


Dopamine-Depleted Dopamine Transporter Knockout (DDD) Mice: Dyskinesia with L-DOPA and Dopamine D1 Agonists

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Drugs
2.3. Apparati
2.4. Procedures
2.5. Statistics
3. Results
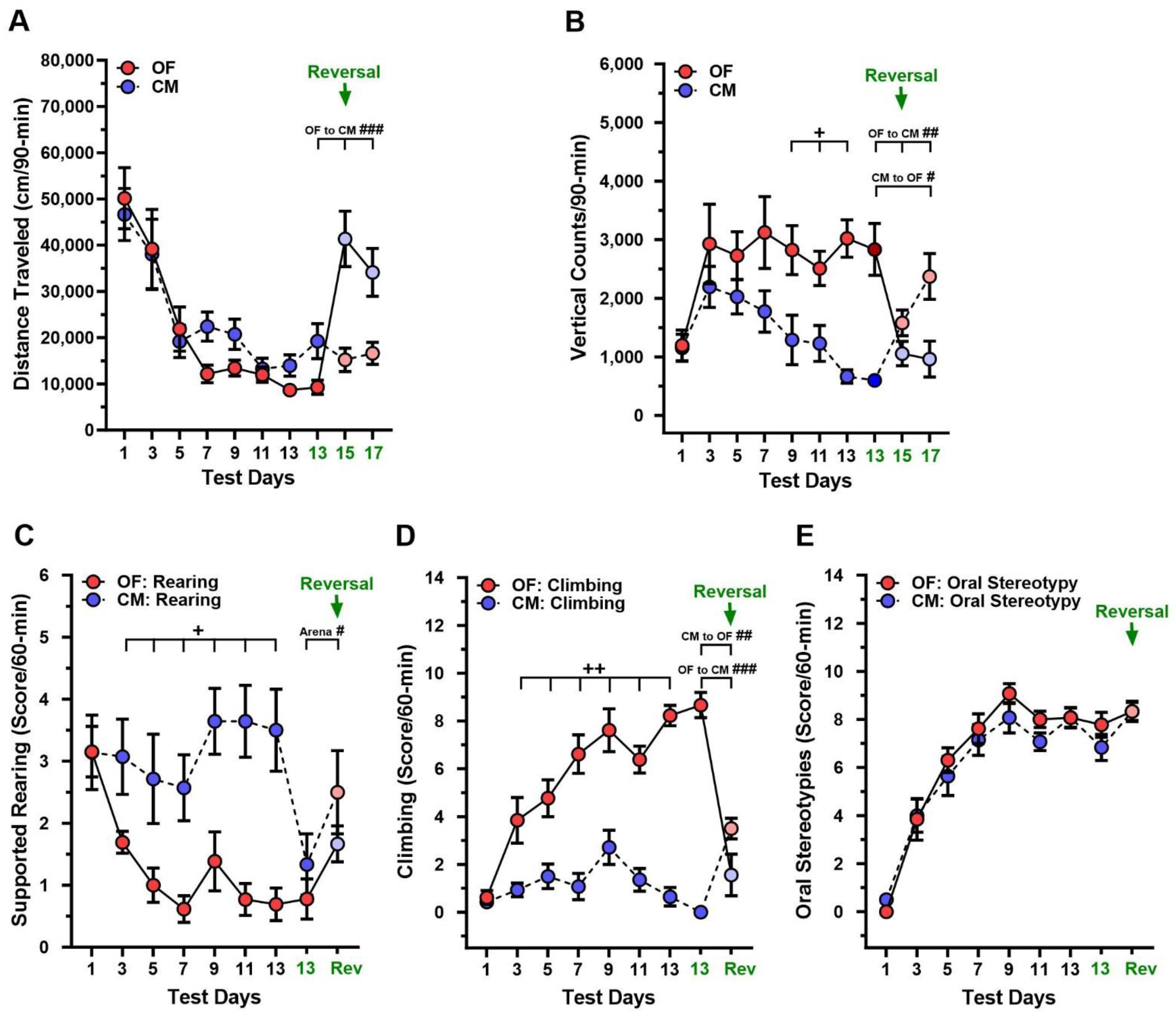
3.1. L-DOPA Sensitization in DDD Mice
3.2. Changes in Test Context during L-DOPA Sensitization in DDD Mice
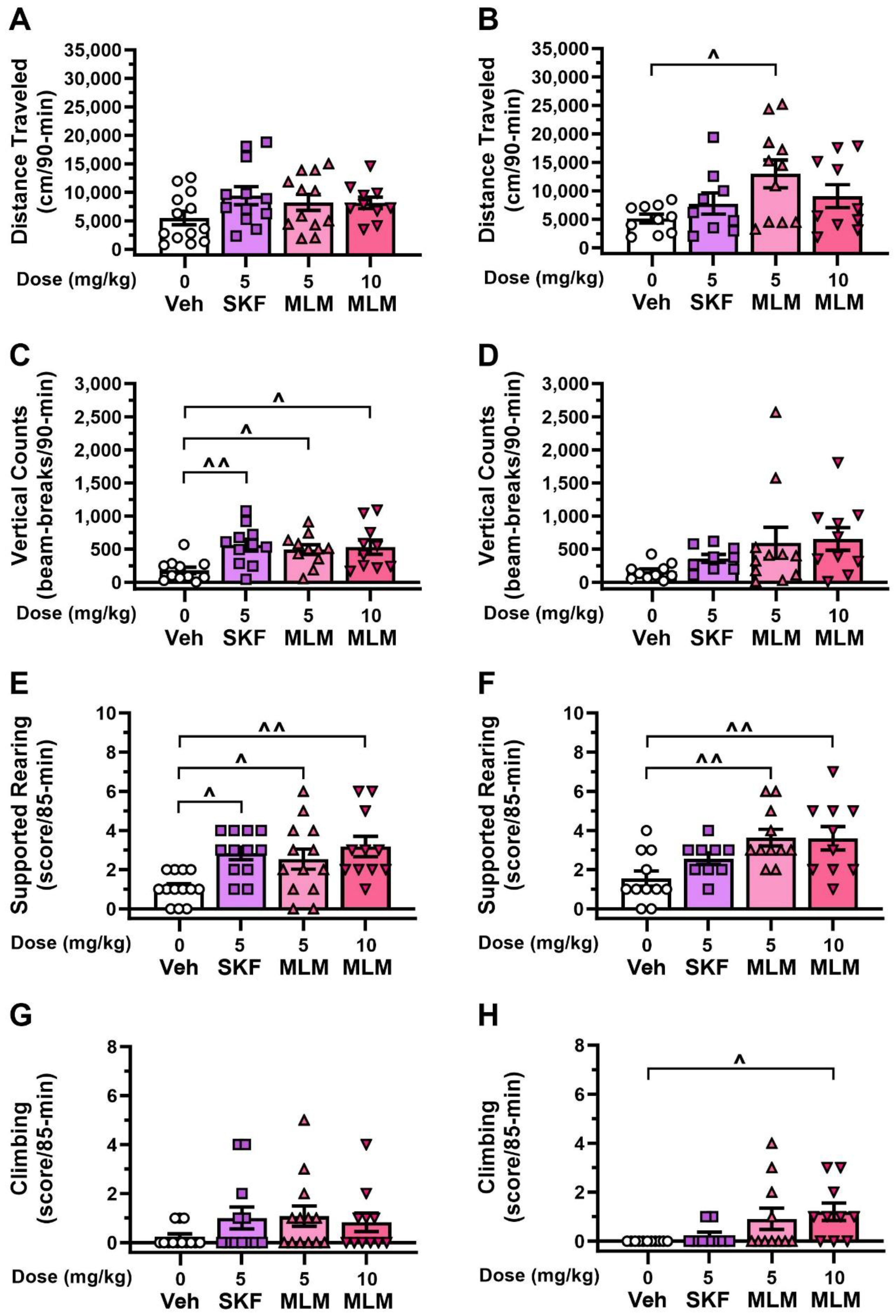
3.3. Effects of D1R Agonists on Stereotypic Behaviors in DDD Mice in Two Test Contexts
3.4. Effects of D1R Compounds in the DDD Model without L-DOPA
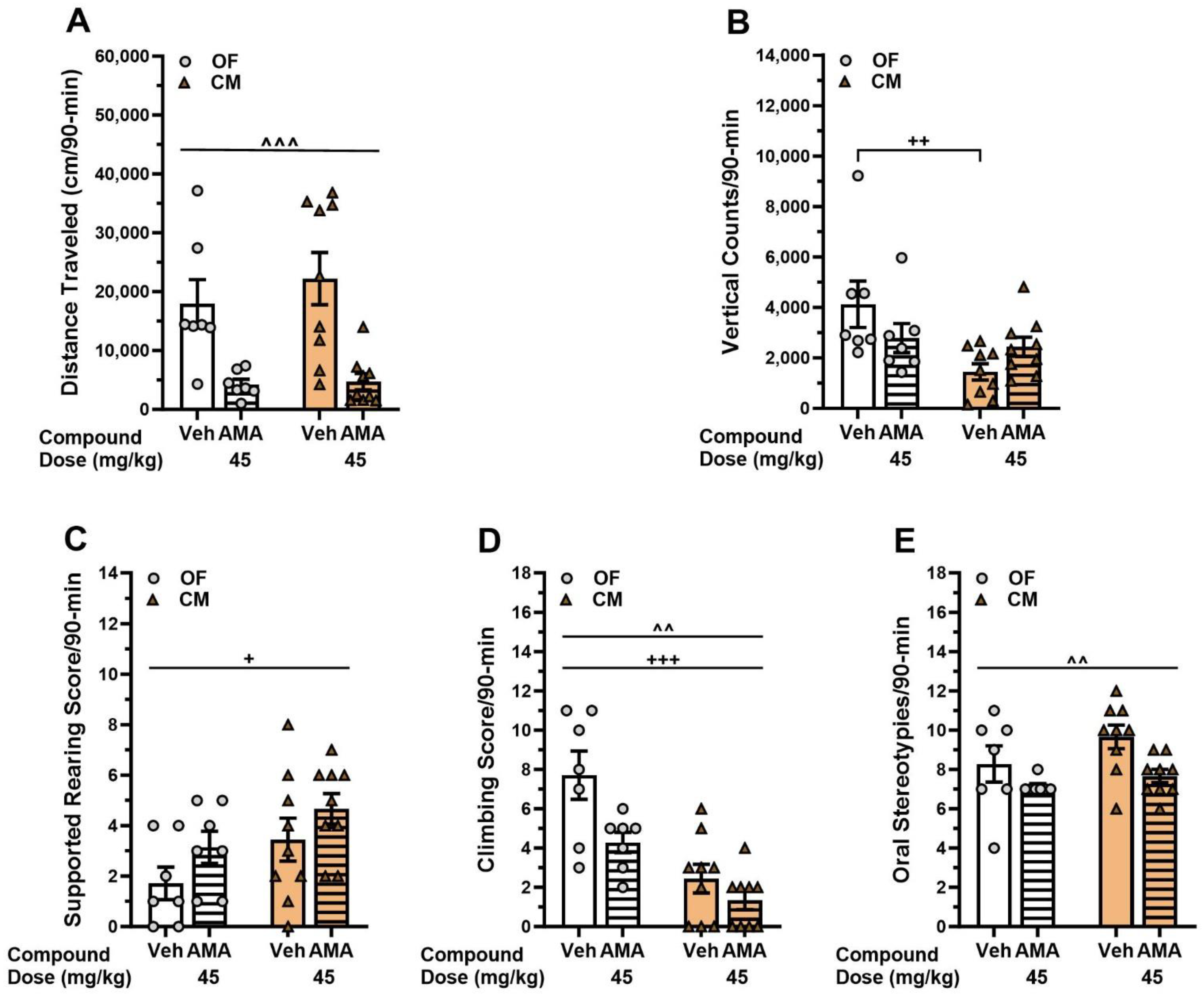
3.5. Effects of Amantadine in the DDD Mice Treated with L-DOPA
4. Discussion
4.1. Sensitization in DDD Mice
4.2. Stereotypy and LID in the DDD Mice during Changes in the Test Context
4.3. Responses to D1R Agonists and Amantadine by DDD Mice
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marras, C.; Beck, J.C.; Bower, J.H.; Roberts, E.; Ritz, B.; Ross, G.W.; Abbott, R.D.; Savica, R.; Van Den Eeden, S.K.; Willis, A.W.; et al. Prevalence of Parkinson’s disease across North America. NPJ Park. Dis. 2018, 4, 21. [Google Scholar] [CrossRef]
- Balestrino, R.; Schapira, A.H.V. Parkinson disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef]
- Ayano, G. Parkinson’s disease: A concise overview of etiology, epidemiology, diagnosis, comorbidity and management. J. Neurol. Dis. 2016, 4, 6. [Google Scholar] [CrossRef]
- Marras, C.; Chaudhuri, K.R. Nonmotor features of Parkinson’s disease subtypes. Mov. Disord. 2016, 31, 1095–1102. [Google Scholar] [CrossRef]
- Foltynie, T.; Brayne, C.; Barker, R.A. The heterogeneity of idiopathic Parkinson’s disease. J. Neurol. 2002, 249, 138–145. [Google Scholar] [CrossRef]
- Billingsley, K.J.; Bandres-Ciga, S.; Saez-Atienzar, S.; Singleton, A.B. Genetic risk factors in Parkinson’s disease. Cell Tissue Res. 2018, 373, 9–20. [Google Scholar] [CrossRef]
- Lesage, S.; Brice, A. Parkinson’s disease: From monogenic forms to genetic susceptibility factors. Hum. Mol. Genet 2009, 18, R48–R59. [Google Scholar] [CrossRef]
- Reed, X.; Bandrés-Ciga, S.; Blauwendraat, C.; Cookson, M.R. The role of monogenic genes in idiopathic Parkinson’s disease. Neurobiol. Dis. 2019, 124, 230–239. [Google Scholar] [CrossRef]
- Costello, S.; Cockburn, M.; Bronstein, J.; Zhang, X.; Ritz, B. Parkinson’s disease and residential exposure to maneb and paraquat from agricultural applications in the central valley of California. Am. J. Epidemiol. 2009, 169, 919–926. [Google Scholar] [CrossRef]
- Van Maele-Fabry, G.; Hoet, P.; Vilain, F.; Lison, D. Occupational exposure to pesticides and Parkinson’s disease: A systematic review and meta-analysis of cohort studies. Environ. Int. 2012, 46, 30–43. [Google Scholar] [CrossRef]
- Kamel, F. Epidemiology. Paths from pesticides to Parkinson’s. Science 2013, 341, 722–773. [Google Scholar] [CrossRef]
- Berry, C.; La Vecchia, C.; Nicotera, P. Paraquat and Parkinson’s disease. Cell Death Diff. 2010, 17, 1115–1125. [Google Scholar] [CrossRef]
- van der Mark, M.; Vermeulen, R.; Nijssen, P.C.; Mulleners, W.M.; Sas, A.M.; van Laar, T.; Huss, A.; Kromhout, H. Occupational exposure to solvents, metals and welding fumes and risk of Parkinson’s disease. Park. Relat. Disord. 2015, 21, 635–639. [Google Scholar] [CrossRef]
- De Miranda, B.R.; Goldman, S.M.; Miller, G.W.; Greenamyre, J.T.; Dorsey, E.R. Preventing Parkinson’s disease: An environmental agenda. J. Park. Dis. 2022, 12, 45–68. [Google Scholar] [CrossRef]
- Bjorklund, G.; Stejskal, V.; Urbina, M.A.; Dadar, M.; Chirumbolo, S.; Mutter, J. Metals and Parkinson’s disease: Mechanisms and biochemical processes. Curr. Med. Chem. 2018, 25, 2198–2214. [Google Scholar] [CrossRef]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef]
- Konnova, E.A.; Swanberg, M. Animal Models of Parkinson’s Disease. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Bisbane, Australia, 2018. [Google Scholar]
- Hwang, D.Y.; Ardayfio, P.; Kang, U.J.; Semina, E.V.; Kim, K.S. Selective loss of dopaminergic neurons in the substantia nigra of Pitx3-deficient aphakia mice. Brain Res. Mol. Brain Res. 2003, 114, 123–131. [Google Scholar] [CrossRef]
- Jiang, C.; Wan, X.; He, Y.; Pan, T.; Jankovic, J.; Le, W. Age-dependent dopaminergic dysfunction in Nurr1 knockout mice. Exp. Neurol. 2005, 191, 154–162. [Google Scholar] [CrossRef]
- Ekstrand, M.I.; Terzioglu, M.; Galter, D.; Zhu, S.; Hofstetter, C.; Lindqvist, E.; Thams, S.; Bergstrand, A.; Hansson, F.S.; Trifunovic, A.; et al. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 1325–1330. [Google Scholar] [CrossRef]
- Sonnier, L.; Le Pen, G.; Hartmann, A.; Bizot, J.C.; Trovero, F.; Krebs, M.O.; Prochiantz, A. Progressive loss of dopaminergic neurons in the ventral midbrain of adult mice heterozygote for Engrailed1. J. Neurosci. 2007, 27, 1063–1071. [Google Scholar] [CrossRef]
- Gonzalez-Reyes, L.E.; Verbitsky, M.; Blesa, J.; Jackson-Lewis, V.; Paredes, D.; Tillack, K.; Phani, S.; Kramer, E.R.; Przedborski, S.; Kottmann, A.H. Sonic hedgehog maintains cellular and neurochemical homeostasis in the adult nigrostriatal circuit. Neuron 2012, 75, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, R.J.; Drukarch, B.; Sahadat, M.C.; Goosen, C.; Wolters, E.C.; Stoof, J.C. The dopamine D1 agonist SKF 81297 and the dopamine D2 agonist LY 171555 act synergistically to stimulate motor behavior of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned parkinsonian rhesus monkeys. Mov. Disord. 1994, 9, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Yasuno, H.; Masuda, Y.; Ozaki, H.; Sano, T.; Shinozawa, T.; Watanabe, T. Identifying the dataset to define the optimal timing of histopathological examination for central nervous system toxicity in MPTP-induced Parkinson’s disease monkey model. J. Toxicol. Pathol. 2023, 36, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Giovanni, A.; Sieber, B.A.; Heikkila, R.E.; Sonsalla, P.K. Studies on species sensitivity to the dopaminergic neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Part 1: Systemic administration. J. Pharmacol. Exp. Ther. 1994, 270, 1000–1007. [Google Scholar]
- Sedelis, M.; Hofele, K.; Auburger, G.W.; Morgan, S.; Huston, J.P.; Schwarting, R.K. MPTP susceptibility in the mouse: Behavioral, neurochemical, and histological analysis of gender and strain differences. Behav. Genet. 2000, 30, 171–182. [Google Scholar] [CrossRef]
- Henderson, J.M.; Watson, S.; Halliday, G.M.; Heinemann, T.; Gerlach, M. Relationships between various behavioural abnormalities and nigrostriatal dopamine depletion in the unilateral 6-OHDA-lesioned rat. Behav. Brain Res. 2003, 139, 105–113. [Google Scholar] [CrossRef]
- Alvarez-Fischer, D.; Henze, C.; Strenzke, C.; Westrich, J.; Ferger, B.; Höglinger, G.U.; Oertel, W.H.; Hartmann, A. Characterization of the striatal 6-OHDA model of Parkinson’s disease in wild type and α-synuclein-deleted mice. Exp. Neurol. 2008, 210, 182–193. [Google Scholar] [CrossRef]
- Emborg, M.E. Nonhuman primate models of Parkinson’s disease. ILAR J. 2007, 48, 339–355. [Google Scholar] [CrossRef]
- Vermilyea, S.C.; Emborg, M.E. α-Synuclein and nonhuman primate models of Parkinson’s disease. J. Neurosci. Methods 2015, 255, 38–51. [Google Scholar] [CrossRef]
- Fox, S.H.; Brotchie, J.M. The MPTP-lesioned non-human primate models of Parkinson’s disease. Past, present, and future. Prog. Brain Res. 2010, 184, 33–357. [Google Scholar]
- Schwarting, R.K.; Sedelis, M.; Hofele, K.; Auburger, G.W.; Huston, J.P. Strain-dependent recovery of open-field behavior and striatal dopamine deficiency in the mouse MPTP model of Parkinson’s disease. Neurotox. Res. 1999, 1, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Sedelis, M.; Schwarting, R.K.; Huston, J.P. Behavioral phenotyping of the MPTP mouse model of Parkinson’s disease. Behav. Brain Res. 2001, 125, 109–125. [Google Scholar] [CrossRef] [PubMed]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2012, 39, 889–909. [Google Scholar] [CrossRef]
- Bove, J.; Perier, C. Neurotoxin-based models of Parkinson’s disease. Neuroscience 2012, 211, 51–76. [Google Scholar] [CrossRef] [PubMed]
- Jackson-Lewis, V.; Blesa, J.; Przedborski, S. Animal models of Parkinson’s disease. Park. Relat. Disord. 2012, 18 (Suppl. S1), S183–S185. [Google Scholar] [CrossRef] [PubMed]
- Willis, G.L.; Kennedy, G.A. The implementation of acute versus chronic animal models for treatment discovery in Parkinson’s disease. Rev. Neurosci. 2004, 15, 75–87. [Google Scholar] [CrossRef]
- Sotnikova, T.D.; Beaulieu, J.M.; Barak, L.S.; Wetsel, W.C.; Caron, M.G.; Gainetdinov, R.R. Dopamine-independent locomotor actions of amphetamines in a novel acute mouse model of Parkinson disease. PLoS Biol. 2005, 3, e271. [Google Scholar] [CrossRef]
- Sotnikova, T.D.; Caron, M.G.; Gainetdinov, R.R. DDD mice, a novel acute mouse model of Parkinson’s disease. Neurology 2006, 67 (Suppl. S2), S12–S17. [Google Scholar] [CrossRef]
- Sotnikova, T.D.; Zorina, O.I.; Ghisi, V.; Caron, M.G.; Gainetdinov, R.R. Trace amine associated receptor 1 and movement control. Park. Relat. Disord. 2008, 14 (Suppl. S2), S99–S102. [Google Scholar] [CrossRef]
- Managò, F.; Espinoza, S.; Salahpour, A.; Sotnikova, T.D.; Caron, M.G.; Premont, R.T.; Gainetdinov, R.R. The role of GRK6 in animal models of Parkinson’s disease and L-Dopa treatment. Sci. Rep. 2012, 2, 301. [Google Scholar] [CrossRef]
- Urs, N.M.; Bido, S.; Peterson, S.M.; Daigle, T.L.; Bass, C.E.; Gainetdinov, R.R.; Bezard, E.; Caron, M.G. Targeting β-arrestin2 in the treatment of L-DOPA-induced dyskinesia in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2015, 112, E2517–E2526. [Google Scholar] [CrossRef] [PubMed]
- Sukhanov, I.; Dorotenko, A.; Fesenko, Z.; Savchenko, A.; Efimova, E.V.; Mor, M.S.; Belozertseva, I.V.; Sotnikova, T.D.; Gainetdinov, R.R. Inhibition of PDE10A in a new rat model of severe dopamine depletion suggests new approach to non-dopamine Parkinson’s disease therapy. Biomolecules 2023, 13, 9. [Google Scholar] [CrossRef] [PubMed]
- Giros, B.; Jaber, M.; Jones, S.R.; Wightman, M.R.; Caron, M.G. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature 1996, 379, 606–612. [Google Scholar] [CrossRef]
- Fahn, S.; Oakes, D.; Shoulson, I.; Kieburtz, K.; Rudolph, A.; Lang, A.; Olanow, C.W.; Tanner, C.; Marek, K.; Parkinson Study Group. Levodopa and the progression of Parkinson’s disease. N. Engl. J. Med. 2004, 351, 2498–2508. [Google Scholar]
- Espay, A.J.; Morgante, F.; Merola, A.; Fasano, A.; Marsili, L.; Fox, S.H.; Bezard, E.; Picconi, B.; Calabresi, P.; Lang, A.E. Levodopa-induced dyskinesia in Parkinson disease: Current and evolving concepts. Ann. Neurol. 2018, 84, 797–811. [Google Scholar] [CrossRef] [PubMed]
- Aquino, C.C.; Fox, S.H. Clinical spectrum of levodopa-induced complications. Mov. Disord. 2015, 30, 80–89. [Google Scholar] [CrossRef]
- Tanaka, H.; Kannari, K.; Maeda, T.; Tomiyama, M.; Suda, T.; Matsunaga, M. Role of serotonergic neurons in L-DOPA-derived extracellular dopamine in the striatum of 6-OHDA-lesioned rats. Neuroreport 1999, 10, 631–634. [Google Scholar] [CrossRef]
- Chotibut, T.; Fields, V.; Salvatore, M.F. Norepinephrine transporter inhibition with desipramine exacerbates L-DOPA-induced dyskinesia: Role for synaptic dopamine regulation in denervated nigrostriatal terminals. Mol. Pharmacol. 2014, 86, 675–685. [Google Scholar] [CrossRef]
- Nishijima, H.; Tomiyama, M. What mechanisms are responsible for the reuptake of levodopa-derived dopamine in parkinsonian striatum? Front. Neurosci. 2016, 10, 575. [Google Scholar] [CrossRef]
- Chagraoui, A.; Boulain, M.; Juvin, L.; Anouar, Y.; Barrière, G.; Deurwaerdère, P. L-DOPA in Parkinson’s 2016, disease: Looking at the “false” neurotransmitters and their meaning. Int. J. Mol. Sci. 2019, 21, 294. [Google Scholar] [CrossRef]
- Viaro, R.; Longo, F.; Vincenzi, F.; Varani, K.; Morari, M. L-DOPA promotes striatal dopamine release through D1 receptors and reversal of dopamine transporter. Brain Res. 2021, 1768, 147583. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Restrepo, J.; Won, L.; Hwang, D.Y.; Kim, K.S.; Kang, U.J. Chronic 3,4-dihydroxyphenylalanine treatment induces dyskinesia in aphakia mice, a novel genetic model of Parkinson’s disease. Neurobiol. Dis. 2007, 27, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Diaz, O.; Zhang, Y.; Ladenheim, B.; Cadet, J.L.; Chiang, Y.H.; Olson, L.; Hoffer, B.J.; Bäckman, C.M. L-DOPA-induced dyskinesias in parkinsonian mice: Disease severity or L-Dopa history. Brain Res. 2015, 1618, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Goetz, C.G.; Poewe, W.; Rascol, O.; Sampaio, C. Evidence-based medical review update: Pharmacological and surgical treatments of Parkinson’s disease: 2001 to 2004. Mov. Disord. 2005, 20, 523–539. [Google Scholar] [CrossRef]
- Isaacson, S.H.; Hauser, R.A.; Pahwa, R.; Gray, D.; Duvvuri, S. Dopamine agonists in Parkinson’s disease: Impact of D1-like or D2-like dopamine receptor subtype selectivity and avenues for future treatment. Clin. Park. Relat. Disord. 2023, 9, 100212. [Google Scholar] [CrossRef]
- Lewis, M.M.; Van Scoy, L.J.; De Jesus, S.; Hakun, J.G.; Eslinger, P.J.; Fernandez-Mendoza, J.; Kong, L.; Yang, Y.; Snyder, B.L.; Loktionova, N.; et al. Dopamine D1 agonists: First potential treatment for late-stage Parkinson’s disease. Biomolecules 2023, 13, 829. [Google Scholar] [CrossRef]
- Darmopil, S.; Martín, A.B.; De Diego, I.R.; Ares, S.; Moratalla, R. Genetic inactivation of dopamine D1 but not D2 receptors inhibits L-DOPA-induced dyskinesia and histone activation. Biol. Psychiatry 2009, 66, 603–661. [Google Scholar] [CrossRef]
- Martini, M.L.; Ray, C.; Yu, X.; Liu, J.; Pogorelov, V.M.; Wetsel, W.C.; Huang, X.P.; McCorvy, J.D.; Caron, M.G.; Jin, J. Designing functionally selective noncatechol dopamine D1 receptor agonists with potent in vivo antiparkinsonian activity. ACS Chem. Neurosci. 2019, 10, 4160–4182. [Google Scholar] [CrossRef]
- Dawson, T.M.; Ko, H.S.; Dawson, V.L. Genetic animal models of Parkinson’s disease. Neuron 2010, 66, 646–661. [Google Scholar] [CrossRef]
- Morin, N.; Jourdain, V.A.; Di Paolo, T. Modeling dyskinesia in animal models of Parkinson disease. Exp. Neurol. 2014, 256, 105–116. [Google Scholar] [CrossRef]
- Ha, A.D.; Jankovic, J. An introduction to dyskinesia--the clinical spectrum. Int. Rev. Neurobiol. 2011, 98, 1–29. [Google Scholar] [PubMed]
- Cenci, M.A.; Whishaw, I.Q.; Schallert, T. Animal models of neurological deficits: How relevant is the rat? Nat. Rev. Neurosci. 2002, 3, 574–579. [Google Scholar] [CrossRef]
- Winkler, C.; Kirik, D.; Björklund, A.; Cenci, M.A. L-DOPA-induced dyskinesia in the intrastriatal 6-hydroxydopamine model of parkinson’s disease: Relation to motor and cellular parameters of nigrostriatal function. Neurobiol. Dis. 2002, 10, 165–186. [Google Scholar] [CrossRef] [PubMed]
- Lundblad, M.; Picconi, B.; Lindgren, H.; Cenci, M.A. A model of L-DOPA-induced dyskinesia in 6-hydroxydopamine lesioned mice: Relation to motor and cellular parameters of nigrostriatal function. Neurobiol. Dis. 2004, 16, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Pogorelov, V.M.; Rodriguiz, R.M.; Insco, M.L.; Caron, M.G.; Wetsel, W.C. Novelty seeking and stereotypic activation of behavior in mice with disruption of the Dat1 gene. Neuropsychopharmacology 2005, 30, 1818–1831. [Google Scholar] [CrossRef] [PubMed]
- Johnston, T.H.; Lee, J.; Gomez-Ramirez, J.; Fox, S.H.; Brotchie, J.M. A simple rodent assay for the in vivo identification of agents with potential to reduce levodopa-induced dyskinesia in Parkinson’s disease. Exp. Neurol. 2005, 191, 243–250. [Google Scholar] [CrossRef]
- Rascol, O.; Nutt, J.G.; Blin, O.; Goetz, C.G.; Trugman, J.M.; Soubrouillard, C.; Carter, J.H.; Currie, L.J.; Fabre, N.; Thalamas, C.; et al. Induction by dopamine D1 receptor agonist ABT-431 of dyskinesia similar to levodopa in patients with Parkinson disease. Arch. Neurol. 2001, 58, 249–254. [Google Scholar] [CrossRef]
- Konta, B.; Frank, W. The treatment of Parkinson’s disease with dopamine agonists. GMS Health Technol. Assess. 2008, 4, Doc05. [Google Scholar]
- Danysz, W.; Parsons, C.G.; Kornhuber, J.; Schmidt, W.J.; Quack, G. Aminoadamantanes as NMDA receptor antagonists and antiparkinsonian agents--preclinical studies. Neurosci. Biobehav. Rev. 1997, 21, 455–468. [Google Scholar] [CrossRef]
- Young, D.; Popiolek, M.; Trapa, P.; Fonseca, K.R.; Brevard, J.; Gray, D.L.; Kozak, R. D1 agonist improved movement of parkinsonian nonhuman primates with limited dyskinesia side effects. ACS Chem. Neurosci. 2020, 11, 560–566. [Google Scholar] [CrossRef]
- Denayer, T.; Stöhr, T.; Roy, M.V. Animal models in translational medicine: Validation and prediction. Eur. J. Mol. Clin. Med. 2014, 2, 5–11. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Behavior a,b,c | Description |
|---|---|
| Supported rearing | Standing on one or two hind legs with the forepaws on the wall |
| Climbing | Standing on the tail and/or one or two hind legs with the forepaws on the wall as if attempting to climb |
| Oral stereotypies | Licking and/or gnawing behaviors (i.e., opening and closing the lower jaw) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pogorelov, V.M.; Martini, M.L.; Jin, J.; Wetsel, W.C.; Caron, M.G. Dopamine-Depleted Dopamine Transporter Knockout (DDD) Mice: Dyskinesia with L-DOPA and Dopamine D1 Agonists. Biomolecules 2023, 13, 1658. https://doi.org/10.3390/biom13111658
Pogorelov VM, Martini ML, Jin J, Wetsel WC, Caron MG. Dopamine-Depleted Dopamine Transporter Knockout (DDD) Mice: Dyskinesia with L-DOPA and Dopamine D1 Agonists. Biomolecules. 2023; 13(11):1658. https://doi.org/10.3390/biom13111658
Chicago/Turabian StylePogorelov, Vladimir M., Michael L. Martini, Jian Jin, William C. Wetsel, and Marc G. Caron. 2023. "Dopamine-Depleted Dopamine Transporter Knockout (DDD) Mice: Dyskinesia with L-DOPA and Dopamine D1 Agonists" Biomolecules 13, no. 11: 1658. https://doi.org/10.3390/biom13111658
APA StylePogorelov, V. M., Martini, M. L., Jin, J., Wetsel, W. C., & Caron, M. G. (2023). Dopamine-Depleted Dopamine Transporter Knockout (DDD) Mice: Dyskinesia with L-DOPA and Dopamine D1 Agonists. Biomolecules, 13(11), 1658. https://doi.org/10.3390/biom13111658