. Rev. Physiol. 2018, 80, 309–326. [Google Scholar] [CrossRef]
- Kuratsune, M.; Masaki, T.; Hirai, T.; Kiribayashi, K.; Yokoyama, Y.; Arakawa, T.; Yorioka, N.; Kohno, N. Signal transducer and activator of transcription 3 involvement in the development of renal interstitial fibrosis after unilateral ureteral obstruction. Nephrology 2007, 12, 565–571. [Google Scholar] [CrossRef]
- Pang, M.; Ma, L.; Gong, R.; Tolbert, E.; Mao, H.; Ponnusamy, M.; Chin, Y.E.; Yan, H.; Dworkin, L.D.; Zhuang, S. A novel STAT3 inhibitor, S3I-201, attenuates renal interstitial fibroblast activation and interstitial fibrosis in obstructive nephropathy. Kidney Int. 2010, 78, 257–268. [Google Scholar] [CrossRef]
- Menke, J.; Sollinger, D.; Schamberger, B.; Heemann, U.; Lutz, J. The effect of ischemia/reperfusion on the kidney graft. Curr. Opin. Organ Transpl. 2014, 19, 395–400. [Google Scholar] [CrossRef]
- Carvajal, G.; Rodriguez-Vita, J.; Rodrigues-Diez, R.; Sanchez-Lopez, E.; Ruperez, M.; Cartier, C.; Esteban, V.; Ortiz, A.; Egido, J.; Mezzano, S.A.; et al. Angiotensin II activates the Smad pathway during epithelial mesenchymal transdifferentiation. Kidney Int. 2008, 74, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Luo, M.; Li, R.; Huang, Y.; Zhang, R.; Wu, Q.; Wang, F.; Li, Y.; Yu, X. Blockage of JAK/STAT signalling attenuates renal ischaemia-reperfusion injury in rat. Nephrol. Dial. Transpl. 2008, 23, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Arany, I.; Megyesi, J.K.; Nelkin, B.D.; Safirstein, R.L. STAT3 attenuates EGFR-mediated ERK activation and cell survival during oxidant stress in mouse proximal tubular cells. Kidney Int. 2006, 70, 669–674. [Google Scholar] [CrossRef]
- Neria, F.; Castilla, M.A.; Sanchez, R.F.; Gonzalez Pacheco, F.R.; Deudero, J.J.; Calabia, O.; Tejedor, A.; Manzarbeitia, F.; Ortiz, A.; Caramelo, C. Inhibition of JAK2 protects renal endothelial and epithelial cells from oxidative stress and cyclosporin A toxicity. Kidney Int. 2009, 75, 227–234. [Google Scholar] [CrossRef]
- Yokota, N.; Burne-Taney, M.; Racusen, L.; Rabb, H. Contrasting roles for STAT4 and STAT6 signal transduction pathways in murine renal ischemia-reperfusion injury. Am. J. Physiol. Ren. Physiol. 2003, 285, F319–F325. [Google Scholar] [CrossRef]
- Hasslacher, C. Diabetic nephropathy: Structural-functional relationships. Contrib. Nephrol. 1989, 73, 24–28; discussion 28–29. [Google Scholar]
- Schrijvers, B.F.; De Vriese, A.S.; Flyvbjerg, A. From hyperglycemia to diabetic kidney disease: The role of metabolic, hemodynamic, intracellular factors and growth factors/cytokines. Endocr. Rev. 2004, 25, 971–1010. [Google Scholar] [CrossRef]
- Marrero, M.B.; Banes-Berceli, A.K.; Stern, D.M.; Eaton, D.C. Role of the JAK/STAT signaling pathway in diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2006, 290, F762–F768. [Google Scholar] [CrossRef]
- Berthier, C.C.; Zhang, H.; Schin, M.; Henger, A.; Nelson, R.G.; Yee, B.; Boucherot, A.; Neusser, M.A.; Cohen, C.D.; Carter-Su, C.; et al. Enhanced expression of Janus kinase-signal transducer and activator of transcription pathway members in human diabetic nephropathy. Diabetes 2009, 58, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Banes, A.K.; Shaw, S.; Jenkins, J.; Redd, H.; Amiri, F.; Pollock, D.M.; Marrero, M.B. Angiotensin II blockade prevents hyperglycemia-induced activation of JAK and STAT proteins in diabetic rat kidney glomeruli. Am. J. Physiol. Ren. Physiol. 2004, 286, F653–F659. [Google Scholar] [CrossRef]
- Lu, T.C.; Wang, Z.H.; Feng, X.; Chuang, P.Y.; Fang, W.; Shen, Y.; Levy, D.E.; Xiong, H.; Chen, N.; He, J.C. Knockdown of Stat3 activity in vivo prevents diabetic glomerulopathy. Kidney Int. 2009, 76, 63–71. [Google Scholar] [CrossRef]
- Amiri, F.; Shaw, S.; Wang, X.; Tang, J.; Waller, J.L.; Eaton, D.C.; Marrero, M.B. Angiotensin II activation of the JAK/STAT pathway in mesangial cells is altered by high glucose. Kidney Int. 2002, 61, 1605–1616. [Google Scholar] [CrossRef]
- Wang, X.; Shaw, S.; Amiri, F.; Eaton, D.C.; Marrero, M.B. Inhibition of the Jak/STAT signaling pathway prevents the high glucose-induced increase in tgf-beta and fibronectin synthesis in mesangial cells. Diabetes 2002, 51, 3505–3509. [Google Scholar] [CrossRef]
- Hirai, T.; Masaki, T.; Kuratsune, M.; Yorioka, N.; Kohno, N. PDGF receptor tyrosine kinase inhibitor suppresses mesangial cell proliferation involving STAT3 activation. Clin. Exp. Immunol. 2006, 144, 353–361. [Google Scholar] [CrossRef]
- Yanagita, M.; Arai, H.; Nakano, T.; Ohashi, K.; Mizuno, K.; Fukatsu, A.; Doi, T.; Kita, T. Gas6 induces mesangial cell proliferation via latent transcription factor STAT3. J. Biol. Chem. 2001, 276, 42364–42369. [Google Scholar] [CrossRef]
- Wang, S.; Yang, N.; Zhang, L.; Huang, B.; Tan, H.; Liang, Y.; Li, Y.; Yu, X. Jak/STAT signaling is involved in the inflammatory infiltration of the kidneys in MRL/lpr mice. Lupus 2010, 19, 1171–1180. [Google Scholar] [CrossRef]
- Matsui, F.; Meldrum, K.K. The role of the Janus kinase family/signal transducer and activator of transcription signaling pathway in fibrotic renal disease. J. Surg. Res. 2012, 178, 339–345. [Google Scholar] [CrossRef]
- Fan, Z.; Gao, Y.; Huang, Z.; Xue, F.; Wu, S.; Yang, J.; Zhu, L.; Fu, L. Protective effect of hydrogen-rich saline on pressure overload-induced cardiac hypertrophyin rats: Possible role of JAK-STAT signaling. BMC Cardiovasc. Disord. 2018, 18, 32. [Google Scholar] [CrossRef]
- Xuan, Y.T.; Guo, Y.; Han, H.; Zhu, Y.; Bolli, R. An essential role of the JAK-STAT pathway in ischemic preconditioning. Proc. Natl. Acad. Sci. USA 2001, 98, 9050–9055. [Google Scholar] [CrossRef]
- Xuan, Y.T.; Guo, Y.; Zhu, Y.; Han, H.; Langenbach, R.; Dawn, B.; Bolli, R. Mechanism of cyclooxygenase-2 upregulation in late preconditioning. J. Mol. Cell. Cardiol. 2003, 35, 525–537. [Google Scholar] [CrossRef]
- Xuan, Y.T.; Guo, Y.; Zhu, Y.; Wang, O.L.; Rokosh, G.; Messing, R.O.; Bolli, R. Role of the protein kinase C-epsilon-Raf-1-MEK-1/2-p44/42 MAPK signaling cascade in the activation of signal transducers and activators of transcription 1 and 3 and induction of cyclooxygenase-2 after ischemic preconditioning. Circulation 2005, 112, 1971–1978. [Google Scholar] [CrossRef] [PubMed]
- Hilfiker-Kleiner, D.; Hilfiker, A.; Fuchs, M.; Kaminski, K.; Schaefer, A.; Schieffer, B.; Hillmer, A.; Schmiedl, A.; Ding, Z.; Podewski, E.; et al. Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ. Res. 2004, 95, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Dai, B.; Cui, M.; Zhu, M.; Su, W.L.; Qiu, M.C.; Zhang, H. STAT1/3 and ERK1/2 synergistically regulate cardiac fibrosis induced by high glucose. Cell Physiol. Biochem. 2013, 32, 960–971. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Surinkaew, S.; Naud, P.; Qi, X.Y.; Gillis, M.A.; Shi, Y.F.; Tardif, J.C.; Dobrev, D.; Nattel, S. JAK-STAT signalling and the atrial fibrillation promoting fibrotic substrate. Cardiovasc. Res. 2017, 113, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Fukuda, K.; Saito, M.; Matsuzaki, J.; Kodama, H.; Sano, M.; Takahashi, T.; Kato, T.; Ogawa, S. Mechanical stretch activates the JAK/STAT pathway in rat cardiomyocytes. Circ. Res. 1999, 84, 1127–1136. [Google Scholar] [CrossRef]
- Hirota, H.; Yoshida, K.; Kishimoto, T.; Taga, T. Continuous activation of gp130, a signal-transducing receptor component for interleukin 6-related cytokines, causes myocardial hypertrophy in mice. Proc. Natl. Acad. Sci. USA 1995, 92, 4862–4866. [Google Scholar] [CrossRef]
- Ancey, C.; Menet, E.; Corbi, P.; Fredj, S.; Garcia, M.; Rucker-Martin, C.; Bescond, J.; Morel, F.; Wijdenes, J.; Lecron, J.C.; et al. Human cardiomyocyte hypertrophy induced in vitro by gp130 stimulation. Cardiovasc. Res. 2003, 59, 78–85. [Google Scholar] [CrossRef]
- Barry, S.P.; Townsend, P.A.; Latchman, D.S.; Stephanou, A. Role of the JAK-STAT pathway in myocardial injury. Trends Mol. Med. 2007, 13, 82–89. [Google Scholar] [CrossRef]
- Tefferi, A.; Lasho, T.L.; Schwager, S.M.; Steensma, D.P.; Mesa, R.A.; Li, C.Y.; Wadleigh, M.; Gary Gilliland, D. The JAK2(V617F) tyrosine kinase mutation in myelofibrosis with myeloid metaplasia: Lineage specificity and clinical correlates. Br. J. Haematol. 2005, 131, 320–328. [Google Scholar] [CrossRef]
- Schieber, M.; Crispino, J.D.; Stein, B. Myelofibrosis in 2019: Moving beyond JAK2 inhibition. Blood Cancer J. 2019, 9, 74. [Google Scholar] [CrossRef]
- Ulich, T.R.; del Castillo, J.; Senaldi, G.; Kinstler, O.; Yin, S.; Kaufman, S.; Tarpley, J.; Choi, E.; Kirley, T.; Hunt, P.; et al. Systemic hematologic effects of PEG-rHuMGDF-induced megakaryocyte hyperplasia in mice. Blood 1996, 87, 5006–5015. [Google Scholar] [CrossRef]
- Yanagida, M.; Ide, Y.; Imai, A.; Toriyama, M.; Aoki, T.; Harada, K.; Izumi, H.; Uzumaki, H.; Kusaka, M.; Tokiwa, T. The role of transforming growth factor-beta in PEG-rHuMGDF-induced reversible myelofibrosis in rats. Br. J. Haematol. 1997, 99, 739–745. [Google Scholar] [CrossRef]
- He, X.; Chen, Z.; Jiang, Y.; Qiu, X.; Zhao, X. Different mutations of the human c-mpl gene indicate distinct haematopoietic diseases. J. Hematol. Oncol. 2013, 6, 11. [Google Scholar] [CrossRef]
- Zahr, A.A.; Salama, M.E.; Carreau, N.; Tremblay, D.; Verstovsek, S.; Mesa, R.; Hoffman, R.; Mascarenhas, J. Bone marrow fibrosis in myelofibrosis: Pathogenesis, prognosis and targeted strategies. Haematologica 2016, 101, 660–671. [Google Scholar] [CrossRef]
- Maher, T.M.; Wells, A.U.; Laurent, G.J. Idiopathic pulmonary fibrosis: Multiple causes and multiple mechanisms? Eur. Respir. J. 2007, 30, 835–839. [Google Scholar] [CrossRef]
- Wilson, M.S.; Wynn, T.A. Pulmonary fibrosis: Pathogenesis, etiology and regulation. Mucosal. Immunol. 2009, 2, 103–121. [Google Scholar] [CrossRef]
- Idiopathic Pulmonary Fibrosis Clinical Research Network; Raghu, G.; Anstrom, K.J.; King, T.E., Jr.; Lasky, J.A.; Martinez, F.J. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N. Engl. J. Med. 2012, 366, 1968–1977. [Google Scholar] [CrossRef]
- Coward, W.R.; Saini, G.; Jenkins, G. The pathogenesis of idiopathic pulmonary fibrosis. Adv. Respir. Dis. 2010, 4, 367–388. [Google Scholar] [CrossRef]
- Wolters, P.J.; Collard, H.R.; Jones, K.D. Pathogenesis of idiopathic pulmonary fibrosis. Annu. Rev. Pathol. 2014, 9, 157–179. [Google Scholar] [CrossRef]
- Milara, J.; Hernandez, G.; Ballester, B.; Morell, A.; Roger, I.; Montero, P.; Escriva, J.; Lloris, J.M.; Molina-Molina, M.; Morcillo, E.; et al. The JAK2 pathway is activated in idiopathic pulmonary fibrosis. Respir. Res. 2018, 19, 24. [Google Scholar] [CrossRef]
- Talotta, R. The rationale for targeting the JAK/STAT pathway in scleroderma-associated interstitial lung disease. Immunotherapy 2021, 13, 241–256. [Google Scholar] [CrossRef]
- Wang, W.; Bhattacharyya, S.; Marangoni, R.G.; Carns, M.; Dennis-Aren, K.; Yeldandi, A.; Wei, J.; Varga, J. The JAK/STAT pathway is activated in systemic sclerosis and is effectively targeted by tofacitinib. J. Scleroderma Relat. Disord. 2020, 5, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Jiang, J.; Ma, T.; Xie, J.; Duan, L.; Chen, R.; Song, P.; Yu, Z.; Liu, C.; Zhu, Q.; et al. Dexamethasone attenuates bleomycin-induced lung fibrosis in mice through TGF-beta, Smad3 and JAK-STAT pathway. Int. J. Clin. Exp. Med. 2014, 7, 2645–2650. [Google Scholar]
- Yang, G.; Lyu, L.; Wang, X.; Bao, L.; Lyu, B.; Lin, Z. Systemic treatment with resveratrol alleviates adjuvant arthritis-interstitial lung disease in rats via modulation of JAK/STAT/RANKL signaling pathway. Pulm Pharm. Ther. 2019, 56, 69–74. [Google Scholar] [CrossRef]
- Wang, F.; Wang, S.; Zhang, C.; Tian, X.; Zhou, Y.; Xuan, W.; Matteson, E.L.; Luo, F.; Tschumperlin, D.; Vassallo, R. Noncanonical JAK1/STAT3 interactions with TGF-β modulate myofibroblast transdifferentiation and fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2022, 323, L698–l714. [Google Scholar] [CrossRef] [PubMed]
- Milara, J.; Ballester, B.; Morell, A.; Ortiz, J.L.; Escriva, J.; Fernandez, E.; Perez-Vizcaino, F.; Cogolludo, A.; Pastor, E.; Artigues, E.; et al. JAK2 mediates lung fibrosis, pulmonary vascular remodelling and hypertension in idiopathic pulmonary fibrosis: An experimental study. Thorax 2018, 73, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zheng, X.; Wang, C.; Sun, X.; Sun, H.; Ma, T.; Li, Y.; Liu, K.; Chen, L.; Ma, X. Synthesis and biological activity of thieno[3,2-d]yrimidines as potent JAK3 inhibitors for the treatment of idiopathic pulmonary fibrosis. Bioorganic Med. Chem. 2020, 28, 115254. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| STAT Proteins | Major Triggers or Cytokines | Possible Roles in Fibrotic Liver |
|---|---|---|
| STAT1 | IFN-α, IFN-β, IFN-γ | Hepatic Stellate Cells→Loss→Inhibited proliferation, increased apoptosis and blocked cell cycle Hepatocytes→Inhibition of liver regeneration, and pro-apoptosis |
| STAT2 | IFN-α, IFN-β, IFN-λ | Promote antiviral responses Protect against hepatic fibrosis |
| STAT3 | IL-6, IL-22 | Hepatocytes→Loss→Increased inflammation in the CCl4-induced chronic model Hepatocytes→Loss→Reduced inflammation in the CCl4-induced acute model |
| STAT4 | IL-12, IFN-γ | Impaired STAT4 phosphorylation→liver inflammation and fibrosis Promotes liver inflammation |
| STAT5 | GH, IL-2, IL-3, IL-5 | Hepatocytes→Loss→Increased TGF-β levels, increased the sensitivity of Kupffer or hepatic stellate cells to TGF-β Hepatocytes→Loss→Reduces proliferation Antifibrotic effects in the mouse model of cholestasis |
| STAT6 | IL-4, IL-13 | Hepatocytes→Loss→Reduced collagen deposition Hepatic Stellate Cells→Promotes liver fibrogenesis Promotes inflammation in hepatitis Protects against ischemia/reperfusion and inflammation of drug-induced liver injuries |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Wang, F.; Luo, F. The Role of JAK/STAT Pathway in Fibrotic Diseases: Molecular and Cellular Mechanisms. Biomolecules 2023, 13, 119. https://doi.org/10.3390/biom13010119
Liu J, Wang F, Luo F. The Role of JAK/STAT Pathway in Fibrotic Diseases: Molecular and Cellular Mechanisms. Biomolecules. 2023; 13(1):119. https://doi.org/10.3390/biom13010119
Chicago/Turabian StyleLiu, Jia, Faping Wang, and Fengming Luo. 2023. "The Role of JAK/STAT Pathway in Fibrotic Diseases: Molecular and Cellular Mechanisms" Biomolecules 13, no. 1: 119. https://doi.org/10.3390/biom13010119
APA StyleLiu, J., Wang, F., & Luo, F. (2023). The Role of JAK/STAT Pathway in Fibrotic Diseases: Molecular and Cellular Mechanisms. Biomolecules, 13(1), 119. https://doi.org/10.3390/biom13010119