
Targeting the Structural Maturation Pathway of HIV-1 Reverse Transcriptase

 ,
,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals Evaluated
2.2. Protein Design and Expression
2.3. Expression of FPC
2.4. Expression of p51
2.5. Expression of FPC Variants for Resonance Assignments
2.6. NMR Studies
2.6.1. STD Experiments
2.6.2. NMR Assignments
2.7. Computational Ligand Identification
2.8. Protein Crystallography/Crystallization
2.9. Dimerization Assay by Size Exclusion Chromatography
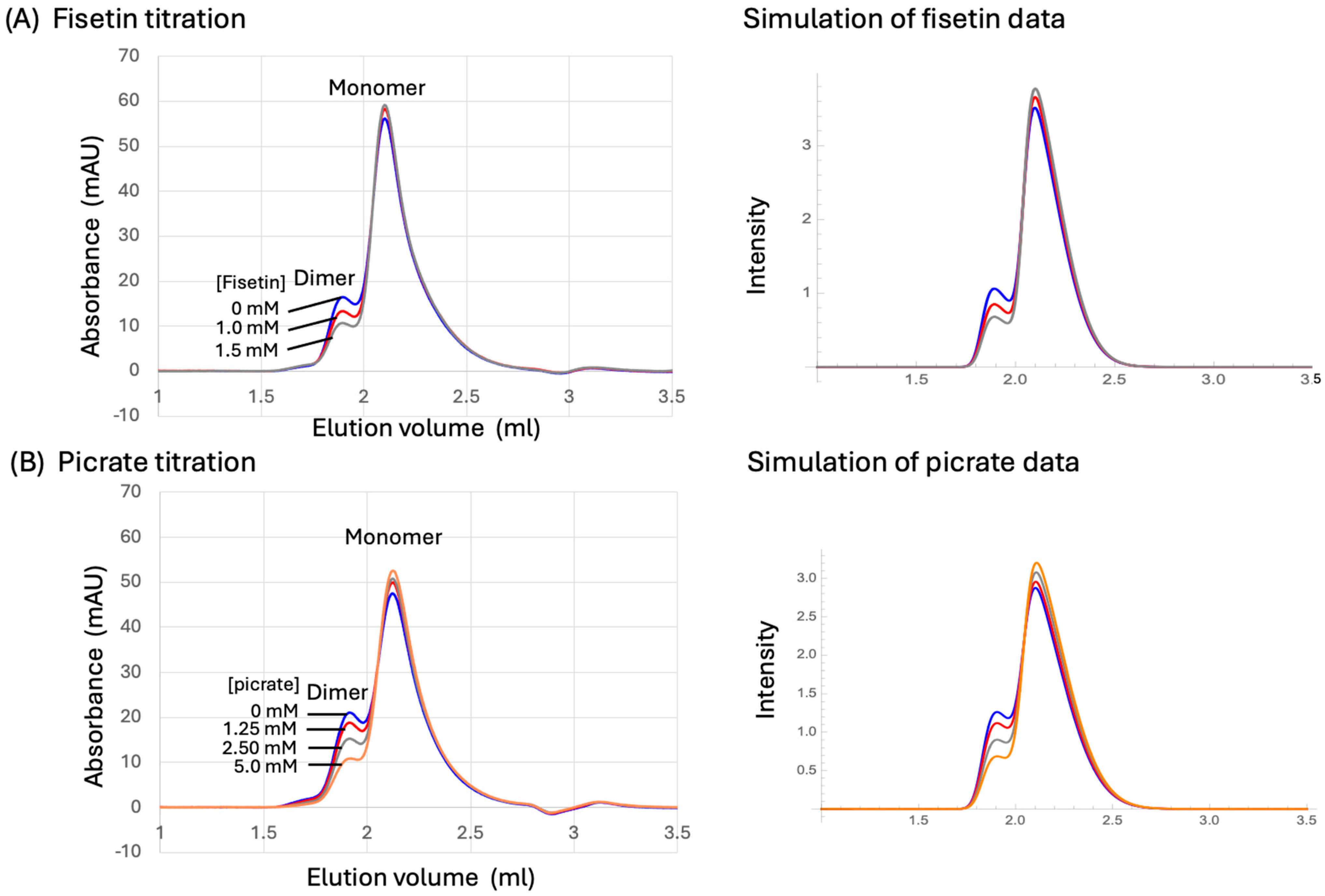
2.10. SEC Data Analysis
3. Results
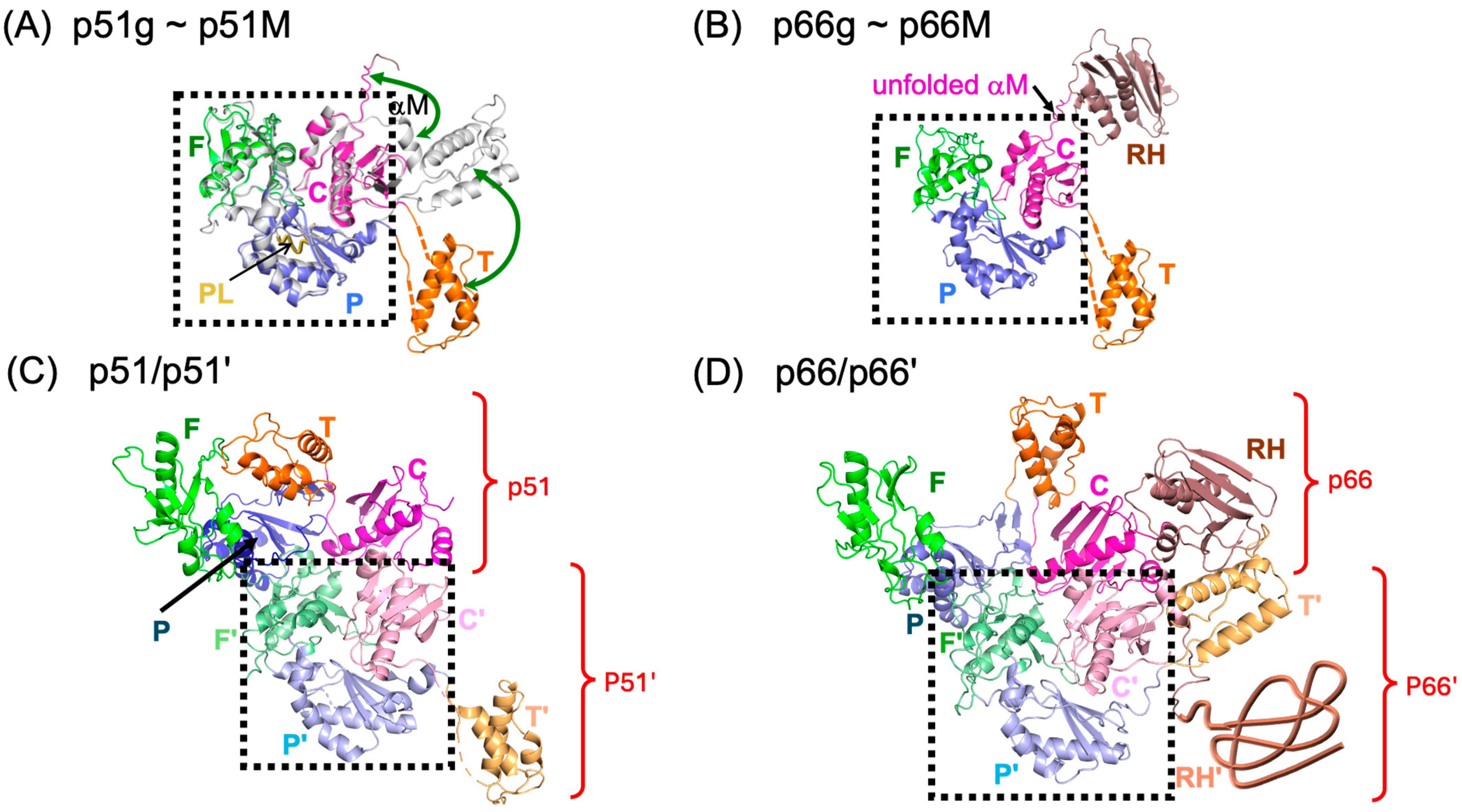
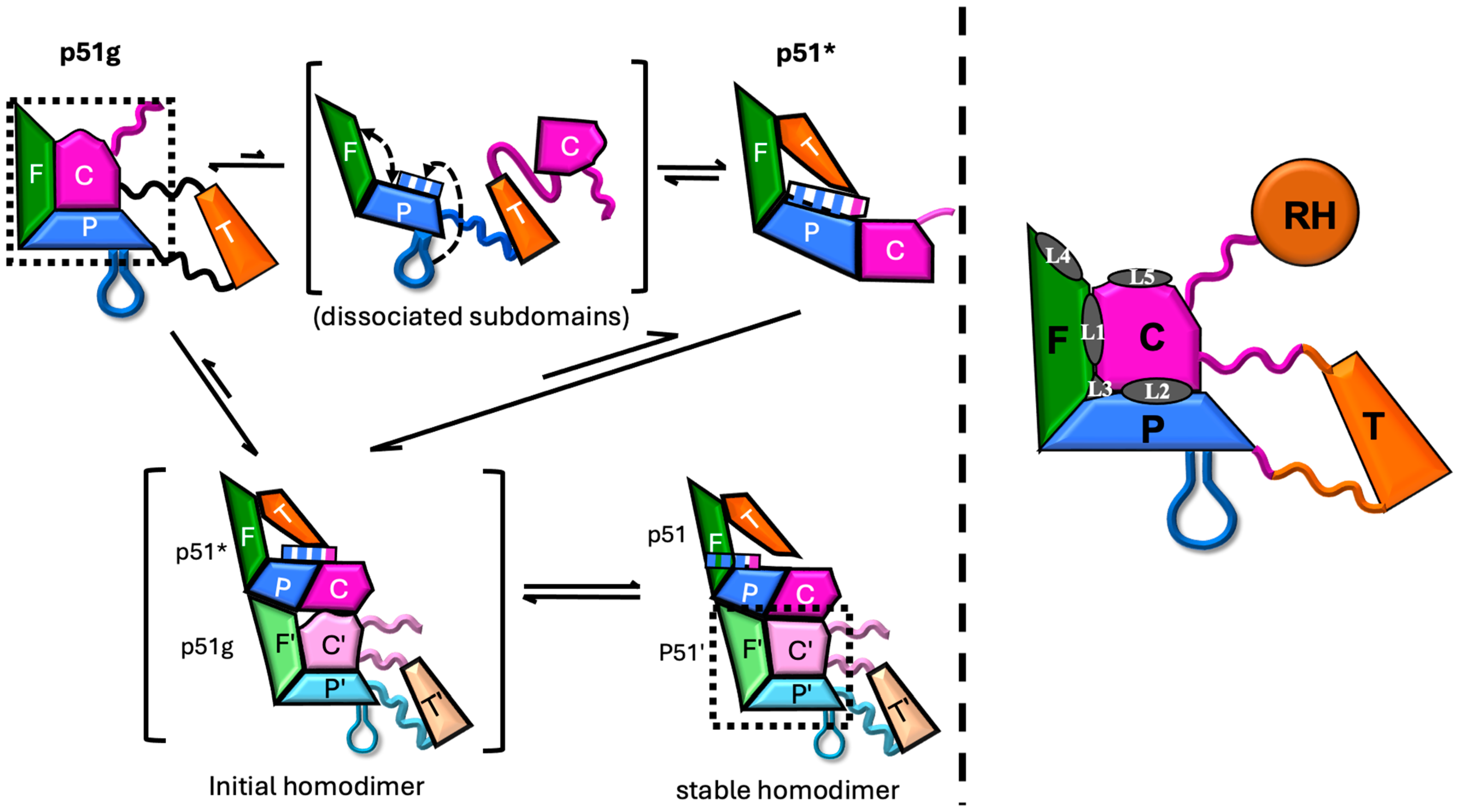
3.1. Structural Maturation Pathway and Inhibition Strategy


3.2. Characterization of Fingers/Palm/Connection (FPC) Constructs
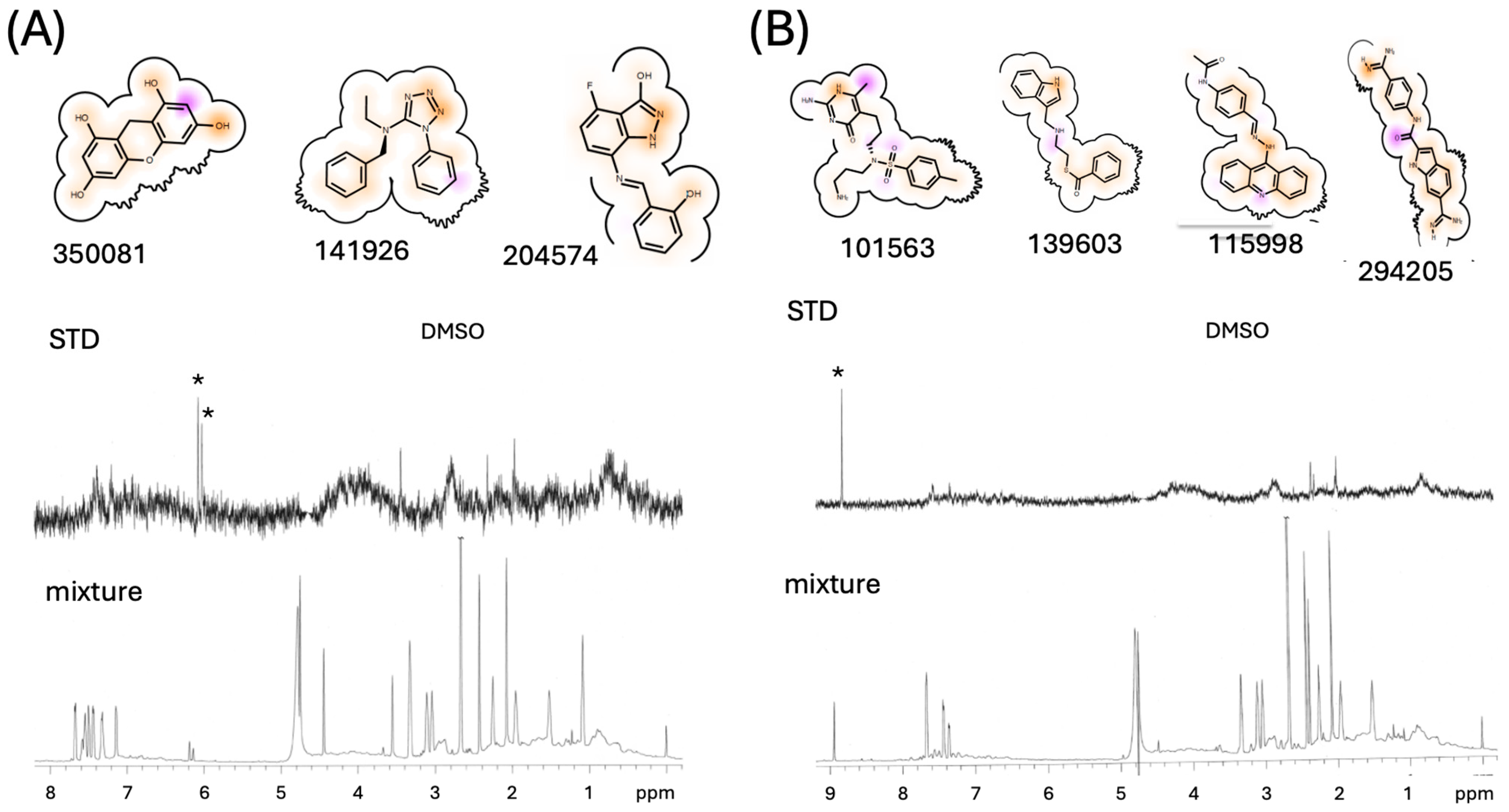
3.3. Initial Ligand Identification
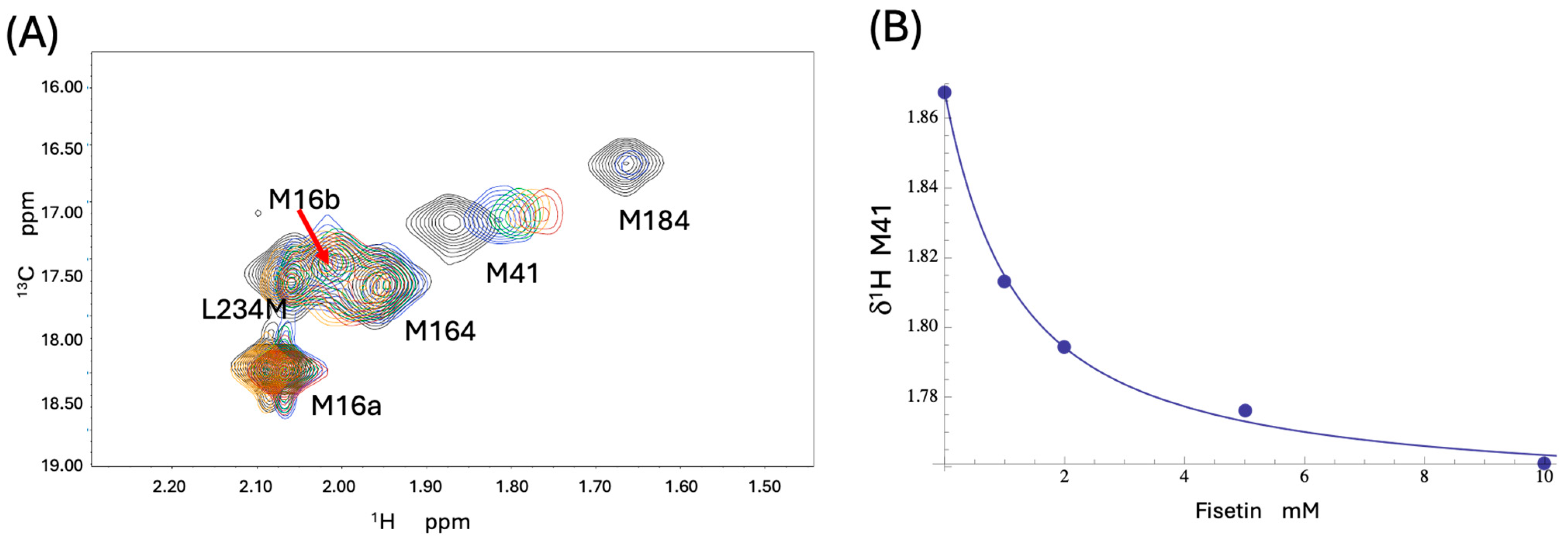
3.4. NMR Studies of [13CH3-Met]FPC
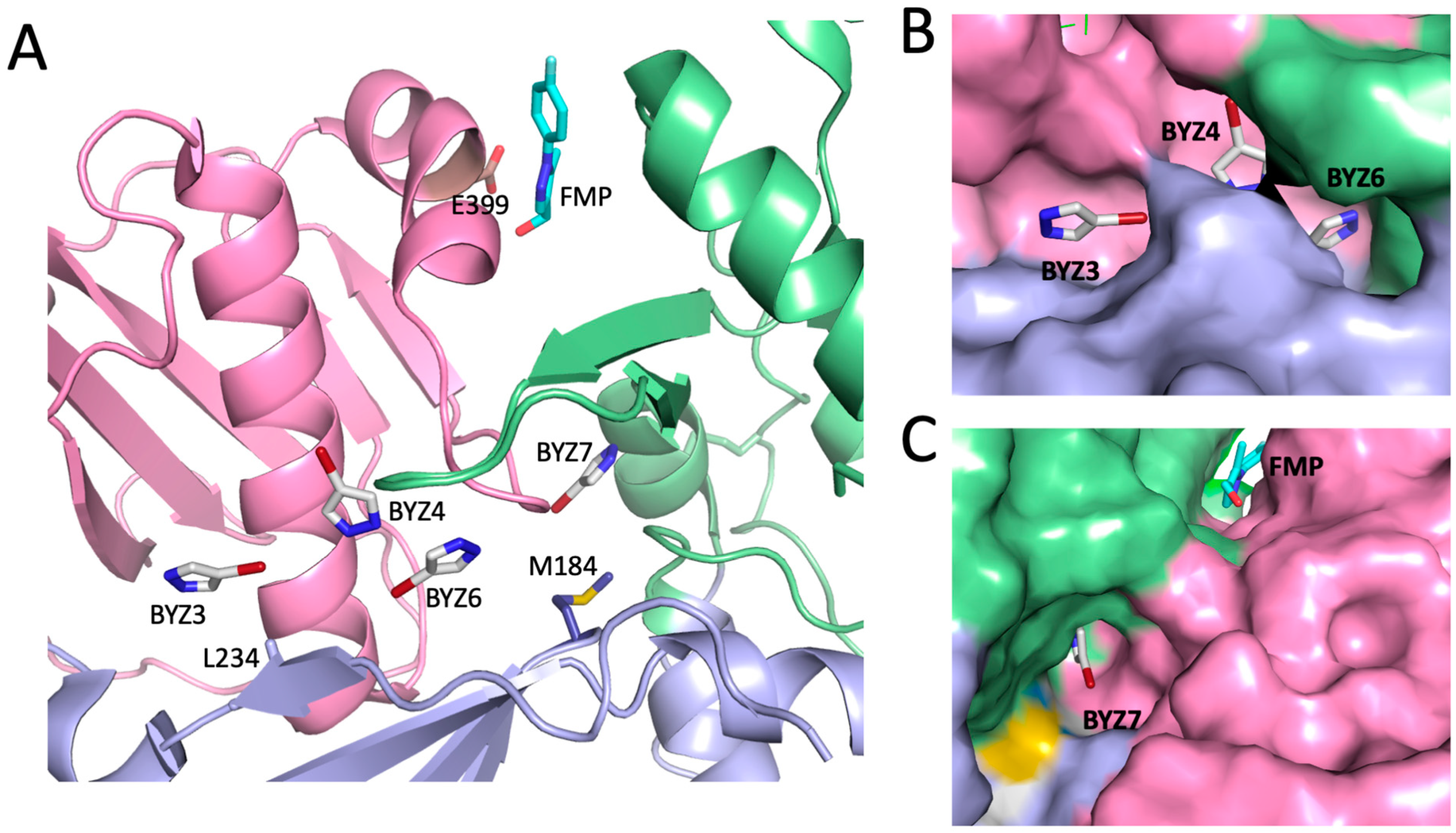
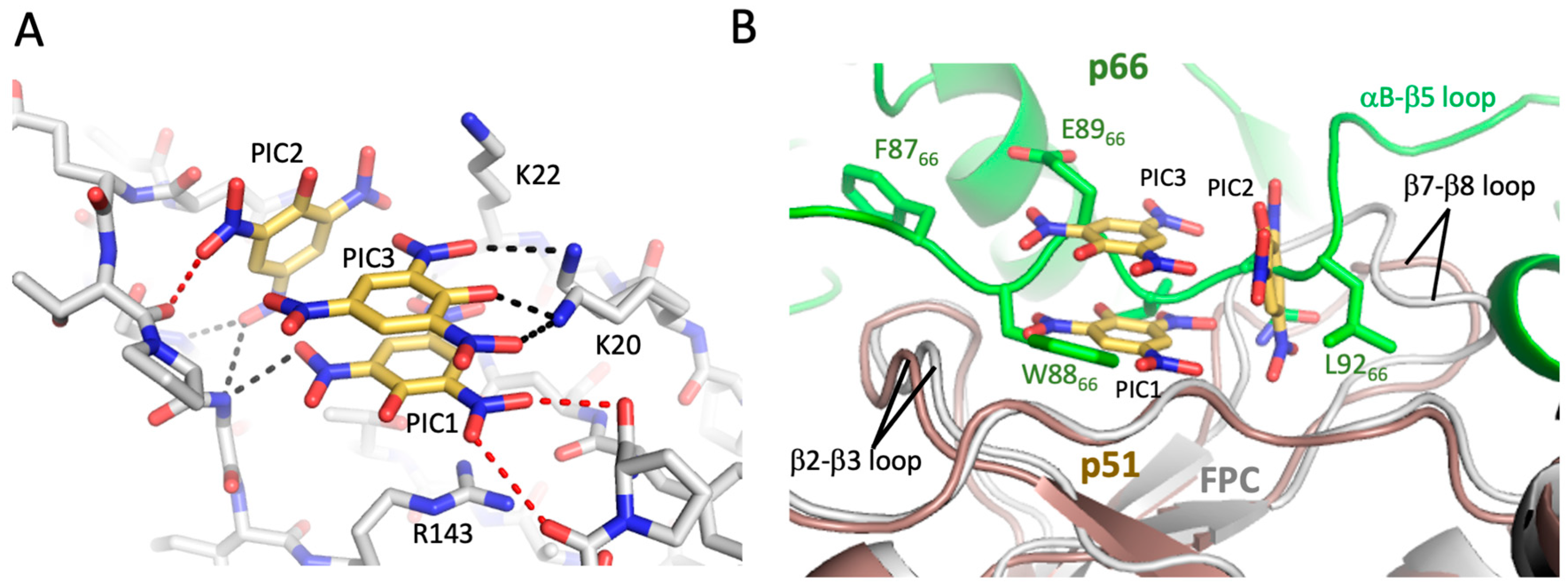
3.5. Crystallographic Characterization of Ligand Binding
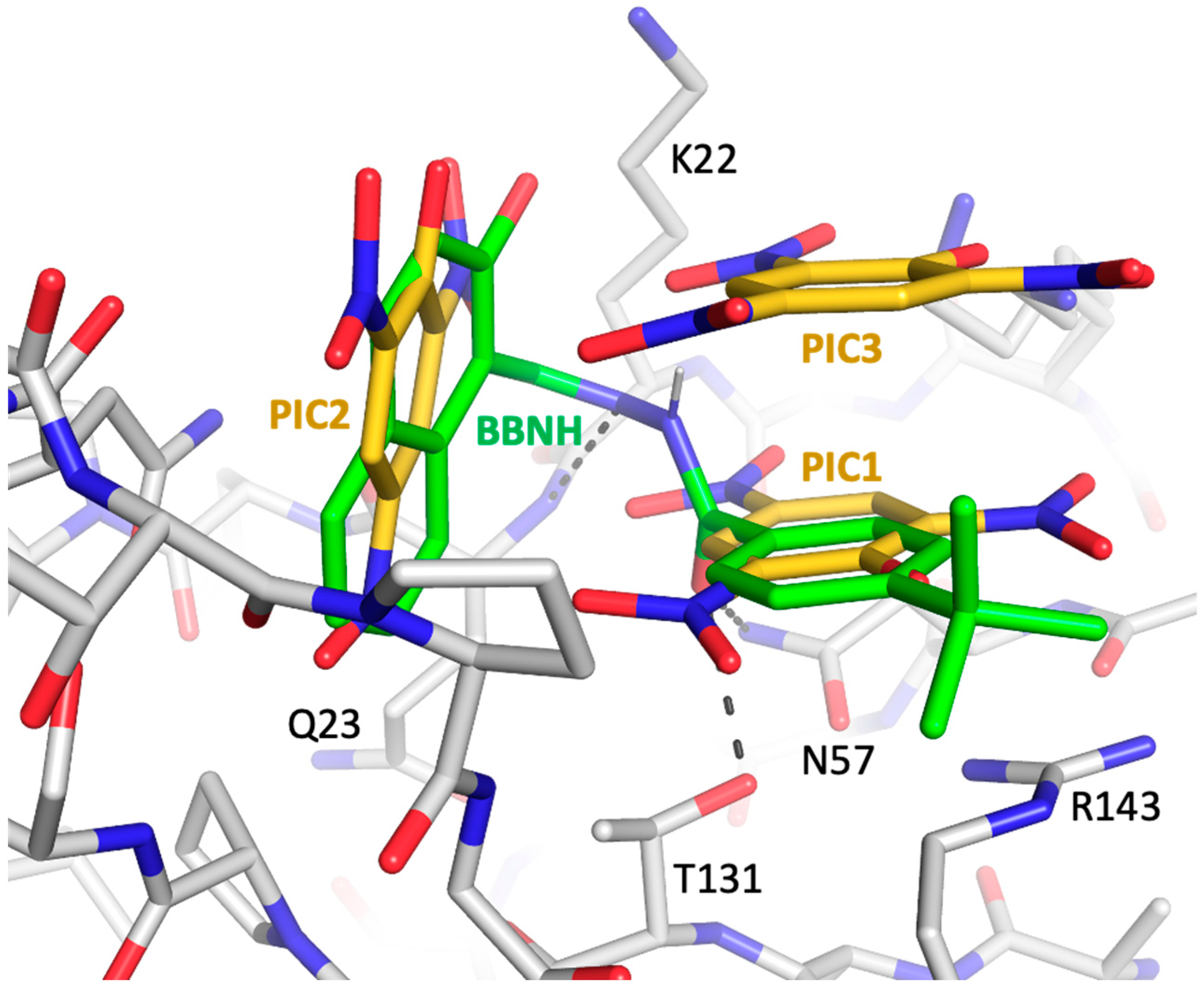
3.6. Comparison with a Previously Identified Dimerization Inhibitor
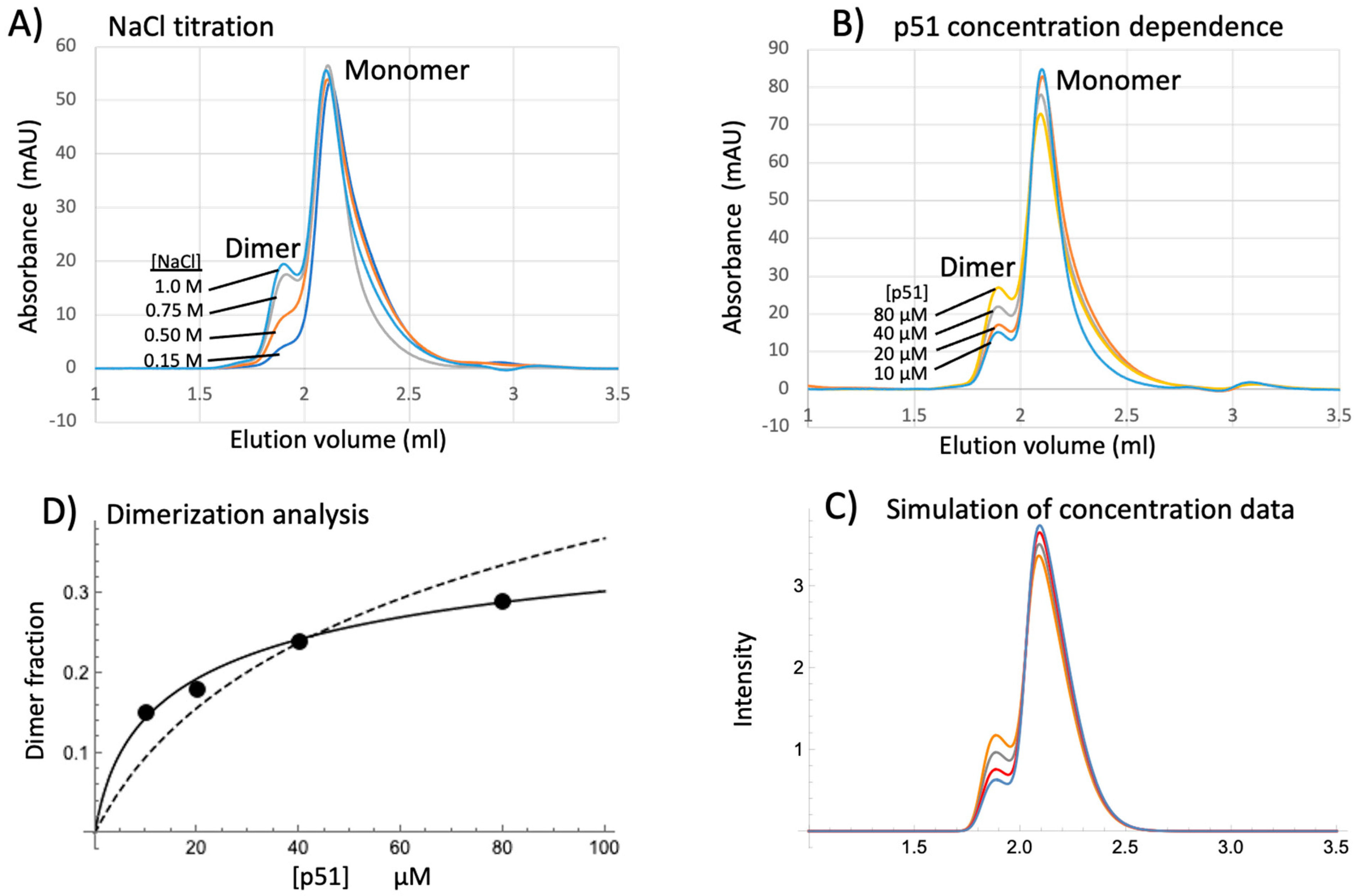
3.7. SEC Analysis of p51 Dimerization
4. Discussion
4.1. Subdomain Interface Binding Sites and Ligand Identification
4.2. Direct Interference with Dimerization
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Agosto, L.M.; Zhong, P.; Munro, J.; Mothes, W. Highly Active Antiretroviral Therapies Are Effective against HIV-1 Cell-to-Cell Transmission. PLoS Pathog. 2014, 10, e1003982. [Google Scholar] [CrossRef] [PubMed]
- Gallant, J.E.; DeJesus, E.; Arribas, J.R.; Pozniak, A.L.; Gazzard, B.; Campo, R.E.; Lu, B.; McColl, D.; Chuck, S.; Enejosa, J.; et al. Tenofovir DF, emtricitabine, and efavirenz vs. zidovudine, lamivudine, and efavirenz for HIV. N. Engl. J. Med. 2006, 354, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, S.; Symons, J.A.; Deval, J. Innovation and trends in the development and approval of antiviral medicines: 1987–2017 and beyond. Antivir. Res. 2018, 155, 76–88. [Google Scholar] [CrossRef]
- Das, K.; Arnold, E. HIV-1 reverse transcriptase and antiviral drug resistance. Part 1. Curr. Opin. Virol. 2013, 3, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Arnold, E. HIV-1 reverse transcriptase and antiviral drug resistance. Part 2. Curr. Opin. Virol. 2013, 3, 119–128. [Google Scholar] [CrossRef]
- Menendez-Arias, L. Targeting HIV: Antiretroviral therapy and development of drug resistance. Trends Pharmacol. Sci. 2002, 23, 381–388. [Google Scholar] [CrossRef]
- de Bethune, M.P. Non-nucleoside reverse transcriptase inhibitors (NNRTIs), their discovery, development, and use in the treatment of HIV-1 infection: A review of the last 20 years (1989–2009). Antivir. Res. 2010, 85, 75–90. [Google Scholar] [CrossRef]
- Rodgers, D.W.; Gamblin, S.J.; Harris, B.A.; Ray, S.; Culp, J.S.; Hellmig, B.; Woolf, D.J.; Debouck, C.; Harrison, S.C. The structure of unliganded reverse transcriptase from the human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA 1995, 92, 1222–1226. [Google Scholar] [CrossRef]
- Hsiou, Y.; Ding, J.; Das, K.; Clark, A.D.; Hughes, S.H.; Arnold, E. Structure of unliganded HIV-1 reverse transcriptase at 2.7 angstrom resolution: Implications of conformational changes for polymerization and inhibition mechanisms. Structure 1996, 4, 853–860. [Google Scholar] [CrossRef]
- Wang, X.; Gao, P.; Menendez-Arias, L.; Liu, X.; Zhan, P. Update on Recent Developments in Small Molecular HIV-1 RNase H Inhibitors (2013–2016): Opportunities and Challenges. Curr. Med. Chem. 2018, 25, 1682–1702. [Google Scholar] [CrossRef]
- Boyer, P.L.; Smith, S.J.; Zhao, X.Z.; Das, K.; Gruber, K.; Arnold, E.; Burke, T.R., Jr.; Hughes, S.H. Developing and Evaluating Inhibitors against the RNase H Active Site of HIV-1 Reverse Transcriptase. J. Virol. 2018, 92, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Pedersen, L.C.; Gabel, S.A.; Mueller, G.A.; DeRose, E.F.; London, R.E. Unfolding the HIV-1 reverse transcriptase RNase H domain—How to lose a molecular tug-of-war. Nucleic Acids Res. 2016, 44, 1776–1788. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Pedersen, L.C.; Gabel, S.A.; Mueller, G.A.; Cuneo, M.J.; DeRose, E.F.; Krahn, J.M.; London, R.E. Selective unfolding of one Ribonuclease H domain of HIV reverse transcriptase is linked to homodimer formation. Nucleic Acids Res. 2014, 42, 5361–5377. [Google Scholar] [CrossRef]
- London, R.E. Structural Maturation of HIV-1 Reverse Transcriptase-A Metamorphic Solution to Genomic Instability. Viruses 2016, 8, 260. [Google Scholar] [CrossRef]
- Sharma, S.K.; Fan, N.S.; Evans, D.B. Human-Immunodeficiency-Virus Type-1 (Hiv-1) Recombinant Reverse-Transcriptase—Asymmetry in P66 Subunits of the P66/P66 Homodimer. FEBS Lett. 1994, 343, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Fan, N.; Rank, K.B.; Leone, J.W.; Heinrikson, R.L.; Bannow, C.A.; Smith, C.W.; Evans, D.B.; Poppe, S.M.; Tarpley, W.G.; Rothrock, D.J.; et al. The differential processing of homodimers of reverse transcriptases from human immunodeficiency viruses type 1 and 2 is a consequence of the distinct specificities of the viral proteases. J. Biol. Chem. 1995, 270, 13573–13579. [Google Scholar] [CrossRef]
- Lewis, W.; Day, B.J.; Copeland, W.C. Mitochondrial toxicity of NRTI antiviral drugs: An integrated cellular perspective. Nat. Rev. Drug Discov. 2003, 2, 812–822. [Google Scholar] [CrossRef]
- Esposito, F.; Corona, A.; Tramontano, E. HIV-1 Reverse Transcriptase Still Remains a New Drug Target: Structure, Function, Classical Inhibitors, and New Inhibitors with Innovative Mechanisms of Actions. Mol. Biol. Int. 2012, 2012, 586401. [Google Scholar] [CrossRef]
- Waheed, A.A.; Tachedjian, G. Why Do We Need New Drug Classes for HIV Treatment and Prevention? Curr. Top. Med. Chem. 2016, 16, 1343–1349. [Google Scholar] [CrossRef]
- Sluis-Cremer, N.; Kempner, E.; Parniak, M.A. Structure-activity relationships in HIV-1 reverse transcriptase revealed by radiation target analysis. Protein Sci. 2003, 12, 2081–2086. [Google Scholar] [CrossRef]
- Tachedjian, G.; Radzio, J.; Sluis-Cremer, N. Relationship between enzyme activity and dimeric structure of recombinant HIV-1 reverse transcriptase. Proteins-Struct. Funct. Bioinform. 2005, 60, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Bauman, J.D.; Harrison, J.J.; Arnold, E. Rapid experimental SAD phasing and hot-spot identification with halogenated fragments. IUCrJ 2016, 3 Pt 1, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Bauman, J.D.; Patel, D.; Dharia, C.; Fromer, M.W.; Ahmed, S.; Frenkel, Y.; Vijayan, R.S.; Eck, J.T.; Ho, W.C.; Das, K.; et al. Detecting allosteric sites of HIV-1 reverse transcriptase by X-ray crystallographic fragment screening. J. Med. Chem. 2013, 56, 2738–2746. [Google Scholar] [CrossRef]
- Himmel, D.M.; Sarafianos, S.G.; Dharmasena, S.; Hossain, M.M.; McCoy-Simandle, K.; Ilina, T.; Clark, A.D., Jr.; Knight, J.L.; Julias, J.G.; Clark, P.K.; et al. HIV-1 reverse transcriptase structure with RNase H inhibitor dihydroxy benzoyl naphthyl hydrazone bound at a novel site. ACS Chem. Biol. 2006, 1, 702–712. [Google Scholar] [CrossRef] [PubMed]
- Thammaporn, R.; Yagi-Utsumi, M.; Yamaguchi, T.; Boonsri, P.; Saparpakorn, P.; Choowongkomon, K.; Techasakul, S.; Kato, K.; Hannongbua, S. NMR characterization of HIV-1 reverse transcriptase binding to various non-nucleoside reverse transcriptase inhibitors with different activities. Sci. Rep. 2015, 5, 15806. [Google Scholar] [CrossRef]
- Zheng, X.; Mueller, G.A.; DeRose, E.F.; London, R.E. Solution characterization of [methyl-(13)C]methionine HIV-1 reverse transcriptase by NMR spectroscopy. Antivir. Res. 2009, 84, 205–214. [Google Scholar] [CrossRef]
- Studier, F.W. Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef]
- Mayer, M.; Meyer, B. Characterization of Ligand Binding by Saturation Transfer Difference NMR Spectroscopy. Angew. Chem. Int. Ed. Engl. 1999, 38, 1784–1788. [Google Scholar] [CrossRef]
- Hwang, T.L.; Shaka, A.J. Water Suppression That Works—Excitation Sculpting Using Arbitrary Wave-Forms and Pulsed-Field Gradients. J. Magn. Reson. Ser. A 1995, 112, 275–279. [Google Scholar] [CrossRef]
- Smallcombe, S.H.; Patt, S.L.; Keifer, P.A. WET solvent suppression and its applications to LC NMR and high-resolution NMR spectroscopy. J. Magn. Reson. Ser. A 1995, 117, 295–303. [Google Scholar] [CrossRef]
- Monga, M.; Sausville, E.A. Developmental therapeutics program at the NCI: Molecular target and drug discovery process. Leukemia 2002, 16, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Smerdon, S.J.; Jager, J.; Kohlstaedt, L.A.; Rice, P.A.; Friedman, J.M.; Steitz, T.A. Structural Basis of Asymmetry in the Human-Immunodeficiency-Virus Type-1 Reverse-Transcriptase Heterodimer. Proc. Natl. Acad. Sci. USA 1994, 91, 7242–7246. [Google Scholar] [CrossRef] [PubMed]
- Minor, W.; Cymborowski, M.; Otwinowski, Z.; Chruszcz, M. HKL-3000: The integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 2006, 62 Pt 8, 859–866. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D-Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D-Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D-Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Zwart, P.H.; Afonine, P.V.; Grosse-Kunstleve, R.W.; Hung, L.W.; Ioerger, T.R.; McCoy, A.J.; McKee, E.; Moriarty, N.W.; Read, R.J.; Sacchettini, J.C.; et al. Automated structure solution with the PHENIX suite. Methods Mol. Biol. 2008, 426, 419–435. [Google Scholar]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D-Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef]
- Zheng, X.; Perera, L.; Mueller, G.A.; DeRose, E.F.; London, R.E. Asymmetric conformational maturation of HIV-1 reverse transcriptase. Elife 2015, 4, e06359. [Google Scholar] [CrossRef]
- Zheng, X.H.; Mueller, G.A.; Cuneo, M.J.; DeRose, E.F.; London, R.E. Homodimerization of the p51 Subunit of HIV-1 Reverse Transcriptase. Biochemistry 2010, 49, 2821–2833. [Google Scholar] [CrossRef]
- Zheng, X.; Mueller, G.A.; Kim, K.; Perera, L.; DeRose, E.F.; London, R.E. Identification of drivers for the metamorphic transition of HIV-1 reverse transcriptase. Biochem. J. 2017, 474, 3321–3338. [Google Scholar] [CrossRef] [PubMed]
- Dufour, E.; El Dirani-Diab, R.; Boulme, F.; Fournier, M.; Nevinsky, G.; Tarrago-Litvak, L.; Litvak, S.; Andreola, M.L. p66/p51 and p51/p51 recombinant forms of reverse transcriptase from human immunodeficiency virus type 1—Interactions with primer tRNA(Lys3), initiation of cDNA synthesis, and effect of inhibitors. Eur. J. Biochem. 1998, 251, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Bavand, M.R.; Wagner, R.; Richmond, T.J. Hiv-1 Reverse-Transcriptase—Polymerization Properties of the P51 Homodimer Compared to the P66/P51 Heterodimer. Biochemistry 1993, 32, 10543–10552. [Google Scholar] [CrossRef] [PubMed]
- London, R.E. HIV-1 Reverse Transcriptase: A Metamorphic Protein with Three Stable States. Structure 2019, 27, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Butterfoss, G.L.; DeRose, E.F.; Gabel, S.A.; Perera, L.; Krahn, J.M.; Mueller, G.A.; Zheng, X.; London, R.E. Conformational dependence of 13C shielding and coupling constants for methionine methyl groups. J. Biomol. NMR 2010, 48, 31–47. [Google Scholar] [CrossRef]
- Liu, Y.; Grimm, M.; Dai, W.T.; Hou, M.C.; Xiao, Z.X.; Cao, Y. CB-Dock: A web server for cavity detection-guided protein-ligand blind docking. Acta Pharmacol. Sin. 2020, 41, 138–144. [Google Scholar] [CrossRef]
- Unge, T.; Knight, S.; Bhikhabhai, R.; Lovgren, S.; Dauter, Z.; Wilson, K.; Strandberg, B. 2.2 Å resolution structure of the amino-terminal half of HIV-1 reverse transcriptase (fingers and palm subdomains). Structure 1994, 2, 953–961. [Google Scholar] [CrossRef]
- Upadhyay, A.; Pandey, N.; Mishra, C.A.; Talele, T.T.; Pandey, V.N. A Single Deletion at Position 134, 135, or 136 in the Beta 7-Beta 8 Loop of the p51 Subunit of HIV-1 RT Disrupts the Formation of Heterodimeric Enzyme. J. Cell. Biochem. 2010, 109, 598–605. [Google Scholar] [CrossRef]
- Sanchez-Murcia, P.A.; de Castro, S.; Garcia-Aparicio, C.; Jimenez, M.A.; Corona, A.; Tramontano, E.; Sluis-Cremer, N.; Menendez-Arias, L.; Velazquez, S.; Gago, F.; et al. Peptides Mimicking the beta7/beta8 Loop of HIV-1 Reverse Transcriptase p51 as “Hotspot-Targeted” Dimerization Inhibitors. ACS Med. Chem. Lett. 2020, 11, 811–817. [Google Scholar] [CrossRef]
- Sluis-Cremer, N.; Arion, D.; Parniak, M.A. Destabilization of the HIV-1 reverse transcriptase dimer upon interaction with N-acyl hydrazone inhibitors. Mol. Pharmacol. 2002, 62, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Valley, C.C.; Cembran, A.; Perlmutter, J.D.; Lewis, A.K.; Labello, N.P.; Gao, J.; Sachs, J.N. The methionine-aromatic motif plays a unique role in stabilizing protein structure. J. Biol. Chem. 2012, 287, 34979–34991. [Google Scholar] [CrossRef] [PubMed]
- Cabodevilla, J.F.; Odriozola, L.; Santiago, E.; Martinez-Irujo, J.J. Factors affecting the dimerization of the p66 form of HIV-1 reverse transcriptase. Eur. J. Biochem. 2001, 268, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Rowley, G.L.; Ma, Q.F.; Bathurst, I.C.; Barr, P.J.; Kenyon, G.L. Stabilization and Activation of Recombinant Human Immunodeficiency Virus-1 Reverse Transcriptase-P66. Biochem. Biophys. Res. Commun. 1990, 167, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Tachedjian, G.; Moore, K.L.; Goff, S.P.; Sluis-Cremer, N. Efavirenz enhances the proteolytic processing of an HIV-1 pol polyprotein precursor and reverse transcriptase homodimer formation. FEBS Lett. 2005, 579, 379–384. [Google Scholar] [CrossRef]
- Tachedjian, G.; Orlova, M.; Sarafianos, S.G.; Arnold, E.; Goff, S.P. Nonnucleoside reverse transcriptase inhibitors are chemical enhancers of dimerization of the HIV type 1 reverse transcriptase. Proc. Natl. Acad. Sci. USA 2001, 98, 7188–7193. [Google Scholar] [CrossRef]
- Divita, G.; Rittinger, K.; Geourjon, C.; Deleage, G.; Goody, R.S. Dimerization Kinetics of Hiv-1 and Hiv-2 Reverse-Transcriptase—A 2-Step Process. J. Mol. Biol. 1995, 245, 508–521. [Google Scholar] [CrossRef]
- Ruiz, F.X.; Arnold, E. Evolving understanding of HIV-1 reverse transcriptase structure, function, inhibition, and resistance. Curr. Opin. Struct. Biol. 2020, 61, 113–123. [Google Scholar] [CrossRef]
- Hostomsky, Z.; Hostomska, Z.; Fu, T.B.; Taylor, J. Reverse transcriptase of human immunodeficiency virus type 1: Functionality of subunits of the heterodimer in DNA synthesis. J. Virol. 1992, 66, 3179–3182. [Google Scholar] [CrossRef]
- Hajduk, P.J.; Huth, J.R.; Fesik, S.W. Druggability indices for protein targets derived from NMR-based screening data. J. Med. Chem. 2005, 48, 2518–2525. [Google Scholar] [CrossRef]
- Fauman, E.B.; Rai, B.K.; Huang, E.S. Structure-based druggability assessment--identifying suitable targets for small molecule therapeutics. Curr. Opin. Chem. Biol. 2011, 15, 463–468. [Google Scholar] [CrossRef]
- Morris, M.C.; Robert-Hebmann, V.; Chaloin, L.; Mery, J.; Heitz, F.; Devaux, C.; Goody, R.S.; Divita, G. A new potent HIV-1 reverse transcriptase inhibitor—A synthetic peptide derived from the interface subunit domains. J. Biol. Chem. 1999, 274, 24941–24946. [Google Scholar] [CrossRef]
- Divita, G.; Restle, T.; Goody, R.S.; Chermann, J.C.; Baillon, J.G. Inhibition of Human-Immunodeficiency-Virus Type-1 Reverse-Transcriptase Dimerization Using Synthetic Peptides Derived from the Connection Domain. J. Biol. Chem. 1994, 269, 13080–13083. [Google Scholar] [CrossRef]
- Restle, T.; Pawlita, M.; Sczakiel, G.; Muller, B.; Goody, R.S. Structure-function relationships of HIV-1 reverse transcriptase determined using monoclonal antibodies. J. Biol. Chem. 1992, 267, 14654–14661. [Google Scholar] [CrossRef] [PubMed]
- Camarasa, M.J.; Velazquez, S.; San-Felix, A.; Perez-Perez, M.J.; Bonache, M.C.; De Castro, S. TSAO derivatives, inhibitors of HIV-1 reverse transcriptase dimerization: Recent progress. Curr. Pharm. Des. 2006, 12, 1895–1907. [Google Scholar] [CrossRef] [PubMed]
- Camarasa, M.J.; Velazquez, S.; San-Felix, A.; Perez-Perez, M.J.; Gago, F. Dimerization inhibitors of HIV-1 reverse transcriptase, protease and integrase: A single mode of inhibition for the three HIV enzymes? Antivir. Res. 2006, 71, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Sluis-Cremer, N.; Tachedjian, G. Modulation of the oligomeric structures of HIV-1 retroviral enzymes by synthetic peptides and small molecules. Eur. J. Biochem. 2002, 269, 5103–5111. [Google Scholar] [CrossRef]
- Sluis-Cremer, N.; Arion, D.; Abram, M.E.; Parniak, M.A. Proteolytic processing of an HIV-1 pol polyprotein precursor: Insights into the mechanism of reverse transcriptase p66/p51 heterodimer formation. Int. J. Biochem. Cell Biol. 2004, 36, 1836–1847. [Google Scholar] [CrossRef]
- Venezia, C.F.; Meany, B.J.; Braz, V.A.; Barkley, M.D. Kinetics of Association and Dissociation of HIV-1 Reverse Transcriptase Subunits. Biochemistry 2009, 48, 9084–9093. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kirby, T.W.; Gabel, S.A.; DeRose, E.F.; Perera, L.; Krahn, J.M.; Pedersen, L.C.; London, R.E. Targeting the Structural Maturation Pathway of HIV-1 Reverse Transcriptase. Biomolecules 2023, 13, 1603. https://doi.org/10.3390/biom13111603
Kirby TW, Gabel SA, DeRose EF, Perera L, Krahn JM, Pedersen LC, London RE. Targeting the Structural Maturation Pathway of HIV-1 Reverse Transcriptase. Biomolecules. 2023; 13(11):1603. https://doi.org/10.3390/biom13111603
Chicago/Turabian StyleKirby, Thomas W., Scott A. Gabel, Eugene F. DeRose, Lalith Perera, Juno M. Krahn, Lars C. Pedersen, and Robert E. London. 2023. "Targeting the Structural Maturation Pathway of HIV-1 Reverse Transcriptase" Biomolecules 13, no. 11: 1603. https://doi.org/10.3390/biom13111603
APA StyleKirby, T. W., Gabel, S. A., DeRose, E. F., Perera, L., Krahn, J. M., Pedersen, L. C., & London, R. E. (2023). Targeting the Structural Maturation Pathway of HIV-1 Reverse Transcriptase. Biomolecules, 13(11), 1603. https://doi.org/10.3390/biom13111603