

Glutamate Receptor Dysregulation and Platelet Glutamate Dynamics in Alzheimer’s and Parkinson’s Diseases: Insights into Current Medications
 ,
,  , and
, and
Abstract
:1. Introduction
2. Alzheimer’s Disease
3. Parkinson’s Disease
4. Glutamate Receptors: Key Players in Neurotransmission, Learning and Brain Health
4.1. Ionotropic Receptors
4.1.1. N-Methyl-D-aspartate Receptor (NMDAR)
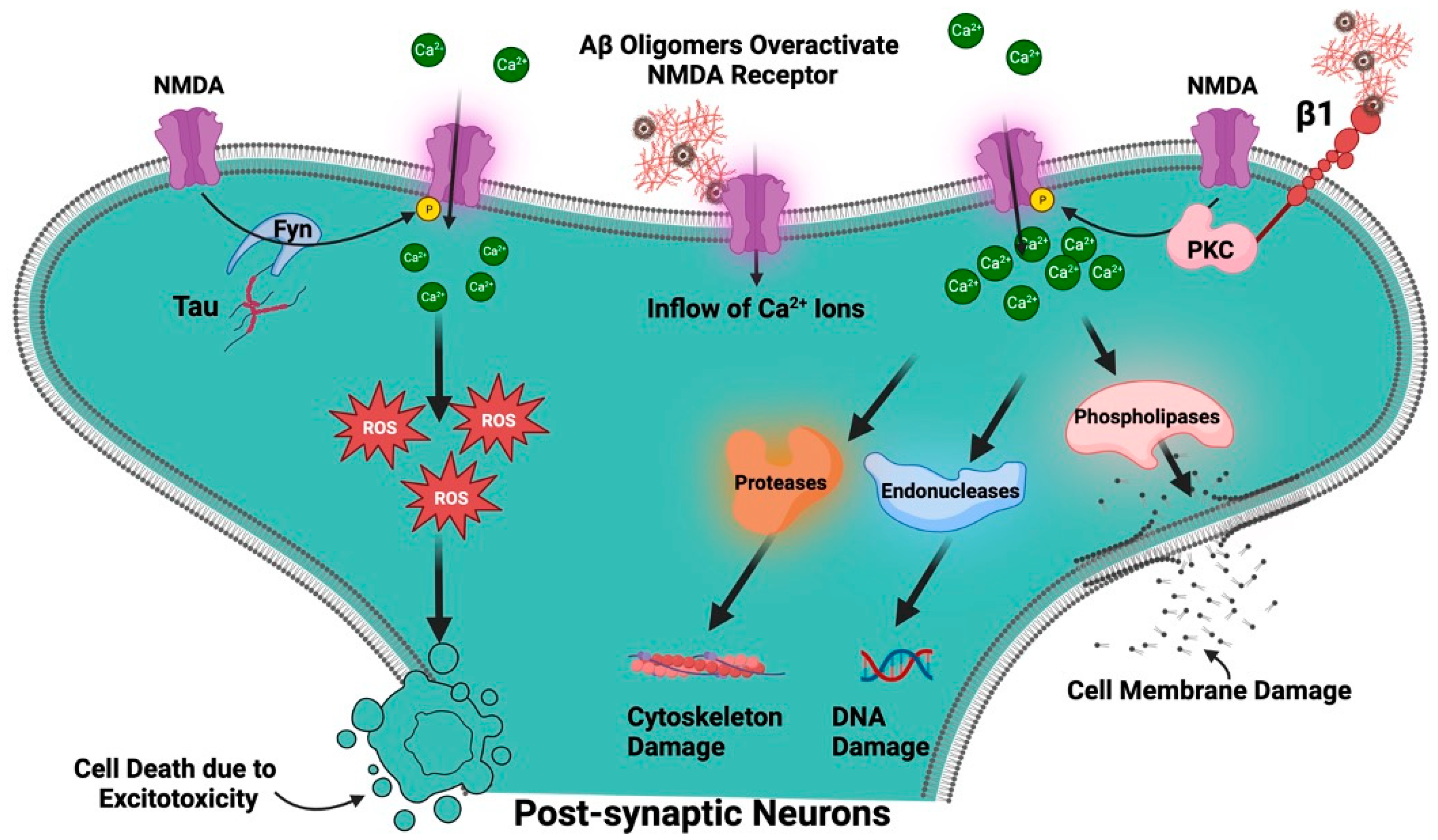
Role of NMDA in Alzheimer’s Disease
Role of NMDAR in Parkinson’s Disease
4.1.2. α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic Acid Receptor (AMPAR)
Role of AMPAR in Alzheimer’s Disease
Role of AMPAR in Parkinson’s Disease
4.1.3. Kainate Receptor (KAR)
Role of Kainate Receptor in Alzheimer’s Disease
Role of Kainate Receptor in Parkinson’s Disease
4.2. Metabotropic Glutamate Receptors
4.2.1. Role of Metabotropic Receptor in Alzheimer’s Disease
4.2.2. Role of Metabotropic Glutamate Receptor in Parkinson’s Disease
5. Platelets as a Cellular Model for Understanding Neuronal Mechanism and Neurological Disorders
The Role of Platelet’s Glutamate and Its Transporter in Alzheimer’s and Parkinson’s Disease
6. Current Pharmacological Treatments for Alzheimer’s and Parkinson’s Disease
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gatto, C.L.; Broadie, K. Genetic controls balancing excitatory and inhibitory synaptogenesis in neurodevelopmental disorder models. Front. Synaptic Neurosci. 2010, 2, 1553. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Duszkiewicz, A.J.; Morris, R.G.M. The synaptic plasticity and memory hypothesis: Encoding, storage and persistence. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130288. [Google Scholar] [CrossRef] [PubMed]
- Reiner, A.; Levitz, J. Glutamatergic Signaling in the Central Nervous System: Ionotropic and Metabotropic Receptors in Concert. Neuron 2018, 98, 1080–1098. [Google Scholar] [CrossRef] [PubMed]
- Beitz, J.M. Parkinson s disease: A review. Front. Biosci. 2014, S6, 65–74. [Google Scholar] [CrossRef]
- Blandini, F.; Porter, R.H.P.; Greenamyre, J.T. Glutamate and Parkinson’s Disease. Mol. Neurobiol. 1996, 12, 73–94. [Google Scholar] [CrossRef]
- Lamptey, R.N.L.; Chaulagain, B.; Trivedi, R.; Gothwal, A.; Layek, B.; Singh, J. A Review of the Common Neurodegenerative Disorders: Current Therapeutic Approaches and the Potential Role of Nanotherapeutics. Int. J. Mol. Sci. 2022, 23, 1851. [Google Scholar] [CrossRef]
- Marttinen, M.; Kurkinen, K.M.; Soininen, H.; Haapasalo, A.; Hiltunen, M. Synaptic dysfunction and septin protein family members in neurodegenerative diseases. Mol. Neurodegener. 2015, 10, 16. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef]
- Mark, L.P.; Prost, R.W.; Ulmer, J.L.; Smith, M.M.; Daniels, D.L.; Strottmann, J.M.; Brown, W.D.; Hacein-Bey, L. Pictorial review of glutamate excitotoxicity: Fundamental concepts for neuroimaging. Am. J. Neuroradiol. 2001, 22, 1813–1824. [Google Scholar]
- Dong, X.-X.; Wang, Y.; Qin, Z.-H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef]
- Bukke, V.N.; Archana, M.; Villani, R.; Romano, A.D.; Wawrzyniak, A.; Balawender, K.; Orkisz, S.; Beggiato, S.; Serviddio, G.; Cassano, T. The Dual Role of Glutamatergic Neurotransmission in Alzheimer’s Disease: From Pathophysiology to Pharmacotherapy. Int. J. Mol. Sci. 2020, 21, 7452. [Google Scholar] [CrossRef] [PubMed]
- Ramalho, J.; Castillo, M. Dementia resulting from traumatic brain injury. Dement. Neuropsychol. 2015, 9, 356–368. [Google Scholar] [CrossRef]
- Ramos-Cejudo, J.; Wisniewski, T.; Marmar, C.; Zetterberg, H.; Blennow, K.; de Leon, M.J.; Fossati, S. Traumatic Brain Injury and Alzheimer’s Disease: The Cerebrovascular Link. EBioMedicine 2018, 28, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Tarawneh, R.; Holtzman, D.M. The Clinical Problem of Symptomatic Alzheimer Disease and Mild Cognitive Impairment. Cold Spring Harb. Perspect. Med. 2012, 2, a006148. [Google Scholar] [CrossRef] [PubMed]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef] [PubMed]
- De Ture, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological Alterations in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef]
- Moloney, C.M.; Lowe, V.J.; Murray, M.E. Visualization of neurofibrillary tangle maturity in Alzheimer’s disease: A clinicopathologic perspective for biomarker research. Alzheimer’s Dement. 2021, 17, 1554–1574. [Google Scholar] [CrossRef]
- Le, R.; Cruz, L.; Urbanc, B.; Knowles, R.B.; Hsiao-Ashe, K.; Duff, K.; Irizarry, M.C.; Stanley, H.E.; Hyman, B.T. Plaque-Induced Abnormalities in Neurite Geometry in Transgenic Models of Alzheimer Disease: Implications for Neural System Disruption. J. Neuropathol. Exp. Neurol. 2001, 60, 753–758. [Google Scholar] [CrossRef]
- Murphy, M.P.; LeVine, H., 3rd. Alzheimer’s Disease and the Amyloid-β Peptide. J. Alzheimer’s Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef]
- O’Brien, R.J.; Wong, P.C. Amyloid Precursor Protein Processing and Alzheimer’s Disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [Google Scholar] [CrossRef] [PubMed]
- Chow, V.W.; Mattson, M.P.; Wong, P.C.; Gleichmann, M. An Overview of APP Processing Enzymes and Products. NeuroMolecular Med. 2010, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ow, S.-Y.; Dunstan, D.E. A brief overview of amyloids and Alzheimer’s disease. Protein Sci. 2014, 23, 1315–1331. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, R.; Baglietto-Vargas, D.; LaFerla, F.M. The Role of Tau in Alzheimer’s Disease and Related Disorders. CNS Neurosci. Ther. 2011, 17, 514–524. [Google Scholar] [CrossRef]
- Lanoiselée, H.-M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.-C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017, 14, e1002270. [Google Scholar] [CrossRef]
- Bekris, L.M.; Yu, C.-E.; Bird, T.D.; Tsuang, D.W. Review Article: Genetics of Alzheimer Disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef]
- Wolfe, C.M.; Fitz, N.F.; Nam, K.N.; Lefterov, I.; Koldamova, R. The Role of APOE and TREM2 in Alzheimer′s Disease—Current Understanding and Perspectives. Int. J. Mol. Sci. 2018, 20, 81. [Google Scholar] [CrossRef]
- Jendresen, C.; Årskog, V.; Daws, M.R.; Nilsson, L.N.G. The Alzheimer’s disease risk factors apolipoprotein E and TREM2 are linked in a receptor signaling pathway. J. Neuroinflamm. 2017, 14, 59. [Google Scholar] [CrossRef]
- Wiseman, F.K.; Pulford, L.J.; Barkus, C.; Liao, F.; Portelius, E.; Webb, R.; Chávez-Gutiérrez, L.; Cleverley, K.; Noy, S.; Sheppard, O.; et al. Trisomy of human chromosome 21 enhances amyloid-β deposition independently of an extra copy of APP. Brain 2018, 141, 2457–2474. [Google Scholar] [CrossRef]
- Han, W.; Li, C. Linking type 2 diabetes and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 6557–6558. [Google Scholar] [CrossRef]
- Carlsson, C.M. Type 2 Diabetes Mellitus, Dyslipidemia, and Alzheimer’s Disease. J. Alzheimer’s Dis. 2010, 20, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Sartori, A.C.; Vance, D.E.; Slater, L.Z.; Crowe, M. The Impact of Inflammation on Cognitive Function in Older Adults. J. Neurosci. Nurs. 2012, 44, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Bakulski, K.M.; Seo, Y.A.; Hickman, R.C.; Brandt, D.; Vadari, H.S.; Hu, H.; Park, S.K. Heavy Metals Exposure and Alzheimer’s Disease and Related Dementias. J. Alzheimer’s Dis. 2020, 76, 1215–1242. [Google Scholar] [CrossRef] [PubMed]
- Emamzadeh, F.N.; Surguchov, A. Parkinson’s Disease: Biomarkers, Treatment, and Risk Factors. Front. Neurosci. 2018, 12, 612. [Google Scholar] [CrossRef] [PubMed]
- Magrinelli, F.; Picelli, A.; Tocco, P.; Federico, A.; Roncari, L.; Smania, N.; Zanette, G.; Tamburin, S. Pathophysiology of Motor Dysfunction in Parkinson’s Disease as the Rationale for Drug Treatment and Rehabilitation. Park. Dis. 2016, 2016, 9832839. [Google Scholar] [CrossRef] [PubMed]
- Outeiro, T.F.; Koss, D.J.; Erskine, D.; Walker, L.; Kurzawa-Akanbi, M.; Burn, D.; Donaghy, P.; Morris, C.; Taylor, J.-P.; Thomas, A.; et al. Dementia with Lewy bodies: An update and outlook. Mol. Neurodegener. 2019, 14, 5. [Google Scholar] [CrossRef]
- Stefanis, L. α-Synuclein in Parkinson’s Disease. Cold Spring Harb. Perspect. Med. 2011, 2, a009399. [Google Scholar] [CrossRef]
- Ball, N.; Teo, W.-P.; Chandra, S.; Chapman, J. Parkinson’s Disease and the Environment. Front. Neurol. 2019, 10, 218. [Google Scholar] [CrossRef]
- Cerri, S.; Mus, L.; Blandini, F. Parkinson’s Disease in Women and Men: What’s the Difference? J. Park. Dis. 2019, 9, 501–515. [Google Scholar] [CrossRef]
- Haaxma, C.A.; Bloem, B.R.; Borm, G.F.; Oyen, W.J.G.; Leenders, K.L.; Eshuis, S.; Booij, J.; Dluzen, D.E.; Horstink, M.W.I.M. Gender differences in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2007, 78, 819–824. [Google Scholar] [CrossRef]
- Cui, H.; Kong, Y.; Zhang, H. Oxidative Stress, Mitochondrial Dysfunction, and Aging. J. Signal Transduct. 2012, 2012, 646354. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Danbolt, N.C. Glutamate as a neurotransmitter in the healthy brain. J. Neural Transm. 2014, 121, 799–817. [Google Scholar] [CrossRef]
- Gautam, D.; Tiwari, A.; Chaurasia, R.N.; Dash, D. Glutamate induces synthesis of thrombogenic peptides and extracellular vesicle release from human platelets. Sci. Rep. 2019, 9, 8346. [Google Scholar] [CrossRef] [PubMed]
- Citri, A.; Malenka, R.C. Synaptic plasticity: Multiple forms, functions, and mechanisms. Neuropsychopharmacology 2008, 33, 18–41. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef]
- Twomey, E.C.; Sobolevsky, A.I. Structural Mechanisms of Gating in Ionotropic Glutamate Receptors. Biochemistry 2018, 57, 267–276. [Google Scholar] [CrossRef]
- Collingridge, G.L.; Kehl, S.J.; McLennan, H. Excitatory amino acids in synaptic transmission in the Schaffer collateral-commissural pathway of the rat hippocampus. J. Physiol. 1983, 334, 33–46. [Google Scholar] [CrossRef]
- Newcomer, J.W.; Farber, N.B.; Olney, J.W. NMDA receptor function, memory, and brain aging. Dialogues Clin. Neurosci. 2000, 2, 219–232. [Google Scholar] [CrossRef]
- Lüscher, C.; Malenka, R.C. NMDA Receptor-Dependent Long-Term Potentiation and Long-Term Depression (LTP/LTD). Cold Spring Harb. Perspect. Biol. 2012, 4, a005710. [Google Scholar] [CrossRef]
- Lee, C.-H.; Lü, W.; Michel, J.C.; Goehring, A.; Du, J.; Song, X.; Gouaux, E. NMDA receptor structures reveal subunit arrangement and pore architecture. Nature 2014, 511, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, H.; Gouaux, E. Mechanisms of activation, inhibition and specificity: Crystal structures of the NMDA receptor NR1 ligand-binding core. EMBO J. 2003, 22, 2873–2885. [Google Scholar] [CrossRef] [PubMed]
- Brothwell, S.L.C.; Barber, J.L.; Monaghan, D.T.; Jane, D.E.; Gibb, A.J.; Jones, S. NR2B- and NR2D-containing synaptic NMDA receptors in developing rat substantia nigra pars compacta dopaminergic neurones. J. Physiol. 2008, 586, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.; Carter, S.R.; Rao, V.R.; Wang, J.; Finkbeiner, S. Splice Variants of the NR1 Subunit Differentially Induce NMDA Receptor-Dependent Gene Expression. J. Neurosci. 2006, 26, 1065–1076. [Google Scholar] [CrossRef]
- Yu, A.; Lau, A.Y. Glutamate and Glycine Binding to the NMDA Receptor. Structure 2018, 26, 1035–1043.e2. [Google Scholar] [CrossRef]
- Henson, M.A.; Roberts, A.C.; Pérez-Otaño, I.; Philpot, B.D. Influence of the NR3A subunit on NMDA receptor functions. Prog. Neurobiol. 2010, 91, 23–37. [Google Scholar] [CrossRef]
- Ortiz-Sanz, C.; Balantzategi, U.; Quintela-López, T.; Ruiz, A.; Luchena, C.; Zuazo-Ibarra, J.; Capetillo-Zarate, E.; Matute, C.; Zugaza, J.L.; Alberdi, E. Amyloid β/PKC-dependent alterations in NMDA receptor composition are detected in early stages of Alzheimer’s disease. Cell Death Dis. 2022, 13, 253. [Google Scholar] [CrossRef]
- Hansen, K.B.; Yi, F.; Perszyk, R.E.; Menniti, F.S.; Traynelis, S.F. NMDA Receptors in the Central Nervous System. In NMDA Receptors. Methods in Molecular Biology; Humana Press: New York, NY, USA, 2017; pp. 1–80. [Google Scholar] [CrossRef]
- Danysz, W.; Parsons, C.G. Alzheimer’s disease, β-amyloid, glutamate, NMDA receptors and memantine-Searching for the connections. Br. J. Pharmacol. 2012, 167, 324–352. [Google Scholar] [CrossRef]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef]
- Liu, J.; Chang, L.; Song, Y.; Li, H.; Wu, Y. The Role of NMDA Receptors in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 43. [Google Scholar] [CrossRef]
- Li, S.; Jin, M.; Koeglsperger, T.; Shepardson, N.E.; Shankar, G.M.; Selkoe, D.J. Soluble Aβ Oligomers Inhibit Long-Term Potentiation through a Mechanism Involving Excessive Activation of Extrasynaptic NR2B-Containing NMDA Receptors. J. Neurosci. 2011, 31, 6627–6638. [Google Scholar] [CrossRef]
- Kass, G.E.N.; Orrenius, S. Calcium Signaling and Cytotoxicity. Environ. Health Perspect. 1999, 107, 25. [Google Scholar] [CrossRef] [PubMed]
- Marambaud, P.; Dreses-Werringloer, U.; Vingtdeux, V. Calcium signaling in neurodegeneration. Mol. Neurodegener. 2009, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Yeung, J.H.Y.; Walby, J.L.; Palpagama, T.H.; Turner, C.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. Glutamatergic receptor expression changes in the Alzheimer’s disease hippocampus and entorhinal cortex. Brain Pathol. 2021, 31, e13005. [Google Scholar] [CrossRef]
- Yu, S.P.; Jiang, M.Q.; Shim, S.S.; Pourkhodadad, S.; Wei, L. Extrasynaptic NMDA receptors in acute and chronic excitotoxicity: Implications for preventive treatments of ischemic stroke and late-onset Alzheimer’s disease. Mol. Neurodegener. 2023, 18, 43. [Google Scholar] [CrossRef] [PubMed]
- Yeung, J.H.Y.; Palpagama, T.H.; Tate, W.P.; Peppercorn, K.; Waldvogel, H.J.; Faull, R.L.M.; Kwakowsky, A. The Acute Effects of Amyloid-Beta1–42 on Glutamatergic Receptor and Transporter Expression in the Mouse Hippocampus. Front. Neurosci. 2020, 13, 1427. [Google Scholar] [CrossRef] [PubMed]
- Bloom, G.S. Amyloid-β and Tau: The Trigger and Bullet in Alzheimer Disease Pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef]
- Zhang, H.; Wei, W.; Zhao, M.; Ma, L.; Jiang, X.; Pei, H.; Cao, Y.; Li, H. Interaction between Aβ and Tau in the Pathogenesis of Alzheimer’s Disease. Int. J. Biol. Sci. 2021, 17, 2181–2192. [Google Scholar] [CrossRef]
- Trepanier, C.H.; Jackson, M.F.; MacDonald, J.F. Regulation of NMDA receptors by the tyrosine kinase Fyn. FEBS J. 2012, 279, 12–19. [Google Scholar] [CrossRef]
- Matrone, C.; Petrillo, F.; Nasso, R.; Ferretti, G. Fyn Tyrosine Kinase as Harmonizing Factor in Neuronal Functions and Dysfunctions. Int. J. Mol. Sci. 2020, 21, 4444. [Google Scholar] [CrossRef]
- Pallas-Bazarra, N.; Draffin, J.; Cuadros, R.; Esteban, J.A.; Avila, J. Tau is required for the function of extrasynaptic NMDA receptors. Sci. Rep. 2019, 9, 9116. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, F.; Mai, D.; Qu, S. Molecular Mechanisms of Glutamate Toxicity in Parkinson’s Disease. Front. Neurosci. 2020, 14, 585584. [Google Scholar] [CrossRef] [PubMed]
- Iovino, L.; Tremblay, M.; Civiero, L. Glutamate-induced excitotoxicity in Parkinson’s disease: The role of glial cells. J. Pharmacol. Sci. 2020, 144, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Mabrouk, O.S.; Mela, F.; Calcagno, M.; Budri, M.; Viaro, R.; Dekundy, A.; Parsons, C.G.; Auberson, Y.P.; Morari, M. GluN2A and GluN2B NMDA Receptor Subunits Differentially Modulate Striatal Output Pathways and Contribute to Levodopa-Induced Abnormal Involuntary Movements in Dyskinetic Rats. ACS Chem. Neurosci. 2013, 4, 808–816. [Google Scholar] [CrossRef]
- Mellone, M.; Stanic, J.; Hernandez, L.F.; Iglesias, E.; Zianni, E.; Longhi, A.; Prigent, A.; Picconi, B.; Calabresi, P.; Hirsch, E.C.; et al. NMDA receptor GluN2A/GluN2B subunit ratio as synaptic trait of levodopa-induced dyskinesias: From experimental models to patients. Front. Cell. Neurosci. 2015, 9, 245. [Google Scholar] [CrossRef]
- Ahmed, I.; Bose, S.K.; Pavese, N.; Ramlackhansingh, A.; Turkheimer, F.; Hotton, G.; Hammers, A.; Brooks, D.J. Glutamate NMDA receptor dysregulation in Parkinson’s disease with dyskinesias. Brain 2011, 134, 979–986. [Google Scholar] [CrossRef]
- Greenamyre, J.T.; O’brien, C.F. N-Methyl-D-Aspartate Antagonists in the Treatment of Parkinson’s Disease. Arch. Neurol. 1991, 48, 977–981. [Google Scholar] [CrossRef]
- Loopuijt, L.D.; Schmidt, W.J. The role of NMDA receptors in the slow neuronal degeneration of Parkinson’s disease. Amino Acids 1998, 14, 17–23. [Google Scholar] [CrossRef]
- Gan, J.; Qi, C.; Mao, L.-M.; Liu, Z. Changes in surface expression of N-methyl-D-aspartate receptors in the striatum in a rat model of Parkinson’s disease. Drug Des. Dev. Ther. 2014, 8, 165–173. [Google Scholar] [CrossRef]
- Gibson, L.L.; Pollak, T.A.; Hart, M.; Heslegrave, A.; Hye, A.; Church, A.J.; Lakdawala, N.; Nicholson, T.R.; Batzu, L.; Rota, S.; et al. NMDA Receptor Antibodies and Neuropsychiatric Symptoms in Parkinson’s Disease. J. Neuropsychiatry Clin. Neurosci. 2023, 35, 236–243. [Google Scholar] [CrossRef]
- Henley, J.M.; Wilkinson, K.A. Synaptic AMPA receptor composition in development, plasticity and disease. Nat. Rev. Neurosci. 2016, 17, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Chater, T.E.; Goda, Y. The role of AMPA receptors in postsynaptic mechanisms of synaptic plasticity. Front. Cell. Neurosci. 2014, 8, 401. [Google Scholar] [CrossRef] [PubMed]
- Gan, Q.; Salussolia, C.L.; Wollmuth, L.P. Assembly of AMPA receptors: Mechanisms and regulation. J. Physiol. 2015, 593, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Greger, I.H.; Watson, J.F.; Cull-Candy, S.G. Structural and Functional Architecture of AMPA-Type Glutamate Receptors and Their Auxiliary Proteins. Neuron 2017, 94, 713–730. [Google Scholar] [CrossRef]
- Graves, A.R.; Roth, R.H.; Tan, H.L.; Zhu, Q.; Bygrave, A.M.; Lopez-Ortega, E.; Hong, I.; Spiegel, A.C.; Johnson, R.C.; Vogelstein, J.T.; et al. Visualizing synaptic plasticity in vivo by large-scale imaging of endogenous AMPA receptors. eLife 2021, 10, 66809. [Google Scholar] [CrossRef]
- Zhang, H.; Bramham, C.R. Bidirectional Dysregulation of AMPA Receptor-Mediated Synaptic Transmission and Plasticity in Brain Disorders. Front. Synaptic Neurosci. 2020, 12, 26. [Google Scholar] [CrossRef]
- Guntupalli, S.; Widagdo, J.; Anggono, V. Amyloid-β-Induced Dysregulation of AMPA Receptor Trafficking. Neural Plast. 2016, 2016, 3204519. [Google Scholar] [CrossRef]
- Hettinger, J.C.; Lee, H.; Bu, G.; Holtzman, D.M.; Cirrito, J.R. AMPA-ergic regulation of amyloid-β levels in an Alzheimer’s disease mouse model. Mol. Neurodegener. 2018, 13, 22. [Google Scholar] [CrossRef]
- Chang, E.H.; Savage, M.J.; Flood, D.G.; Thomas, J.M.; Levy, R.B.; Mahadomrongkul, V.; Shirao, T.; Aoki, C.; Huerta, P.T. AMPA receptor downscaling at the onset of Alzheimer’s disease pathology in double knockin mice. Proc. Natl. Acad. Sci. USA 2006, 103, 3410–3415. [Google Scholar] [CrossRef]
- Alfaro-Ruiz, R.; Aguado, C.; Martín-Belmonte, A.; Moreno-Martínez, A.E.; Merchán-Rubira, J.; Hernández, F.; Ávila, J.; Fukazawa, Y.; Luján, R. Alteration in the Synaptic and Extrasynaptic Organization of AMPA Receptors in the Hippocampus of P301S Tau Transgenic Mice. Int. J. Mol. Sci. 2022, 23, 13527. [Google Scholar] [CrossRef]
- Martín-Belmonte, A.; Aguado, C.; Alfaro-Ruíz, R.; Itakura, M.; Moreno-Martínez, A.E.; de la Ossa, L.; Molnár, E.; Fukazawa, Y.; Luján, R. Age-Dependent Shift of AMPA Receptors From Synapses to Intracellular Compartments in Alzheimer’s Disease: Immunocytochemical Analysis of the CA1 Hippocampal Region in APP/PS1 Transgenic Mouse Model. Front. Aging Neurosci. 2020, 12, 577996. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.; Vissel, B. The essential role of AMPA receptor GluA2 subunit RNA editing in the normal and diseased brain. Front. Mol. Neurosci. 2012, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Wright, A. The role of AMPA Receptor GluA2 Subunit Q/R Site RNA Editing in the Normal and Alzheimer’s Diseased Brain. Ph.D. Thesis, UNSW Sydney, Sydney, Australia, 2014. [Google Scholar] [CrossRef]
- Ouattara, B.; Hoyer, D.; Grégoire, L.; Morissette, M.; Gasparini, F.; Gomez-Mancilla, B.; Di Paolo, T. Changes of AMPA receptors in MPTP monkeys with levodopa-induced dyskinesias. Neuroscience 2010, 167, 1160–1167. [Google Scholar] [CrossRef]
- Kwon, D.K.; Kwatra, M.; Wang, J.; Ko, H.S. Levodopa-Induced Dyskinesia in Parkinson’s Disease: Pathogenesis and Emerging Treatment Strategies. Cells 2022, 11, 3736. [Google Scholar] [CrossRef] [PubMed]
- Calon, F.; Rajput, A.H.; Hornykiewicz, O.; Bédard, P.J.; Di Paolo, T. Levodopa-induced motor complications are associated with alterations of glutamate receptors in Parkinson’s disease. Neurobiol. Dis. 2003, 14, 404–416. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, X.; Bernardi, R.E.; Ju, J.; Wei, S.; Gong, Z. Activation of AMPA Receptors in the Lateral Habenula Produces Anxiolytic Effects in a Rat Model of Parkinson’s Disease. Front. Pharmacol. 2022, 13, 821975. [Google Scholar] [CrossRef]
- Nakajima, S.; Saeki, N.; Tamano, H.; Nishio, R.; Katahira, M.; Takeuchi, A.; Takeda, A. Age-related vulnerability to nigral dopaminergic degeneration in rats via Zn2+-permeable GluR2-lacking AMPA receptor activation. NeuroToxicology 2021, 83, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Tamura, H.; Nishio, R.; Saeki, N.; Katahira, M.; Morioka, H.; Tamano, H.; Takeda, A. Paraquat-induced intracellular Zn2+ dysregulation causes dopaminergic degeneration in the substantia nigra, but not in the striatum. NeuroToxicology 2022, 90, 136–144. [Google Scholar] [CrossRef]
- Chang, Y.; Du, C.; Han, L.; Lv, S.; Zhang, J.; Bian, G.; Tang, G.; Liu, Y.; Chen, T.; Liu, J. Enhanced AMPA receptor-mediated excitatory transmission in the rodent rostromedial tegmental nucleus following lesion of the nigrostriatal pathway. Neurochem. Int. 2019, 122, 85–93. [Google Scholar] [CrossRef]
- Nair, J.D.; Wilkinson, K.A.; Henley, J.M.; Mellor, J.R. Kainate receptors and synaptic plasticity. Neuropharmacology 2021, 196, 108540. [Google Scholar] [CrossRef]
- Lerma, J. Roles and rules of kainate receptors in synaptic transmission. Nat. Rev. Neurosci. 2003, 4, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.; Fisher, J. Contributions of different kainate receptor subunits to the properties of recombinant homomeric and heteromeric receptors. Neuroscience 2014, 278, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Bettler, B.; Mullet, C. Review: Neurotransmitter Receptors II AMPA and Kainate Receptors. Neuropharmacology 1995, 34, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Khanra, N.; Brown, P.M.G.E.; Perozzo, A.M.; Bowie, D.; Meyerson, J.R. Architecture and structural dynamics of the heteromeric GluK2/K5 kainate receptor. eLife 2021, 10, e66097. [Google Scholar] [CrossRef]
- Hollmann, M.; Heinemann, S. Cloned Glutamate Receptors. Annu. Rev. Neurosci. 1994, 17, 31–108. [Google Scholar] [CrossRef]
- Darstein, M.; Petralia, R.S.; Swanson, G.T.; Wenthold, R.J.; Heinemann, S.F. Distribution of Kainate Receptor Subunits at Hippocampal Mossy Fiber Synapses. J. Neurosci. 2003, 23, 8013–8019. [Google Scholar] [CrossRef]
- Carta, M.; Fièvre, S.; Gorlewicz, A.; Mulle, C. Kainate receptors in the hippocampus. Eur. J. Neurosci. 2014, 39, 1835–1844. [Google Scholar] [CrossRef]
- Mulle, C.; Crépel, V. Regulation and dysregulation of neuronal circuits by KARs. Neuropharmacology 2021, 197, 108699. [Google Scholar] [CrossRef]
- Barthet, G.; Moreira-De-Sá, A.; Zhang, P.; Deforges, S.; Castanheira, J.; Gorlewicz, A.; Mulle, C. Presenilin and APP Regulate Synaptic Kainate Receptors. J. Neurosci. 2022, 42, 9253–9262. [Google Scholar] [CrossRef]
- Zhang, X.-M.; Zhu, J. Kainic Acid-Induced Neurotoxicity: Targeting Glial Responses and Glia-Derived Cytokines. Curr. Neuropharmacol. 2011, 9, 388–398. [Google Scholar] [CrossRef]
- Cho, I.-H.; Hong, J.; Suh, E.C.; Kim, J.H.; Lee, H.; Lee, J.E.; Lee, S.; Kim, C.-H.; Kim, D.W.; Jo, E.-K.; et al. Role of microglial IKKβ in kainic acid-induced hippocampal neuronal cell death. Brain 2008, 131, 3019–3033. [Google Scholar] [CrossRef] [PubMed]
- Penkowa, M.; Molinero, A.; Carrasco, J.; Hidalgo, J. Interleukin-6 deficiency reduces the brain inflammatory response and increases oxidative stress and neurodegeneration after kainic acid-induced seizures. Neuroscience 2001, 102, 805–818. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Y.; Qiu, X.; Lv, Y.-D.; Dong, D.; Wu, X.-J.; Zhu, J.; Zheng, X.-Y. Kainic acid Induces production and aggregation of amyloid β-protein and memory deficits by activating inflammasomes in NLRP3- and NF-κB-stimulated pathways. Aging 2019, 11, 3795–3810. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, S.; Fu, P.; Zhang, Z.; Lin, K.; Ko, J.K.-S.; Yung, K.K.-L. Roles of Glutamate Receptors in Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 4391. [Google Scholar] [CrossRef]
- Maraschi, A.; Ciammola, A.; Folci, A.; Sassone, F.; Ronzitti, G.; Cappelletti, G.; Silani, V.; Sato, S.; Hattori, N.; Mazzanti, M.; et al. Parkin regulates kainate receptors by interacting with the GluK2 subunit. Nat. Commun. 2014, 5, 5182. [Google Scholar] [CrossRef]
- Stayte, S.; Laloli, K.J.; Rentsch, P.; Lowth, A.; Li, K.M.; Pickford, R.; Vissel, B. The kainate receptor antagonist UBP310 but not single deletion of GluK1, GluK2, or GluK3 subunits, inhibits MPTP-induced degeneration in the mouse midbrain. Exp. Neurol. 2020, 323, 113062. [Google Scholar] [CrossRef]
- Jin, X.-T.; Smith, Y. Localization and Functions of Kainate Receptors in the Basal Ganglia. In Kainate Receptors. Advances in Experimental Medicine and Biology; Springer: Boston, MA, USA, 2011; pp. 27–37. [Google Scholar] [CrossRef]
- Su, L.-D.; Wang, N.; Han, J.; Shen, Y. Group 1 Metabotropic Glutamate Receptors in Neurological and Psychiatric Diseases: Mechanisms and Prospective. Neuroscientist 2022, 28, 453–468. [Google Scholar] [CrossRef]
- Maiese, K.; Chong, Z.Z.; Shang, Y.C.; Hou, J. Therapeutic Promise and Principles: Metabotropic Glutamate Receptors. Oxidative Med. Cell. Longev. 2008, 1, 890561. [Google Scholar] [CrossRef]
- Trepanier, C.; Lei, G.; Xie, Y.-F.; MacDonald, J.F. Group II metabotropic glutamate receptors modify N-methyl-D-aspartate receptors via Src kinase. Sci. Rep. 2013, 3, 926. [Google Scholar] [CrossRef]
- Crupi, R.; Impellizzeri, D.; Cuzzocrea, S. Role of metabotropic glutamate receptors in neurological disorders. Front. Mol. Neurosci. 2019, 12, 20. [Google Scholar] [CrossRef]
- Dasgupta, A.; Lim, Y.J.; Kumar, K.; Baby, N.; Pang, K.L.K.; Benoy, A.; Behnisch, T.; Sajikumar, S. Group III metabotropic glutamate receptors gate long-term potentiation and synaptic tagging/capture in rat hippocampal area CA2. eLife 2020, 9, e55344. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.T.; Eriksson, J.; Antoni, G.; Yngve, U.; Cato, L.; Lannfelt, L.; Sehlin, D.; Syvänen, S. Brain mGluR5 in mice with amyloid beta pathology studied with in vivo [11C]ABP688 PET imaging and ex vivo immunoblotting. Neuropharmacology 2017, 113, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Brody, A.H.; Strittmatter, S.M. Synaptotoxic Signaling by Amyloid Beta Oligomers in Alzheimer’s Disease through Prion Protein and mGluR5. Adv. Pharmacol. 2018, 82, 293–323. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Dhull, D.K.; Mishra, P.S. Therapeutic potential of mGluR5 targeting in Alzheimer’s disease. Front. Neurosci. 2015, 9, 215. [Google Scholar] [CrossRef]
- Abd-Elrahman, K.S.; Hamilton, A.; Albaker, A.; Ferguson, S.S.G. mGluR5 Contribution to Neuropathology in Alzheimer Mice Is Disease Stage-Dependent. ACS Pharmacol. Transl. Sci. 2020, 3, 334–344. [Google Scholar] [CrossRef]
- Kitazawa, M.; Medeiros, R.; LaFerla, F.M. Transgenic Mouse Models of Alzheimer Disease: Developing a Better Model as a Tool for Therapeutic Interventions. Curr. Pharm. Des. 2012, 18, 1131–1147. [Google Scholar] [CrossRef]
- Kanemoto, S.; Griffin, J.; Markham-Coultes, K.; Aubert, I.; Tandon, A.; George-Hyslop, P.; Fraser, P. Proliferation, differentiation and amyloid-β production in neural progenitor cells isolated from TgCRND8 mice. Neuroscience 2014, 261, 52–59. [Google Scholar] [CrossRef]
- Lovasic, L.; Bauschke, H.; Janus, C. Working memory impairment in a transgenic amyloid precursor protein TgCRND8 mouse model of Alzheimer’s disease. Genes Brain Behav. 2005, 4, 197–208. [Google Scholar] [CrossRef]
- Kim, S.H.; Fraser, P.E.; Westaway, D.; George-Hyslop, P.H.S.; Ehrlich, M.E.; Gandy, S. Group II Metabotropic Glutamate Receptor Stimulation Triggers Production and Release of Alzheimer’s Amyloid β42 from Isolated Intact Nerve Terminals. J. Neurosci. 2010, 30, 3870–3875. [Google Scholar] [CrossRef]
- Tyszkiewicz, J.P.; Gu, Z.; Wang, X.; Cai, X.; Yan, Z. Group II metabotropic glutamate receptors enhance NMDA receptor currents via a protein kinase C-dependent mechanism in pyramidal neurones of rat prefrontal cortex. J. Physiol. 2004, 554, 765–777. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.W.; Shen, K.-Z. Group II metabotropic glutamate receptor modulation of excitatory transmission in rat subthalamic nucleus. J. Physiol. 2003, 553, 489–496. [Google Scholar] [CrossRef]
- Tyszkiewicz, J.P.; Yan, Z. β-Amyloid Peptides Impair PKC-Dependent Functions of Metabotropic Glutamate Receptors in Prefrontal Cortical Neurons. J. Neurophysiol. 2005, 93, 3102–3111. [Google Scholar] [CrossRef]
- Taylor, D.L.; Diemel, L.T.; Pocock, J.M. Activation of Microglial Group III Metabotropic Glutamate Receptors Protects Neurons against Microglial Neurotoxicity. J. Neurosci. 2003, 23, 2150–2160. [Google Scholar] [CrossRef] [PubMed]
- Masilamoni, G.J.; Smith, Y. Metabotropic glutamate receptors: Targets for neuroprotective therapies in Parkinson disease. Curr. Opin. Pharmacol. 2018, 38, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.A.; Conn, P.J.; Niswender, C.M. Glutamate Receptors as Therapeutic Targets for Parkinsons Disease. CNS Neurol. Disord.-Drug Targets 2009, 8, 475–491. [Google Scholar] [CrossRef]
- Matta, J.A.; Ashby, M.C.; Sanz-Clemente, A.; Roche, K.W.; Isaac, J.T.R. mGluR5 and NMDA Receptors Drive the Experience- and Activity-Dependent NMDA Receptor NR2B to NR2A Subunit Switch. Neuron 2011, 70, 339–351. [Google Scholar] [CrossRef]
- Lannes, N.; Eppler, E.; Etemad, S.; Yotovski, P.; Filgueira, L. Microglia at center stage: A comprehensive review about the versatile and unique residential macrophages of the central nervous system. Oncotarget 2017, 8, 114393–114413. [Google Scholar] [CrossRef]
- Zhang, Y.-N.; Fan, J.-K.; Gu, L.; Yang, H.-M.; Zhan, S.-Q.; Zhang, H. Metabotropic glutamate receptor 5 inhibits α-synuclein-induced microglia inflammation to protect from neurotoxicity in Parkinson’s disease. J. Neuroinflamm. 2021, 18, 23. [Google Scholar] [CrossRef]
- Bradley, S.R.; Marino, M.J.; Wittmann, M.; Rouse, S.T.; Awad, H.; Levey, A.I.; Conn, P.J. Activation of Group II Metabotropic Glutamate Receptors Inhibits Synaptic Excitation of the Substantia Nigra Pars Reticulata. J. Neurosci. 2000, 20, 3085–3094. [Google Scholar] [CrossRef]
- Valenti, O.; Marino, M.J.; Wittmann, M.; Lis, E.; DiLella, A.G.; Kinney, G.G.; Conn, P.J. Group III Metabotropic Glutamate Receptor-Mediated Modulation of the Striatopallidal Synapse. J. Neurosci. 2003, 23, 7218–7226. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, C.R.; Lindsley, C.W.; Niswender, C.M. mGluR4-positive allosteric modulation as potential treatment for Parkinson’s disease. Future Med. Chem. 2009, 1, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Dreux, C.; Launay, J.M. Blood platelets: Neuronal model in psychiatric disorders. L’encephale 1985, 11, 57–64. [Google Scholar] [PubMed]
- Asor, E.; Ben-Shachar, D. Platelets: A possible glance into brain biological processes in schizophrenia. World J. Psychiatry 2012, 2, 124–133. [Google Scholar] [CrossRef]
- Ehrlich, D.; Humpel, C. Platelets in psychiatric disorders. World J. Psychiatry 2012, 2, 91–94. [Google Scholar] [CrossRef]
- Stahl, S.M.; Meltzer, H.Y. A kinetic and pharmacologic analysis of 5-hydroxytryptamine transport by human platelets and platelet storage granules: Comparison with central serotonergic neurons. J. Pharmacol. Exp. Ther. 1978, 205, 118–132. [Google Scholar]
- Gill, S.S.; Pulido, O.M. Review Article: Glutamate Receptors in Peripheral Tissues: Current Knowledge, Future Research, and Implications for Toxicology. Toxicol. Pathol. 2001, 29, 208–223. [Google Scholar] [CrossRef]
- Zeller, J.A.; Frahm, K.; Baron, R.; Stingele, R.; Deuschl, G. Platelet-leukocyte interaction and platelet activation in migraine: A link to ischemic stroke? J. Neurol. Neurosurg. Psychiatry 2004, 75, 984–987. [Google Scholar] [CrossRef]
- Beura, S.K.; Panigrahi, A.R.; Yadav, P.; Singh, S.K. Role of platelet in Parkinson’s disease: Insights into pathophysiology & theranostic solutions. Ageing Res. Rev. 2022, 80, 101681. [Google Scholar] [CrossRef]
- Ritvo, E.R.; Yuwiler, A.; Geller, E.; Ornitz, E.M.; Saeger, K.; Plotkin, S. Increased Blood Serotonin and Platelets in Early Infantile Autism. Arch. Gen. Psychiatry 1970, 23, 566–572. [Google Scholar] [CrossRef]
- Wyatt, R.J.; Murphy, D.L. Low Platelet Monoamine Oxidase Activity and Schizophrenia*. Schizophr. Bull. 1976, 2, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Sevush, S.; Jy, W.; Horstman, L.L.; Mao, W.-W.; Kolodny, L.; Ahn, Y.S. Platelet Activation in Alzheimer Disease. Arch. Neurol. 1998, 55, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Mallick, R.L.; Kumari, S.; Singh, N.; Sonkar, V.K.; Dash, D. Prion protein fragment (106–126) induces prothrombotic state by raising platelet intracellular calcium and microparticle release. Cell Calcium 2015, 57, 300–311. [Google Scholar] [CrossRef] [PubMed]
- Gautam, D.; Kailashiya, J.; Tiwari, A.; Chaurasia, R.N.; Annarapu, G.K.; Guchhait, P.; Dash, D. Fibrinogen Mitigates Prion-Mediated Platelet Activation and Neuronal Cell Toxicity. Front. Cell Dev. Biol. 2022, 10, 834016. [Google Scholar] [CrossRef]
- Koçer, A.; Yaman, A.; Niftaliyev, E.; Dürüyen, H.; Eryılmaz, M.; Koçer, E. Assessment of Platelet Indices in Patients with Neurodegenerative Diseases: Mean Platelet Volume Was Increased in Patients with Parkinson’s Disease. Curr. Gerontol. Geriatr. Res. 2013, 2013, 986254. [Google Scholar] [CrossRef]
- Sala, G.; Brighina, L.; Saracchi, E.; Fermi, S.; Riva, C.; Carrozza, V.; Pirovano, M.; Ferrarese, C. Vesicular monoamine transporter 2 mRNA levels are reduced in platelets from patients with Parkinson’s disease. J. Neural Transm. 2010, 117, 1093–1098. [Google Scholar] [CrossRef]
- Haas, R.H.; Nasirian, F.; Nakano, K.; Ward, D.; Pay, M.; Hill, R.; Shults, C.W. Low platelet mitochondrial complex I and complex II/III activity in early untreated parkinson’s disease. Ann. Neurol. 1995, 37, 714–722. [Google Scholar] [CrossRef]
- Benecke, R.; Strümper, P.; Weiss, H. Electron transfer complexes I and IV of platelets are abnormal in Parkinson’s disease but normal in Parkinson-plus syndromes. Brain 1993, 116, 1451–1463. [Google Scholar] [CrossRef]
- Parker, W.D., Jr.; Parks, J.K.; Swerdlow, R.H. Complex I Deficiency in Parkinson’s disease frontal cortex. Brain Res. 2008, 1189, 215–218. [Google Scholar] [CrossRef]
- Arnoldussen, I.A.C.; Witkamp, R.F. Effects of nutrients on platelet function: A modifiable link between metabolic syndrome and neurodegeneration? Biomolecules 2021, 11, 1455. [Google Scholar] [CrossRef]
- Periayah, M.H.; Halim, A.S.; Zaharil, A.; Saad, M. Mechanism Action of Platelets and Crucial Blood Coagulation Pathways in Hemostasis. Int. J. Hematol.-Oncol. Stem Cell Res. 2017, 11, 319. [Google Scholar]
- Begni, B.; Tremolizzo, L.; D’Orlando, C.; Bono, M.S.; Garofolo, R.; Longoni, M.; Ferrarese, C. Substrate-induced modulation of glutamate uptake in human platelets. Br. J. Pharmacol. 2005, 145, 792–799. [Google Scholar] [CrossRef]
- Zoia, C.; Cogliati, T.; Tagliabue, E.; Cavaletti, G.; Sala, G.; Galimberti, G.; Rivolta, I.; Rossi, V.; Frattola, L.; Ferrarese, C. Glutamate transporters in platelets: EAAT1 decrease in aging and in Alzheimer’s disease. Neurobiol. Aging 2004, 25, 149–157. [Google Scholar] [CrossRef]
- Ferrarese, C.; Zoia, C.; Pecora, N.; Piolti, R.; Frigo, M.; Bianchi, G.; Sala, G.; Begni, B.; Riva, R.; Frattola, L. Reduced platelet glutamate uptake in Parkinson’s disease. J. Neural Transm. 1999, 106, 685–692. [Google Scholar] [CrossRef]
- Bell, J.D.; Thomas, T.C.; Lass, E.; Ai, J.; Wan, H.; Lifshitz, J.; Baker, A.J.; Macdonald, R.L. Platelet-mediated changes to neuronal glutamate receptor expression at sites of microthrombosis following experimental subarachnoid hemorrhage. J. Neurosurg. 2014, 121, 1424–1431. [Google Scholar] [CrossRef]
- Deepa, T.A.; Chaurasia, R.N.; Dash, D. Role of Platelets in Glutamate Mediated Excitotoxicity: An Overview. J. Neurol. Neurophysiol. 2015, 6, 5–7. [Google Scholar]
- Chopade, P.; Chopade, N.; Zhao, Z.; Mitragotri, S.; Liao, R.; Suja, V.C. Alzheimer’s and Parkinson’s disease therapies in the clinic. Bioeng. Transl. Med. 2023, 8, e10367. [Google Scholar] [CrossRef]
- Szeto, J.Y.; Lewis, S.J. Current Treatment Options for Alzheimer’s Disease and Parkinson’s Disease Dementia. Curr. Neuropharmacol. 2016, 14, 326–338. [Google Scholar] [CrossRef]
- Briggs, R.; Kennelly, S.P.; O’Neill, D. Drug treatments in Alzheimer’s disease. Clin. Med. 2016, 16, 247–253. [Google Scholar] [CrossRef]
- Honig, L.S.; Boyd, C.D. Treatment of Alzheimer’s Disease: Current Management and Experimental Therapeutics. Curr. Transl. Geriatr. Exp. Gerontol. Rep. 2013, 2, 174–181. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | Drug Class | Mode of Action | Approved Indications | Common Side Effects | Manufacturer and Year of Approval |
|---|---|---|---|---|---|
| Donepezil | Acetylcholinest-erase inhibitor | Increases acetylcholine levels in the brain by inhibiting its breakdown | Mild to severe AD | Nausea, diarrhea, insomnia, vomiting, muscle cramps, fatigue | Eisai Co. (brand name: Aricept), 1996 |
| Rivastigmine | Acetylcholinest-erase inhibitor | Increase acetycholine and butyrylcholine levels in the brain by inhibiting their breakdown | Mild to moderate AD and PD dementia | Nausea, vomiting, loss of appetite, dizziness, weight loss | Novartis AG (brand name: Exelon), 2000 |
| Galantamine | Acetylcholinest-erase inhibitor | Increases acetylcholine levels by inhibiting its breakdown and modulating nicotinic acetylcholine receptors | Mild to moderate AD | Nausea, vomiting, diarrhea, weight loss, loss of appetite | Janssen Pharmaceuticals (brand name: Razadyne), 2001 |
| Memantine (Namenda) | NMDAR antagonist | Modulate the activity of glutamate, an excitatory neurotransmitter, by blocking NMDAR | Moderate to severe AD | Dizziness, confusion, headache, constipation, cough | Allergan (brand name: Namenda), 2003 |
| Memantine+ Donepezil | Combination of NMDAR antagonist and acetylcholinesterase inhibitor | Combines the actions of Memantine andDonepezil | Moderate to severe AD | Headache, diarrhea, dizziness, flu symptoms, cough | Allergan and Eisai Co. (brand name: Namzaric), 2014 |
| Leqembi (Lecanemab) | IgG1 monoclonal antibody | Binds with high affinity to Aβ-soluble protofibrils and helps break them down | Early stage of AD | Amyloid-related imaging abnormalities (ARIA), a temporary swelling and/or bleeding in certain areas of the brain that usually resolves with time. ARIA risk may increase in those who have two copies of the well-known Alzheimer’s risk gene | Eisai/Biogen, 2023 |
| Donenmab | Monoclonal Ab, anti-Aβ | Binds with high affinity to Aβ-soluble protofibrils and helps break them down | Early stage of AD | Headaches, reactions to intravenous drip, swelling and microbleed in the brain | Eli Lilly (it is in the large final stage trial, called TRAILBLAIZER-ALZ2) |
| Drug Name | Drug Class | Mode of Action | Approved Indications | Common Side Effects | Manufacturer and Year of Approval |
|---|---|---|---|---|---|
| Levodopa/Carbidopa (Sinemet) | Dopamine precursor | Increases brain levels of dopamine. Carbidopa prevents peripheral breakdown of levodopa | PD, parkinsonism | Nausea, vomiting, dyskinesia, orthostatic hypotension | Merck Sharp and Dohme, 1975 |
| Pramipexole (Mirapex) | Dopamine agonist | Dopamine agonist: Mimics the effects dopamine in the brain | PD, restless leg syndrome | Nausea, vomiting, loss of appetite, dizziness, weight loss. | Boehringer Ingelheim, 1997 |
| Ropinirole (Requip) | Dopamine agonist | Dopamine agonist: Mimics the effects dopamine in the brain | PD, restless leg syndrome | Fatigue, dizziness, hallucination, nausea | GlaxoSmithKline, 1998 |
| Entacapone (Comtan) | COMT inhibitor | COMT inhibitor prolongs the effects of levodopa by blocking its breakdown in the periphery | PD (adjunct to levodopa/carbidopa) | Nausea, abdominal pain, diarrhea, discoloration of urine | Orion Pharma, 1999 |
| Apomorphine (Apokyn) | Dopamine agonist | Dopamine agonist: Rapidly treats off-episodes by mimicking dopamine | PD (far off episodes) | Yawning, dyskinesias, dizziness, rhinorrhea | Britannia pharmaceuticals, 2004 |
| Rasagiline (Azilect) | MAO-B inhibitor | MAO-B inhibitor: blocks the breakdown of dopamine in the brain | PD | Joint pain, depression, dyskinesias, flu-like symptoms. | Teva pharmaceuticals, 2006 |
| Safinamide (Xadago) | MAO-B inhibitor and glutamate release inhibitor | MAO-B inhibitor and glutamate release inhibitor: enhances dopaminergic activity and reduces glutamate | PD (add-on treatment to levodopa/ carbidopa) | Dyskinesia, hypertension, hallucinations, falls. | Zambon, 2017 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gautam, D.; Naik, U.P.; Naik, M.U.; Yadav, S.K.; Chaurasia, R.N.; Dash, D. Glutamate Receptor Dysregulation and Platelet Glutamate Dynamics in Alzheimer’s and Parkinson’s Diseases: Insights into Current Medications. Biomolecules 2023, 13, 1609. https://doi.org/10.3390/biom13111609
Gautam D, Naik UP, Naik MU, Yadav SK, Chaurasia RN, Dash D. Glutamate Receptor Dysregulation and Platelet Glutamate Dynamics in Alzheimer’s and Parkinson’s Diseases: Insights into Current Medications. Biomolecules. 2023; 13(11):1609. https://doi.org/10.3390/biom13111609
Chicago/Turabian StyleGautam, Deepa, Ulhas P. Naik, Meghna U. Naik, Santosh K. Yadav, Rameshwar Nath Chaurasia, and Debabrata Dash. 2023. "Glutamate Receptor Dysregulation and Platelet Glutamate Dynamics in Alzheimer’s and Parkinson’s Diseases: Insights into Current Medications" Biomolecules 13, no. 11: 1609. https://doi.org/10.3390/biom13111609
APA StyleGautam, D., Naik, U. P., Naik, M. U., Yadav, S. K., Chaurasia, R. N., & Dash, D. (2023). Glutamate Receptor Dysregulation and Platelet Glutamate Dynamics in Alzheimer’s and Parkinson’s Diseases: Insights into Current Medications. Biomolecules, 13(11), 1609. https://doi.org/10.3390/biom13111609