
HIF1A Knockout by Biallelic and Selection-Free CRISPR Gene Editing in Human Primary Endothelial Cells with Ribonucleoprotein Complexes

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation and Culturing of Primary HUVECs
2.2. Preparation of EGFP mRNA and EGFP Plasmid
2.3. Nucleofection of HUVECs with EGFP mRNA Titration
2.4. Nucleofection of HUVECs with EGFP mRNA and EGFP Plasmid
2.5. CRISPR-Cas9 gRNA Design
2.6. Evaluation of CRISPR-Cas9 Gene Editing Efficiencies on HIF1A in HUVECs
2.7. FC Analysis of HIF1A KO and WT HUVECs
2.8. LDL Uptake Assay
2.9. Staining of HIF1A/Hoechst/Actin
2.10. Tube Formation Assay
3. Results
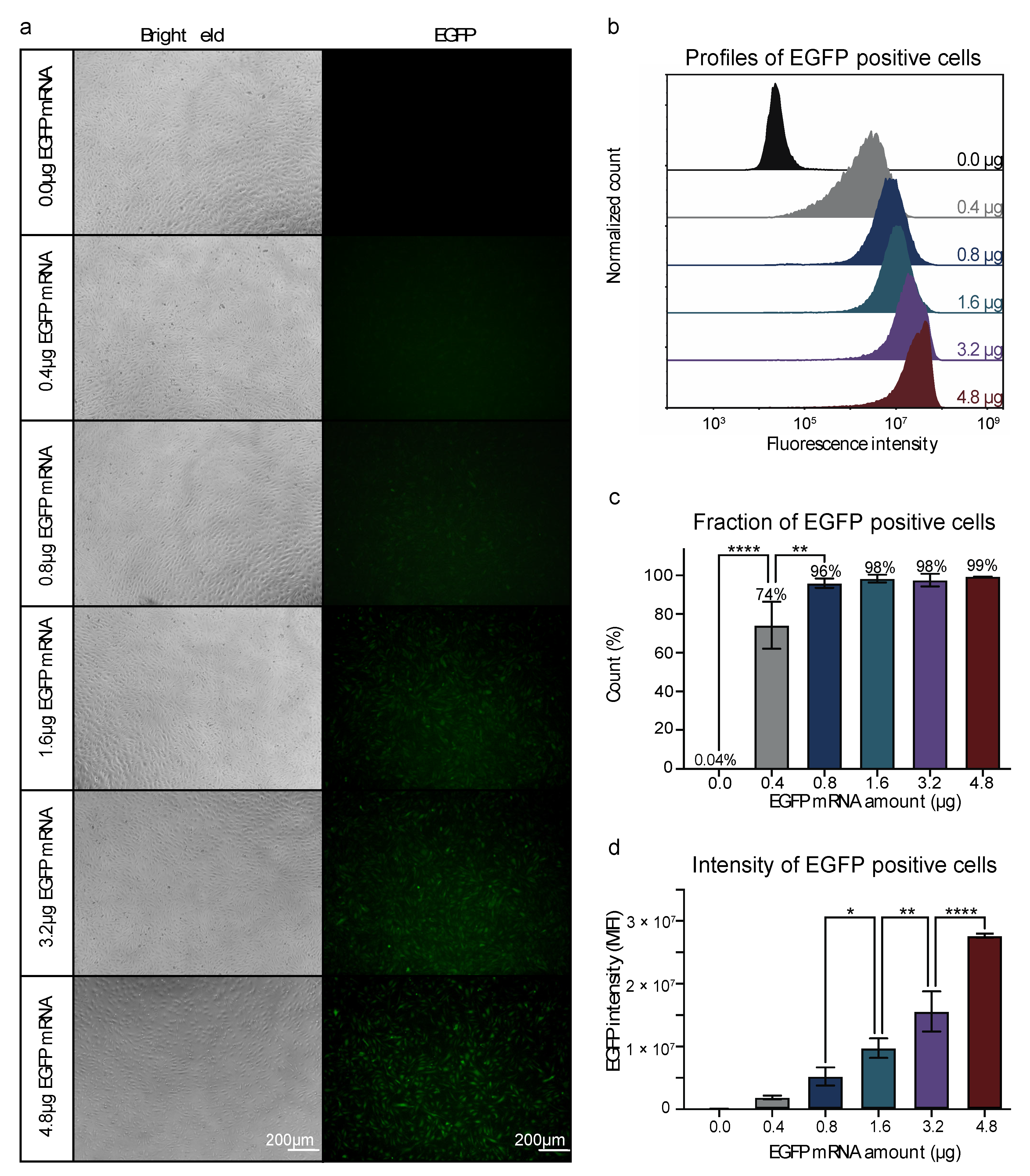
3.1. Efficient mRNA Delivery into HUVECs by Nucleofection
3.2. Efficient CRISPR Gene Editing of Primary HUVECs
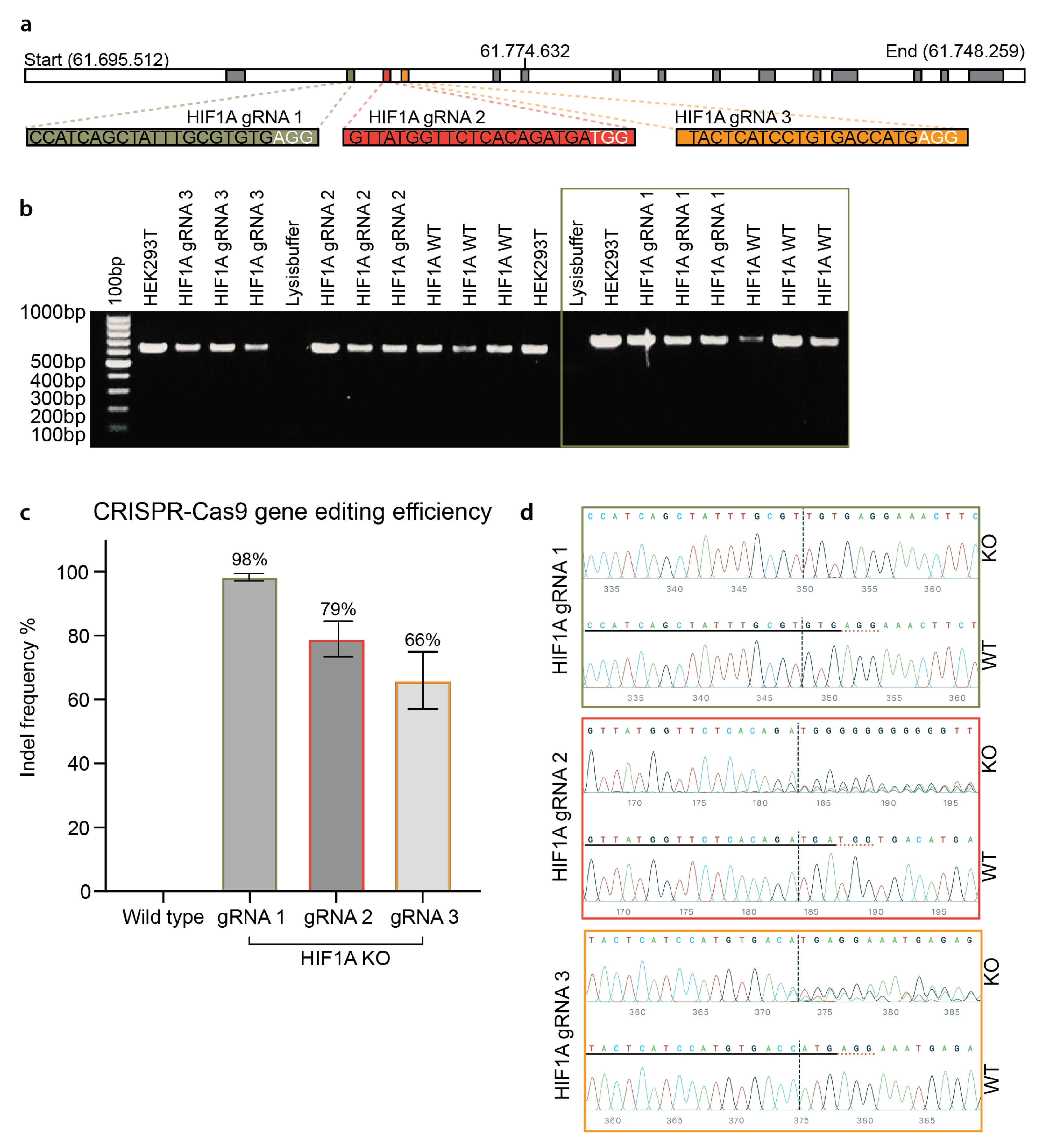
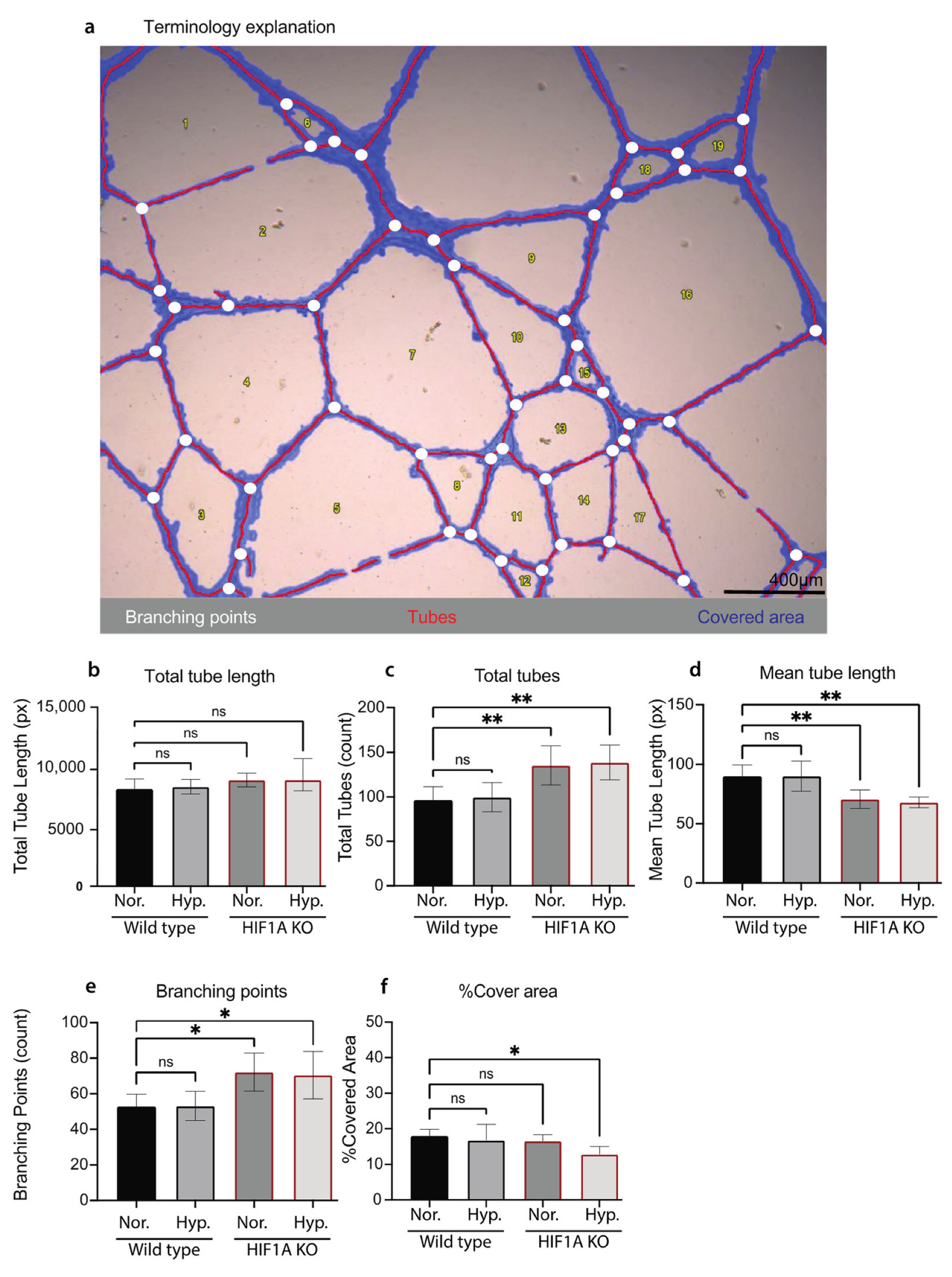
3.3. Functional Validation of HIF1A KO HUVECs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Fishman, A.P. ENDOTHELIUM: A DISTRIBUTED ORGAN OF DIVERSE CAPABILITIES. Ann. N. Y. Acad. Sci. 1982, 401, 1–8. [Google Scholar] [CrossRef]
- Hassan, W. The endothelium and endothelin: Beyond vascular reactivity. Ann. Saudi Med. 2006, 26, 343–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krüger-Genge, A.; Blocki, A.; Franke, R.P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Aspelund, A.; Robciuc, M.R.; Karaman, S.; Makinen, T.; Alitalo, K. Lymphatic System in Cardiovascular Medicine. Circ. Res. 2016, 118, 515–530. [Google Scholar] [CrossRef]
- Eelen, G.; de Zeeuw, P.; Treps, L.; Harjes, U.; Wong, B.W.; Carmeliet, P. Endothelial Cell Metabolism. Physiol. Rev. 2018, 98, 3–58. [Google Scholar] [CrossRef] [Green Version]
- Dumas, S.J.; Meta, E.; Borri, M.; Goveia, J.; Rohlenova, K.; Conchinha, N.V.; Falkenberg, K.; Teuwen, L.A.; de Rooij, L.; Kalucka, J.; et al. Single-Cell RNA Sequencing Reveals Renal Endothelium Heterogeneity and Metabolic Adaptation to Water Deprivation. J. Am. Soc. Nephrol. 2020, 31, 118–138. [Google Scholar] [CrossRef]
- Kalucka, J.; de Rooij, L.; Goveia, J.; Rohlenova, K.; Dumas, S.J.; Meta, E.; Conchinha, N.V.; Taverna, F.; Teuwen, L.A.; Veys, K.; et al. Single-Cell Transcriptome Atlas of Murine Endothelial Cells. Cell 2020, 180, 764–779.e20. [Google Scholar] [CrossRef]
- Wang, F.; Ding, P.; Liang, X.; Ding, X.; Brandt, C.B.; Sjöstedt, E.; Zhu, J.; Bolund, S.; Zhang, L.; de Rooij, L.P.M.H.; et al. Endothelial cell heterogeneity and microglia regulons revealed by a pig cell landscape at single-cell level. Nat. Commun. 2022, 13, 3620. [Google Scholar] [CrossRef]
- Abrahimi, P.; Chang, W.G.; Kluger, M.S.; Qyang, Y.; Tellides, G.; Saltzman, W.M.; Pober, J.S. Efficient gene disruption in cultured primary human endothelial cells by CRISPR/Cas9. Circ. Res. 2015, 117, 121–128. [Google Scholar] [CrossRef]
- Dahlman, J.E.; Barnes, C.; Khan, O.F.; Thiriot, A.; Jhunjunwala, S.; Shaw, T.E.; Xing, Y.; Sager, H.B.; Sahay, G.; Speciner, L.; et al. In vivo endothelial siRNA delivery using polymeric nanoparticles with low molecular weight. Nat. Nanotechnol. 2014, 9, 648–655. [Google Scholar] [CrossRef]
- Davidson, B.L.; McCray, P.B. Current prospects for RNA interference-based therapies. Nat. Rev. Genet. 2011, 12, 329–340. [Google Scholar] [CrossRef]
- Aagaard, L.; Rossi, J.J. RNAi therapeutics: Principles, prospects and challenges. Adv. Drug Deliv. Rev. 2007, 59, 75–86. [Google Scholar] [CrossRef] [Green Version]
- Mangeot, P.E.; Risson, V.; Fusil, F.; Marnef, A.; Laurent, E.; Blin, J.; Mournetas, V.; Massouridès, E.; Sohier, T.J.M.; Corbin, A.; et al. Genome editing in primary cells and in vivo using viral-derived Nanoblades loaded with Cas9-sgRNA ribonucleoproteins. Nat. Commun. 2019, 10, 45. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.; Duan, Y.; Ma, G.; Zhou, G.; Park-Windhol, C.; D’Amore, P.A.; Lei, H. AAV-CRISPR/Cas9-Mediated Depletion of VEGFR2 Blocks Angiogenesis In Vitro. Investig. Ophthalmol. Vis. Sci. 2017, 58, 6082–6090. [Google Scholar] [CrossRef] [Green Version]
- Schwefel, K.; Spiegler, S.; Much, C.D.; Felbor, U.; Rath, M. CRISPR/Cas9-mediated Generation of Human Endothelial Cell Knockout Models of CCM Disease. Methods Mol. Biol. 2020, 2152, 169–177. [Google Scholar] [CrossRef]
- Liao, H.; He, H.; Chen, Y.; Zeng, F.; Huang, J.; Wu, L.; Chen, Y. Effects of long-term serial cell passaging on cell spreading, migration, and cell-surface ultrastructures of cultured vascular endothelial cells. Cytotechnology 2014, 66, 229–238. [Google Scholar] [CrossRef] [Green Version]
- Kok, F.O.; Lawson, N.D. A platform for reverse genetics in endothelial cells. Circ. Res. 2015, 117, 107–108. [Google Scholar] [CrossRef] [Green Version]
- van Beijnum, J.R.; van der Linden, E.; Griffioen, A.W. Angiogenic profiling and comparison of immortalized endothelial cells for functional genomics. Exp. Cell Res. 2008, 314, 264–272. [Google Scholar] [CrossRef]
- Huebert, R.C.; Jagavelu, K.; Liebl, A.F.; Huang, B.Q.; Splinter, P.L.; LaRusso, N.F.; Urrutia, R.A.; Shah, V.H. Immortalized liver endothelial cells: A cell culture model for studies of motility and angiogenesis. Lab. Investig. 2010, 90, 1770–1781. [Google Scholar] [CrossRef]
- Bouïs, D.; Hospers, G.A.P.; Meijer, C.; Molema, G.; Mulder, N.H. Endothelium in vitro: A review of human vascular endothelial cell lines for blood vessel-related research. Angiogenesis 2001, 4, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Schwefel, K.; Spiegler, S.; Ameling, S.; Much, C.D.; Pilz, R.A.; Otto, O.; Völker, U.; Felbor, U.; Rath, M. Biallelic CCM3 mutations cause a clonogenic survival advantage and endothelial cell stiffening. J. Cell. Mol. Med. 2019, 23, 1771–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Jin, H.; Huang, X.; Chaurasiya, B.; Dong, D.; Shanley, T.P.; Zhao, Y.Y. Robust genome editing in adult vascular endothelium by nanoparticle delivery of CRISPR-Cas9 plasmid DNA. Cell Rep. 2022, 38, 110196. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Liu, M.; Klomp, J.; Merrill, B.J.; Rehman, J.; Malik, A.B. Method for Dual Viral Vector Mediated CRISPR-Cas9 Gene Disruption in Primary Human Endothelial Cells. Sci. Rep. 2017, 7, 42127. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Chen, L.; Tian, Z.; Shen, X.; Wang, X.; Wu, H.; Wang, Y.; Zou, J.; Liang, J. CRISPR-Cas9 mediated gene knockout in human coronary artery endothelial cells reveals a pro-inflammatory role of TLR2. Cell Biol. Int. 2018, 42, 187–193. [Google Scholar] [CrossRef]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A review of the challenges and approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Shen, J.; Li, D.; Cheng, Y. Strategies in the delivery of Cas9 ribonucleoprotein for CRISPR/Cas9 genome editing. Theranostics 2021, 11, 614–648. [Google Scholar] [CrossRef]
- Hendel, A.; Bak, R.O.; Clark, J.T.; Kennedy, A.B.; Ryan, D.E.; Roy, S.; Steinfeld, I.; Lunstad, B.D.; Kaiser, R.J.; Wilkens, A.B.; et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015, 33, 985–989. [Google Scholar] [CrossRef]
- Martufi, M.; Good, R.B.; Rapiteanu, R.; Schmidt, T.; Patili, E.; Tvermosegaard, K.; New, M.; Nanthakumar, C.B.; Betts, J.; Blanchard, A.D.; et al. Single-Step, High-Efficiency CRISPR-Cas9 Genome Editing in Primary Human Disease-Derived Fibroblasts. CRISPR J. 2019, 2, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Xiang, X.; Zhao, X.; Pan, X.; Dong, Z.; Yu, J.; Li, S.; Liang, X.; Han, P.; Qu, K.; Jensen, J.B.; et al. Efficient correction of Duchenne muscular dystrophy mutations by SpCas9 and dual gRNAs. Mol. Nucleic Acids 2021, 24, 403–415. [Google Scholar] [CrossRef]
- Riggan, L.; Hildreth, A.D.; Rolot, M.; Wong, Y.-Y.; Satyadi, W.; Sun, R.; Huerta, C.; O’Sullivan, T.E. CRISPR-Cas9 Ribonucleoprotein-Mediated Genomic Editing in Mature Primary Innate Immune Cells. Cell Rep. 2020, 31, 107651. [Google Scholar] [CrossRef]
- Concordet, J.-P.; Haeussler, M. CRISPOR: Intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 2018, 46, W242–W245. [Google Scholar] [CrossRef] [Green Version]
- Xiang, X.; Corsi, G.I.; Anthon, C.; Qu, K.; Pan, X.; Liang, X.; Han, P.; Dong, Z.; Liu, L.; Zhong, J.; et al. Enhancing CRISPR-Cas9 gRNA efficiency prediction by data integration and deep learning. Nat. Commun. 2021, 12, 3238. [Google Scholar] [CrossRef]
- Anthon, C.; Corsi, G.I.; Gorodkin, J. CRISPRon/off: CRISPR/Cas9 on- and off-target gRNA design. Bioinformatics 2022, 38, 5437–5439. [Google Scholar] [CrossRef]
- Mello de Queiroz, F.; Sánchez, A.; Agarwal, J.R.; Stühmer, W.; Pardo, L.A. Nucleofection induces non-specific changes in the metabolic activity of transfected cells. Mol. Biol. Rep. 2012, 39, 2187–2194. [Google Scholar] [CrossRef] [Green Version]
- Logsdon, E.A.; Finley, S.D.; Popel, A.S.; Mac Gabhann, F. A systems biology view of blood vessel growth and remodelling. J. Cell. Mol. Med. 2014, 18, 1491–1508. [Google Scholar] [CrossRef]
- Lee, J.-W.; Bae, S.-H.; Jeong, J.-W.; Kim, S.-H.; Kim, K.-W. Hypoxia-inducible factor (HIF-1)α: Its protein stability and biological functions. Exp. Mol. Med. 2004, 36, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Hewitson, K.S.; Schofield, C.J. The HIF pathway as a therapeutic target. Drug Discov. Today 2004, 9, 704–711. [Google Scholar] [CrossRef]
- Carmeliet, P. Mechanisms of angiogenesis and arteriogenesis. Nat. Med. 2000, 6, 389–395. [Google Scholar] [CrossRef]
- Gerhardt, H.; Golding, M.; Fruttiger, M.; Ruhrberg, C.; Lundkvist, A.; Abramsson, A.; Jeltsch, M.; Mitchell, C.; Alitalo, K.; Shima, D.; et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 2003, 161, 1163–1177. [Google Scholar] [CrossRef]
- Blanco, R.; Gerhardt, H. VEGF and Notch in tip and stalk cell selection. Cold Spring Harb. Perspect. Med. 2013, 3, a006569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpentier, G.; Berndt, S.; Ferratge, S.; Rasband, W.; Cuendet, M.; Uzan, G.; Albanese, P. Angiogenesis Analyzer for ImageJ—A comparative morphometric analysis of “Endothelial Tube Formation Assay” and “Fibrin Bead Assay”. Sci. Rep. 2020, 10, 11568. [Google Scholar] [CrossRef] [PubMed]
- Moradian, H.; Roch, T.; Lendlein, A.; Gossen, M. mRNA Transfection-Induced Activation of Primary Human Monocytes and Macrophages: Dependence on Carrier System and Nucleotide Modification. Sci. Rep. 2020, 10, 4181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunt, M.A.; Currie, M.J.; Robinson, B.A.; Dachs, G.U. Optimizing transfection of primary human umbilical vein endothelial cells using commercially available chemical transfection reagents. J. Biomol. Tech. 2010, 21, 66–72. [Google Scholar] [PubMed]
- Akidil, E.; Albanese, M.; Buschle, A.; Ruhle, A.; Pich, D.; Keppler, O.T.; Hammerschmidt, W. Highly efficient CRISPR-Cas9-mediated gene knockout in primary human B cells for functional genetic studies of Epstein-Barr virus infection. PLOS Pathog. 2021, 17, e1009117. [Google Scholar] [CrossRef] [PubMed]
- Yip, B.H. Recent Advances in CRISPR/Cas9 Delivery Strategies. Biomolecules 2020, 10, 839. [Google Scholar] [CrossRef]
- Guo, T.; Feng, Y.-L.; Xiao, J.-J.; Liu, Q.; Sun, X.-N.; Xiang, J.-F.; Kong, N.; Liu, S.-C.; Chen, G.-Q.; Wang, Y.; et al. Harnessing accurate non-homologous end joining for efficient precise deletion in CRISPR/Cas9-mediated genome editing. Genome Biol. 2018, 19, 170. [Google Scholar] [CrossRef] [Green Version]
- Tang, N.; Wang, L.; Esko, J.; Giordano, F.J.; Huang, Y.; Gerber, H.-P.; Ferrara, N.; Johnson, R.S. Loss of HIF-1α in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell 2004, 6, 485–495. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Kumar, N.; Parachuri, N.; Singh, S.; Bandello, F.; Kuppermann, B.D.; Loewenstein, A. Brolucizumab-related retinal vasculitis: Emerging disconnect between clinical trials and real world. Eye 2021, 35, 1292–1294. [Google Scholar] [CrossRef]
- Gross, J.G.; Glassman, A.R.; Jampol, L.M.; Inusah, S.; Aiello, L.P.; Antoszyk, A.N.; Baker, C.W.; Berger, B.B.; Bressler, N.M.; Browning, D.; et al. Panretinal Photocoagulation vs Intravitreous Ranibizumab for Proliferative Diabetic Retinopathy: A Randomized Clinical Trial. JAMA 2015, 314, 2137–2146. [Google Scholar] [CrossRef]
- Zhao, Y.; Singh, R.P. The role of anti-vascular endothelial growth factor (anti-VEGF) in the management of proliferative diabetic retinopathy. Drugs Context 2018, 7, 212532. [Google Scholar] [CrossRef]
- Itatani, Y.; Kawada, K.; Yamamoto, T.; Sakai, Y. Resistance to Anti-Angiogenic Therapy in Cancer-Alterations to Anti-VEGF Pathway. Int. J. Mol. Sci. 2018, 19, 1232. [Google Scholar] [CrossRef] [Green Version]
- Batioglu, F.; Demirel, S.; Özmert, E.; Abdullayev, A.; Bilici, S. Short-term outcomes of switching anti-VEGF agents in eyes with treatment-resistant wet AMD. BMC Ophthalmol. 2015, 15, 40. [Google Scholar] [CrossRef] [Green Version]
- Holmgaard, A.B.; Askou, A.L.; Jensen, E.G.; Alsing, S.; Bak, R.O.; Mikkelsen, J.G.; Corydon, T.J. Targeted Knockout of the Vegfa Gene in the Retina by Subretinal Injection of RNP Complexes Containing Cas9 Protein and Modified sgRNAs. Mol. Ther. 2021, 29, 191–207. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brandt, C.B.; Fonager, S.V.; Haskó, J.; Helmig, R.B.; Degn, S.; Bolund, L.; Jessen, N.; Lin, L.; Luo, Y. HIF1A Knockout by Biallelic and Selection-Free CRISPR Gene Editing in Human Primary Endothelial Cells with Ribonucleoprotein Complexes. Biomolecules 2023, 13, 23. https://doi.org/10.3390/biom13010023
Brandt CB, Fonager SV, Haskó J, Helmig RB, Degn S, Bolund L, Jessen N, Lin L, Luo Y. HIF1A Knockout by Biallelic and Selection-Free CRISPR Gene Editing in Human Primary Endothelial Cells with Ribonucleoprotein Complexes. Biomolecules. 2023; 13(1):23. https://doi.org/10.3390/biom13010023
Chicago/Turabian StyleBrandt, Camilla Blunk, Sofie Vestergaard Fonager, János Haskó, Rikke Bek Helmig, Søren Degn, Lars Bolund, Niels Jessen, Lin Lin, and Yonglun Luo. 2023. "HIF1A Knockout by Biallelic and Selection-Free CRISPR Gene Editing in Human Primary Endothelial Cells with Ribonucleoprotein Complexes" Biomolecules 13, no. 1: 23. https://doi.org/10.3390/biom13010023
APA StyleBrandt, C. B., Fonager, S. V., Haskó, J., Helmig, R. B., Degn, S., Bolund, L., Jessen, N., Lin, L., & Luo, Y. (2023). HIF1A Knockout by Biallelic and Selection-Free CRISPR Gene Editing in Human Primary Endothelial Cells with Ribonucleoprotein Complexes. Biomolecules, 13(1), 23. https://doi.org/10.3390/biom13010023