

gRNA Design: How Its Evolution Impacted on CRISPR/Cas9 Systems Refinement

 , , and
, , and
Abstract
:1. Introduction
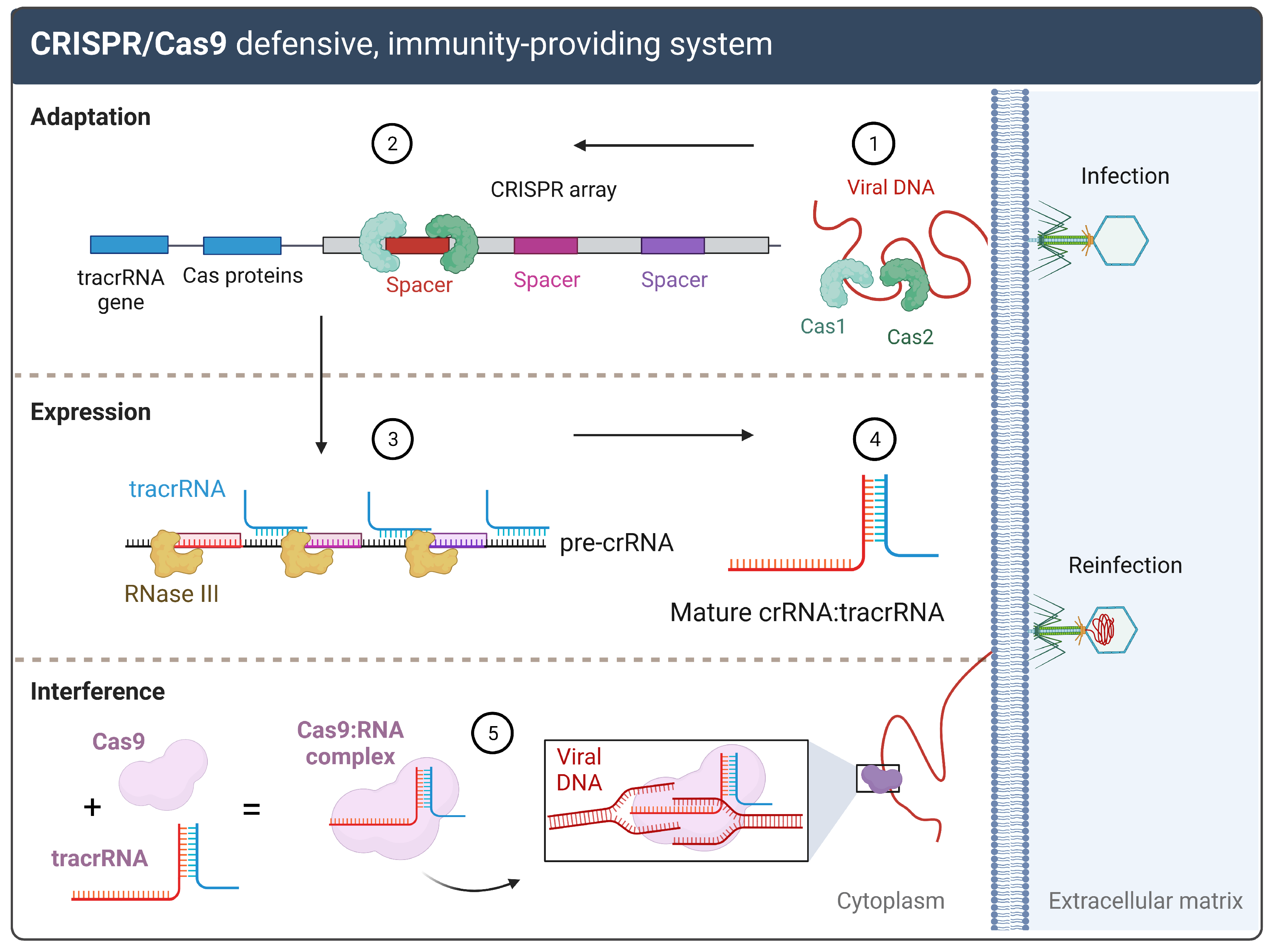
1.1. CRISPR/Cas: From a Bacterial Defense to a Genetic Engineering Tool
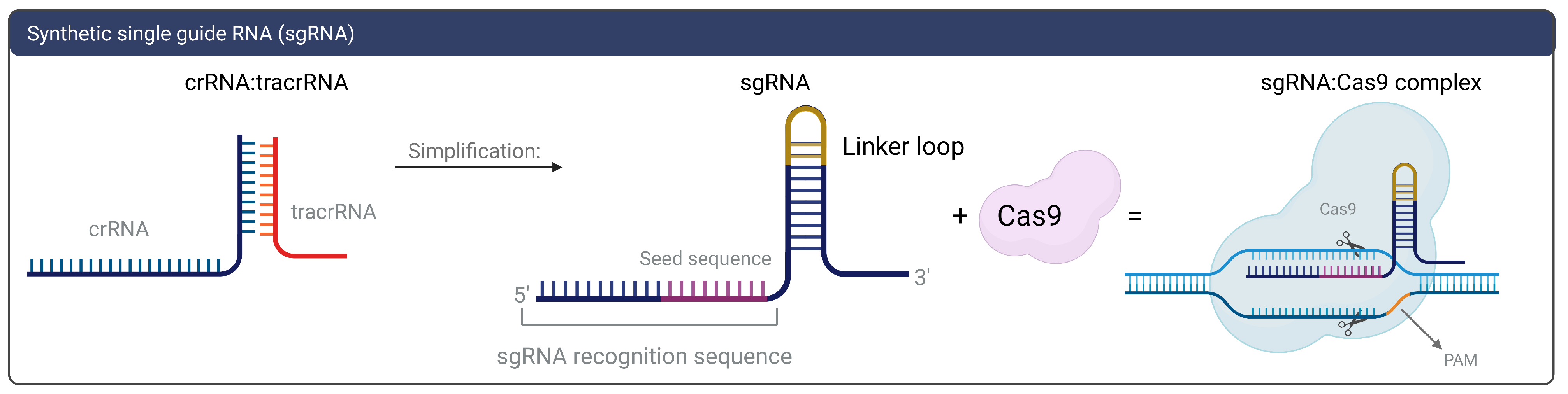
1.2. gRNAs and CRISPR On-and-Off Targets
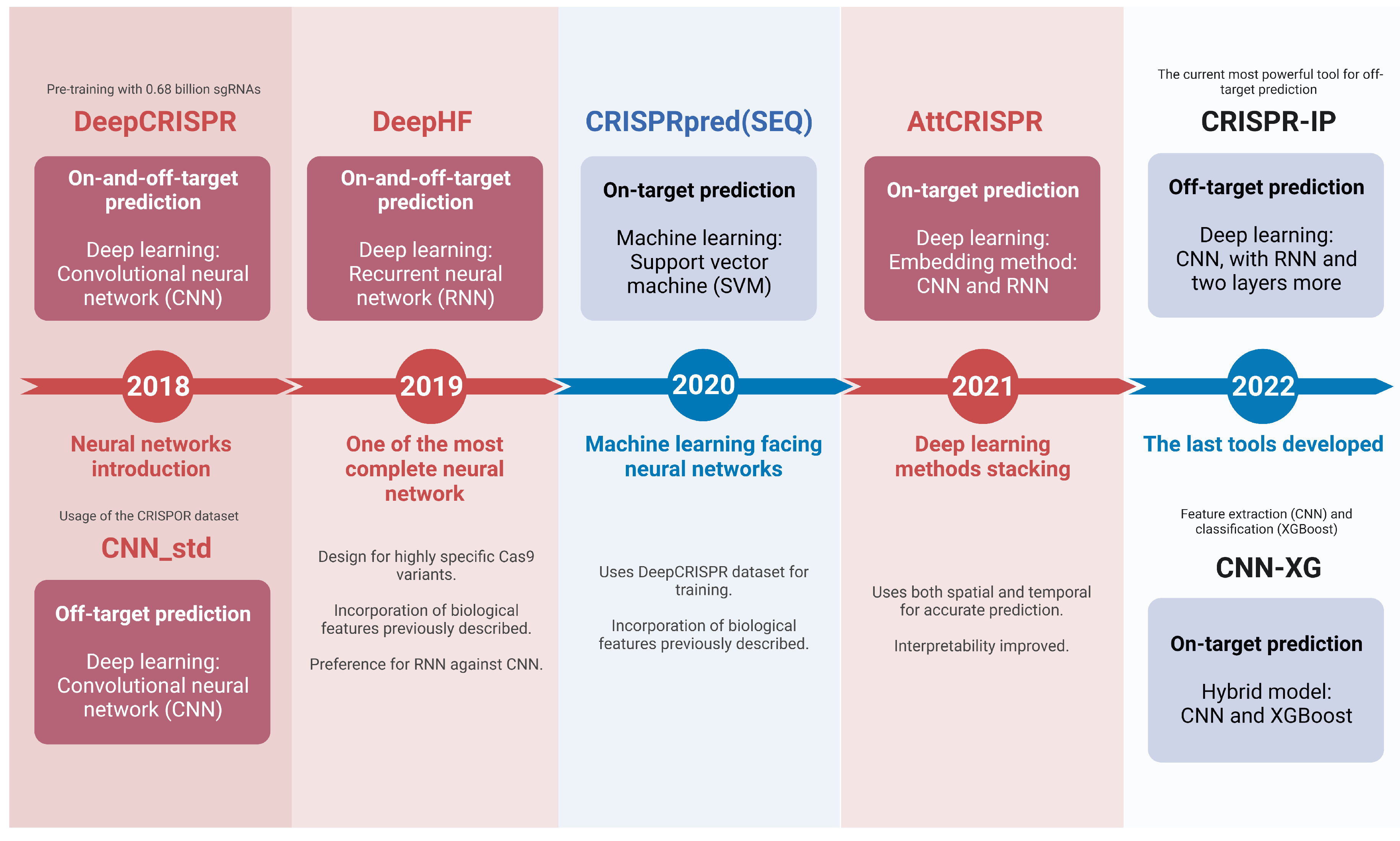
2. Machine Learning in gRNA Design
3. Advances in Neural Networks for gRNA Design
4. Evaluating Model Metrics
5. Reaching Efficiency: A Journey of Optimization
6. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Niazi, S.K. Handbook of Biogeneric Therapeutic Proteins; Taylor & Francis Group: New York, NY, USA, 2006; p. 585. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Ding, Y.; Li, H.; Chen, L.L.; Xie, K. Recent advances in genome editing using CRISPR/Cas9. Front. Plant Sci. 2016, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Weiskittel, T.M.; Correia, C.; Yu, G.T.; Ung, C.Y.; Kaufmann, S.H.; Billadeau, D.D.; Li, H. The trifecta of single-cell, systems-biology, and machine-learning approaches. Genes 2021, 12, 1098. [Google Scholar] [CrossRef] [PubMed]
- Hudson, I.L. Data integration using advances in machine learning in drug discovery and molecular biology. In Artificial Neural Networks; Springer: New York, NY, USA, 2021; pp. 167–184. [Google Scholar]
- Angermueller, C.; Pärnamaa, T.; Parts, L.; Stegle, O. Deep learning for computational biology. Mol. Syst. Biol. 2016, 12, 878. [Google Scholar] [CrossRef]
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakatura, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isoenzyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [CrossRef]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef]
- Mohamadi, S.; Bostanabad, S.Z.; Mirnejad, R. CRISPR arrays: A review on its mechanism. J. Appl. Biotechnol. Rep. 2020, 7, 81–86. [Google Scholar] [CrossRef]
- Wright, A.V.; Liu, J.J.; Knott, G.J.; Doxzen, K.W.; Nogales, E.; Doudna, J.A. Structures of the CRISPR genome integration complex. Science 2017, 357, 1113–1118. [Google Scholar] [CrossRef]
- Nuñez, J.K.; Lee, A.S.; Engelman, A.; Doudna, J.A. Integrase-mediated spacer acquisition during CRISPR-Cas adaptive immunity. Nature 2015, 519, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Ng, S.; Hyun Nam, K.; Ke, A. How type II CRISPR-Cas establish immunity through Cas1-Cas2-mediated spacer integration. Nature 2017, 550, 137–141. [Google Scholar] [CrossRef]
- Rath, D.; Amlinger, L.; Rath, A.; Lundgren, M. The CRISPR-Cas immune system: Biology, mechanisms and applications. Biochimie 2015, 117, 119–128. [Google Scholar] [CrossRef]
- Jinek, M.; Jiang, F.; Taylor, D.W.; Sternberg, S.H.; Kaya, E.; Ma, E.; Anders, C.; Hauer, M.; Zhou, K.; Lin, S.; et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 2014, 343, 1247997. [Google Scholar] [CrossRef] [PubMed]
- Nishimasu, H.; Ran, F.A.; Hsu, P.D.; Konermann, S.; Shehata, S.I.; Dohmae, N.; Ishitani, R.; Zhang, F.; Nureki, O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 2014, 156, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.; Niewoehner, O.; Duerst, A.; Jinek, M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 2014, 513, 569–573. [Google Scholar] [CrossRef]
- Jiang, F.; Zhou, K.; Ma, L.; Gressel, S.; Doudna, J.A. A Cas9-guide RNA complex preorganized for target DNA recognition. Science 2015, 348, 1477–1481. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA–Guided. Science 2012, 337, 816–822. [Google Scholar] [CrossRef]
- Cho, S.W.; Kim, S.; Kim, Y.; Kweon, J.; Kim, H.S.; Bae, S.; Kim, J.S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2013, 24, 132–141. [Google Scholar] [CrossRef]
- Wu, X.; Kriz, A.J.; Sharp, P.A. Target specificity of the CRISPR-Cas9 system. Quant. Biol. 2014, 2, 59–70. [Google Scholar] [CrossRef]
- Zhang, X.H.; Tee, L.Y.; Wang, X.G.; Huang, Q.S.; Yang, S.H. Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol. Ther. Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef]
- Jiang, W.; Bikard, D.; Cox, D.; Zhang, F.; Marraffini, L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013, 31, 233–239. [Google Scholar] [CrossRef]
- Manghwar, H.; Li, B.; Ding, X.; Hussain, A.; Lindsey, K.; Zhang, X.; Jin, S. CRISPR/Cas Systems in Genome Editing: Methodologies and Tools for sgRNA Design, Off-Target Evaluation, and Strategies to Mitigate Off-Target Effects. Adv. Sci. 2020, 7, 1902312. [Google Scholar] [CrossRef]
- Zhang, Z.R.; Jiang, Z.R. Effective use of sequence information to predict CRISPR-Cas9 off-target. Comput. Struct. Biotechnol. J. 2022, 20, 650–661. [Google Scholar] [CrossRef] [PubMed]
- Niu, R.; Peng, J.; Zhang, Z.; Shang, X. R-CRISPR: A deep learning network to predict off-target activities with mismatch, insertion and deletion in CRISPR-Cas9 system. Genes 2021, 12, 1878. [Google Scholar] [CrossRef]
- Borrelli, V.M.; Brambilla, V.; Rogowsky, P.; Marocco, A.; Lanubile, A. The enhancement of plant disease resistance using crispr/cas9 technology. Front. Plant Sci. 2018, 9, 1245. [Google Scholar] [CrossRef]
- Fu, Y.; Sander, J.D.; Reyon, D.; Cascio, V.M.; Joung, J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014, 32, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Doench, J.G.; Hartenian, E.; Graham, D.B.; Tothova, Z.; Hegde, M.; Smith, I.; Sullender, M.; Ebert, B.L.; Xavier, R.J.; Root, D.E. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat. Biotechnol. 2014, 32, 1262–1267. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343, 80–84. [Google Scholar] [CrossRef]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef]
- Pattanayak, V.; Lin, S.; Guilinger, J.P.; Ma, E.; Doudna, J.A.; Liu, D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013, 31, 839–843. [Google Scholar] [CrossRef]
- Konstantakos, V.; Nentidis, A.; Krithara, A.; Paliouras, G. CRISPR-Cas9 gRNA efficiency prediction: An overview of predictive tools and the role of deep learning. Nucleic Acids Res. 2022, 50, 3616–3637. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, C.; Wang, B.; Li, B.; Wang, Q.; Liu, D.; Wang, H.; Zhou, Y.; Shi, L.; Lan, F.; et al. Optimized CRISPR guide RNA design for two high-fidelity Cas9 variants by deep learning. Nat. Commun. 2019, 10, 4284. [Google Scholar] [CrossRef] [PubMed]
- Chollet, F. Deep Learning with Python; Simon and Schuster: New York, NY, USA, 2021. [Google Scholar]
- Volk, M.J.; Lourentzou, I.; Mishra, S.; Vo, L.T.; Zhai, C.; Zhao, H. Biosystems Design by Machine Learning. ACS Synth. Biol. 2020, 9, 1514–1533. [Google Scholar] [CrossRef] [PubMed]
- Alkhnbashi, O.S.; Meier, T.; Mitrofanov, A.; Backofen, R.; Voß, B. CRISPR-Cas bioinformatics. Methods 2020, 172, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Choong, A.C.H.; Lee, N.K. Evaluation of convolutionary neural networks modeling of DNA sequences using ordinal versus one-hot encoding method. In Proceedings of the 2017 International Conference on Computer and Drone Applications (IConDA), Kuching, Malaysia, 9–11 November 2017. [Google Scholar] [CrossRef]
- Lin, J.; Wong, K.C. Off-target predictions in CRISPR-Cas9 gene editing using deep learning. Bioinformatics 2018, 34, i656–i663. [Google Scholar] [CrossRef]
- Yang, K.K.; Wu, Z.; Bedbrook, C.N.; Arnold, F.H. Learned protein embeddings for machine learning. Bioinformatics 2018, 34, 2642–2648. [Google Scholar] [CrossRef]
- Gunasekaran, H.; Ramalakshmi, K.; Arokiaraj, A.R.M.; Kanmani, S.D.; Venkatesan, C.; Dhas, C.S.G. Analysis of DNA Sequence Classification Using CNN and Hybrid Models. Comput. Math. Methods Med. 2021, 2021, 1835056. [Google Scholar] [CrossRef]
- Mathur, I. Predicting Off-Target Potential of CRISPR-Cas9 Single Guide RNA. Master’s Thesis, San Jose State University, San Jose, CA, USA, 2015. [Google Scholar] [CrossRef]
- Wu, X.; Scott, D.A.; Kriz, A.J.; Chiu, A.C.; Hsu, P.D.; Dadon, D.B.; Cheng, A.W.; Trevino, A.E.; Konermann, S.; Chen, S.; et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat. Biotechnol. 2014, 32, 670–676. [Google Scholar] [CrossRef]
- Moreno-Mateos, M.A.; Vejnar, C.E.; Beaudoin, J.D.; Fernandez, J.P.; Mis, E.K.; Khokha, M.K.; Giraldez, A.J. CRISPRscan: Designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat. Methods 2015, 12, 982–988. [Google Scholar] [CrossRef]
- Labuhn, M.; Adams, F.F.; Ng, M.; Knoess, S.; Schambach, A.; Charpentier, E.M.; Schwarzer, A.; Mateo, J.L.; Klusmann, J.H.; Heckl, D. Refined sgRNA efficacy prediction improves largeand small-scale CRISPR-Cas9 applications. Nucleic Acids Res. 2018, 46, 1375–1385. [Google Scholar] [CrossRef]
- Xu, H.; Xiao, T.; Chen, C.H.; Li, W.; Meyer, C.A.; Wu, Q.; Wu, D.; Cong, L.; Zhang, F.; Liu, J.S.; et al. Sequence determinants of improved CRISPR sgRNA design. Genome Res. 2015, 25, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, N.; Khare, S.; Walambe, R.; Kotecha, K. Comparison of machine learning and deep learning techniques in promoter prediction across diverse species. PeerJ Comput. Sci. 2021, 7, e365. [Google Scholar] [CrossRef] [PubMed]
- Abadi, S.; Yan, W.X.; Amar, D.; Mayrose, I. A machine learning approach for predicting CRISPR-Cas9 cleavage efficiencies and patterns underlying its mechanism of action. PLoS Comput. Biol. 2017, 13, e1005807. [Google Scholar] [CrossRef] [PubMed]
- Listgarten, J.; Weinstein, M.; Kleinstiver, B.P.; Sousa, A.A.; Joung, J.K.; Crawford, J.; Gao, K.; Hoang, L.; Elibol, M.; Doench, J.G.; et al. Prediction of off-target activities for the end-to-end design of CRISPR guide RNAs. Nat. Biomed. Eng. 2018, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Lazzarotto, C.R.; Malinin, N.L.; Li, Y.; Zhang, R.; Yang, Y.; Lee, G.H.; Cowley, E.; He, Y.; Lan, X.; Jividen, K.; et al. CHANGE-seq reveals genetic and epigenetic effects on CRISPR–Cas9 genome-wide activity. Nat. Biotechnol. 2020, 38, 1317–1327. [Google Scholar] [CrossRef]
- Wong, N.; Liu, W.; Wang, X. WU-CRISPR: Characteristics of functional guide RNAs for the CRISPR/Cas9 system. Genome Biol. 2015, 16, 1–8. [Google Scholar] [CrossRef]
- Chari, R.; Mali, P.; Moosburner, M.; Church, G.M. Unraveling CRISPR-Cas9 genome engineering parameters via a library-on-library approach. Nat. Methods 2015, 12, 823–826. [Google Scholar] [CrossRef]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef]
- Kaur, K.; Gupta, A.K.; Rajput, A.; Kumar, M. Ge-CRISPR—An integrated pipeline for the prediction and analysis of sgRNAs genome editing efficiency for CRISPR/Cas system. Sci. Rep. 2016, 6, 30870. [Google Scholar] [CrossRef]
- Peng, H.; Zheng, Y.; Zhao, Z.; Liu, T.; Li, J. Recognition of CRISPR/Cas9 off-target sites through ensemble learning of uneven mismatch distributions. Bioinformatics 2018, 34, i757–i765. [Google Scholar] [CrossRef]
- Thongsuwan, S.; Jaiyen, S.; Padcharoen, A.; Agarwal, P. ConvXGB: A new deep learning model for classification problems based on CNN and XGBoost. Nucl. Eng. Technol. 2021, 53, 522–531. [Google Scholar] [CrossRef]
- Dimauro, G.; Barletta, V.S.; Catacchio, C.R.; Colizzi, L.; Maglietta, R.; Ventura, M. A systematic mapping study on machine learning techniques for the prediction of CRISPR/Cas9 sgRNA target cleavage. Comput. Struct. Biotechnol. J. 2022, 20, 5813–5823. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Dai, Z.; Dai, X. C-RNNCrispr: Prediction of CRISPR/Cas9 sgRNA activity using convolutional and recurrent neural networks. Comput. Struct. Biotechnol. J. 2020, 18, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Mukaka, M. Statistics corner: A guide to appropriate use of correlation in medical research. Malawi Med. J. 2012, 24, 69–71. [Google Scholar]
- Thirumalai, C.; Chandhini, S.A.; Vaishnavi, M. Analysing the concrete compressive strength using Pearson and Spearman. In Proceedings of the 2017 International Conference of Electronics, Communication and Aerospace Technology (iCECA), Coimbatore, India, 20–22 April 2017; Volume 2, pp. 215–218. [Google Scholar]
- Zhang, S.; Li, X.; Lin, Q.; Wong, K.C. Synergizing CRISPR/Cas9 off-target predictions for ensemble insights and practical applications. Bioinformatics 2019, 35, 1108–1115. [Google Scholar] [CrossRef]
- Xiao, L.M.; Wan, Y.Q.; Jiang, Z.R. AttCRISPR: A spacetime interpretable model for prediction of sgRNA on-target activity. BMC Bioinform. 2021, 22, 589. [Google Scholar] [CrossRef]
- Xiang, X.; Corsi, G.I.; Anthon, C.; Qu, K.; Pan, X.; Liang, X.; Han, P.; Dong, Z.; Liu, L.; Zhong, J.; et al. Enhancing CRISPR-Cas9 gRNA efficiency prediction by data integration and deep learning. Nat. Commun. 2021, 12, 3238. [Google Scholar] [CrossRef]
- Hancock, J.T.; Khoshgoftaar, T.M.; Johnson, J.M. Evaluating classifier performance with highly imbalanced Big Data. J. Big Data 2023, 10, 42. [Google Scholar] [CrossRef]
- Störtz, F.; Mak, J.; Minary, P. piCRISPR: Physically Informed Features Improve Deep Learning Models for CRISPR/Cas9 Off-Target Cleavage Prediction. bioRxiv 2021. [Google Scholar] [CrossRef]
- Haeussler, M.; Schönig, K.; Eckert, H.; Eschstruth, A.; Mianné, J.; Renaud, J.B.; Schneider-Maunoury, S.; Shkumatava, A.; Teboul, L.; Kent, J.; et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016, 17, 1–12. [Google Scholar] [CrossRef]
- Chuai, G.; Ma, H.; Yan, J.; Chen, M.; Hong, N.; Xue, D.; Zhou, C.; Zhu, C.; Chen, K.; Duan, B.; et al. DeepCRISPR: Optimized CRISPR guide RNA design by deep learning. Genome Biol. 2018, 19, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Dhanjal, J.K.; Dammalapati, S.; Pal, S.; Sundar, D. Evaluation of off-targets predicted by sgRNA design tools. Genomics 2020, 112, 3609–3614. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Zhang, Z.; Zhang, S.; Chen, J.; Wong, K.C. CRISPR-Net: A Recurrent Convolutional Network Quantifies CRISPR Off-Target Activities with Mismatches and Indels. Adv. Sci. 2020, 7, 1903562. [Google Scholar] [CrossRef]
- Li, V.R.; Zhang, Z.; Troyanskaya, O.G. CROTON: An automated and variant-aware deep learning framework for predicting CRISPR/Cas9 editing outcomes. Bioinformatics 2021, 37, i342–i348. [Google Scholar] [CrossRef] [PubMed]
- Chari, R.; Yeo, N.C.; Chavez, A.; Church, G.M. SgRNA Scorer 2.0: A Species-Independent Model to Predict CRISPR/Cas9 Activity. ACS Synth. Biol. 2017, 6, 902–904. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.K.; Rahman, M.S. CRISPRpred: A flexible and efficient tool for sgRNAs on-target activity prediction in CRISPR/Cas9 systems. PLoS ONE 2017, 12, e0181943. [Google Scholar] [CrossRef]
- Kim, H.K.; Min, S.; Song, M.; Jung, S.; Choi, J.W.; Kim, Y.; Lee, S.; Yoon, S.; Kim, H. Deep learning improves prediction of CRISPR-Cpf1 guide RNA activity. Nat. Biotechnol. 2018, 36, 239–241. [Google Scholar] [CrossRef]
- Wilson, L.O.; Reti, D.; O’Brien, A.R.; Dunne, R.A.; Bauer, D.C. High activity target-site identification using phenotypic independent CRISPR-Cas9 core functionality. CRISPR J. 2018, 1, 182–190. [Google Scholar] [CrossRef]
- Rafid, A.H.M.; Toufikuzzaman, M.; Rahman, M.S.; Rahman, M.S. CRISPRpred(SEQ): A sequence-based method for sgRNA on target activity prediction using traditional machine learning. BMC Bioinform. 2020, 21, 223. [Google Scholar] [CrossRef]
- Wang, J.; Xiang, X.; Bolund, L.; Zhang, X.; Cheng, L.; Luo, Y. GNL-Scorer: A generalized model for predicting CRISPR on-target activity by machine learning and featurization. J. Mol. Cell Biol. 2020, 12, 909–911. [Google Scholar] [CrossRef]
- Li, B.; Ai, D.; Liu, X. CNN-XG: A Hybrid Framework for sgRNA On-Target Prediction. Biomolecules 2022, 12, 409. [Google Scholar] [CrossRef] [PubMed]
- Platt, R.J.; Chen, S.; Zhou, Y.; Yim, M.J.; Swiech, L.; Kempton, H.R.; Dahlman, J.E.; Parnas, O.; Eisenhaure, T.M.; Jovanovic, M.; et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 2014, 159, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Chen, S.; Yin, H.; Tammela, T.; Papagiannakopoulos, T.; Joshi, N.S.; Cai, W.; Yang, G.; Bronson, R.; Crowley, D.G.; et al. CRISPR-mediated direct mutation of cancer genes in the mouse liver. Nature 2014, 514, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Montague, T.G.; Cruz, J.M.; Gagnon, J.A.; Church, G.M.; Valen, E. CHOPCHOP: A CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 2014, 42, W401–W407. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, 1–10. [Google Scholar] [CrossRef]
- Lei, Y.; Lu, L.; Liu, H.Y.; Li, S.; Xing, F.; Chen, L.L. CRISPR-P: A web tool for synthetic single-guide RNA design of CRISPR-system in plants. Mol. Plant 2014, 7, 1494–1496. [Google Scholar] [CrossRef]
- Zhang, W.W.; Matlashewski, G. CRISPR-Cas9-mediated genome editing in Leishmania donovani. mBio 2015, 6, e00861-15. [Google Scholar] [CrossRef]
- Paix, A.; Folkmann, A.; Rasoloson, D.; Seydoux, G. High efficiency, homology-directed genome editing in Caenorhabditis elegans using CRISPR-Cas9ribonucleoprotein complexes. Genetics 2015, 201, 47–54. [Google Scholar] [CrossRef]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015, 33, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Radzisheuskaya, A.; Shlyueva, D.; Müller, I.; Helin, K. Optimizing sgRNA position markedly improves the efficiency of CRISPR/dCas9-mediated transcriptional repression. Nucleic Acids Res. 2016, 44, e141. [Google Scholar] [CrossRef]
- Liang, G.; Zhang, H.; Lou, D.; Yu, D. Selection of highly efficient sgRNAs for CRISPR/Cas9-based plant genome editing. Sci. Rep. 2016, 6, 21451. [Google Scholar] [CrossRef] [PubMed]
- Janga, M.R.; Campbell, L.M.; Rathore, K.S. CRISPR/Cas9-mediated targeted mutagenesis in upland cotton (Gossypium hirsutum L.). Plant Mol. Biol. 2017, 94, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Baysal, C.; Bortesi, L.; Zhu, C.; Farré, G.; Schillberg, S.; Christou, P. CRISPR/Cas9 activity in the rice OsBEIIb gene does not induce off-target effects in the closely related paralog OsBEIIa. Mol. Breed. 2016, 36, 1–11. [Google Scholar] [CrossRef]
- Thyme, S.B.; Akhmetova, L.; Montague, T.G.; Valen, E.; Schier, A.F. Internal guide RNA interactions interfere with Cas9-mediated cleavage. Nat. Commun. 2016, 7, 11750. [Google Scholar] [CrossRef]
- Gundry, M.C.; Brunetti, L.; Lin, A.; Mayle, A.E.; Kitano, A.; Wagner, D.; Hsu, J.I.; Hoegenauer, K.A.; Rooney, C.M.; Goodell, M.A.; et al. Highly Efficient Genome Editing of Murine and Human Hematopoietic Progenitor Cells by CRISPR/Cas9. Cell Rep. 2016, 17, 1453–1461. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Morineau, C.; Bellec, Y.; Tellier, F.; Gissot, L.; Kelemen, Z.; Nogué, F.; Faure, J.D. Selective gene dosage by CRISPR-Cas9 genome editing in hexaploid Camelina sativa. Plant Biotechnol. J. 2017, 15, 729–739. [Google Scholar] [CrossRef]
- Shen, J.; Zhou, J.; Chen, G.Q.; Xiu, Z.L. Efficient genome engineering of a virulent Klebsiella bacteriophage using CRISPR-Cas9. J. Virol. 2018, 92, e00534-18. [Google Scholar] [CrossRef]
- Mintz, R.L.; Lao, Y.H.; Chi, C.W.; He, S.; Li, M.; Quek, C.H.; Shao, D.; Chen, B.; Han, J.; Wang, S.; et al. CRISPR/Cas9-mediated mutagenesis to validate the synergy between PARP1 inhibition and chemotherapy in BRCA1-mutated breast cancer cells. Bioeng. Transl. Med. 2020, 5, e10152. [Google Scholar] [CrossRef]
- Gu, X.; Wang, D.; Xu, Z.; Wang, J.; Guo, L.; Chai, R.; Li, G.; Shu, Y.; Li, H. Prevention of acquired sensorineural hearing loss in mice by in vivo Htra2 gene editing. Genome Biol. 2021, 22, 1–23. [Google Scholar] [CrossRef]
- Ferrari Dacrema, M.; Cremonesi, P.; Jannach, D. Are we really making much progress? A worrying analysis of recent neural recommendation approaches. In Proceedings of the 13th ACM Conference on Recommender Systems, Copenhagen, Denmark, 16–20 September 2019; pp. 101–109. [Google Scholar]
- Ren, X.; Guo, H.; Li, S.; Wang, S.; Li, J. A novel image classification method with CNN-XGBoost model. In Proceedings of the Digital Forensics and Watermarking: 16th International Workshop, IWDW 2017, Magdeburg, Germany, 23–25 August 2017; Proceedings 16; Springer: Berlin/Heidelberg, Germany, 2017; pp. 378–390. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Model | Year | Evaluation Metric | Detail | Reference |
|---|---|---|---|---|---|
| CRISPRtool/MIT Design | Conventional non-learning | 2013 | NA | The first tool for gRNA design. | [22] |
| CRISPOR | Self-assembled algorithm | 2016 | Spearman: 0.71–0.77. AUROC: 0.91 | Datasets of gRNA sequences and their off-targets from different studies. | [66] |
| CRISTA | RF | 2017 | Spearman: 0.81. AUROC: 0.96. AUPRC: 0.96 | Assembled dataset for training and validation: GUIDE-Seq, HTGTS, and BLESS. | [48] |
| Predict CRISPR | SVM | 2018 | AUROC: 0.99. AUPRC: 0.45 | One-hot encoding over the Haeussler dataset. | [55] |
| Elevation | DT | 2018 | Spearman: 0.98 | One-hot encoding over GUIDE-seq. Boench V2 and Haeussler. | [49] |
| DeepCRISPR | CNN | 2018 | Spearman: 0.246. AUROC: 0.804, AUPRC: 0.303 | One-hot encoding over GUIDE-seq data. | [67] |
| CNN_std | CNN | 2018 | AUROC: 0.972 | One-hot encoding over CRISPOR dataset and GUIDE-seq dataset. | [39] |
| SynergizingCRISPR | FNN, RF, SVM, DT | 2019 | Spearman: 0.938. AUPRC: 0.299 | GUIDE-Seq and Haeussler dataset. | [61] |
| CHANGE-seq | DT | 2020 | AUROC: 0.995. AUPRC: 0.881 | One-hot encoding. | [50] |
| CRISPcut | LG, RF, DT. | 2020 | Accuracy: 0.9149. AUROC: 0.97 | One-hot encoding over CIRCLE-seq and CRISPcup. | [68] |
| CRISPR-Net | RNN-CNN | 2020 | AUROC: 0.969. AUPRC: 0.477 | One-hot encoding over CIRCLE-Seq and GUIDE-seq datasets. | [69] |
| R-CRISPER | RNN | 2021 | AUROC: 0.991. AUPRC: 0.319 | One-hot encoding over CIRCLE-Seq, SITE and GUIDE datasets. | [26] |
| piCRISPR | RNN-CNN | 2021 | AUROC: 0.995. AUPRC: 0.725 | One-hot encoding over crisprSQL dataset. | [65] |
| CROTON | CNN | 2021 | AUROC: 0.94 | One-hot encoding over FORECasT and SPROUT datasets. | [70] |
| AttCRISPR | Embedding method | 2021 | Spearman: 0.872 | One-hot encoding over DeepHF dataset. | [62] |
| CRISPR-IP | CNN | 2022 | Accuracy: 0.990. AUROC: 0.982. AUPRC: 0.751 | CIRCLE-Seq dataset and SITE-Seq dataset. | [25] |
| Name | Model | Year | Evaluation Metric | Detail | Reference |
|---|---|---|---|---|---|
| Broad GPP | LG | 2014 | Spearman: 0.87 | One-hot encoding over 1841 gRNAs. | [29] |
| WU-CRISPR | SVM | 2015 | AUROC 0.91. Spearman 0.70 | Doench and Chari datasets. | [51] |
| SSC | LG | 2015 | AUROC : 0.729 | Wang, Koike-Yusa, Shalcm, Zhou, Gilbert and Konermann datasets. | [46] |
| CRISPRScan | LR | 2015 | Spearman: 0.68 | 1280 gRNAs in the zebrafish genome. | [44] |
| SgRNAScorer | SVM | 2015 | Accuracy: 0.737. AUPRC: 0.758 | SpCas9 and St1Cas9 datasets. | [52] |
| Azimuth | SVM, LG | 2016 | AUROC: 0.80 | One-hot encoding over Avana and GeCKO libraries. | [53] |
| ge-CRISPR | SVM | 2016 | AUROC: 0.54–0.93 | sgRNAdesigner, CRISPRScan and sgRNAscorer datasets. | [54] |
| CRISPRater | LR | 2017 | Spearman 0.67 | Wang, Koike-Yusa and Xu datasets. | [45] |
| SgRNAScorer 2.0 | SVM | 2017 | Accuracy: 0.737. AUPRC: 0.758 | SpCas9 and St1Cas libraries. | [71] |
| CRISPRpred | SVM | 2017 | AUROC: 0.85. AUPRC: 0.56 | K-mer encoding over Broad GPP. | [72] |
| DeepCRISPR | CNN | 2018 | AUROC: 0.981. AUPR: 0.497. Spearman: 0.406 | One-hot encoding over 15,000 gRNAs from four different cell lines. | [67] |
| DeepCpf1 | CNN | 2018 | Spearman: 0.87. AUROC: 0.89 | One-hot encoding over different datasets oncluding Kleinstiver, Chari and Kim. | [73] |
| TUSCAN | RF | 2018 | Spearman: 0.8. AUC of 0.63 | Chari, Doench, Horlbeck and Moreno-Mateos databases. | [74] |
| DeepHF | RNN | 2019 | Spearman: 0.867 | One-hot encoding over ten public datasets. | [34] |
| CRISPRpred(SEQ) | SVM | 2020 | Spearman: 0.829. AUROC: 0.893 | Haeussler and DeepHF datasets. | [75] |
| GNL-Scorer | FNN | 2020 | Spearman: 0.502 | One-hot encoding over ten public datasets. | [76] |
| C-RNN CRISPR | CNN-RNN | 2020 | Spearman: 0.877. AUROC: 0.976 | One-hot encoding over Chuai dataset. | [58] |
| On-target CRISPRon | CNN | 2021 | Spearman 0.91 | One-hot encoding over 12 K dataset. | [63] |
| CNN-XG | CNN-Tree | 2022 | Spearman 0.7352 AUROC: 0.992 | Ten public datasets. | [77] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Motoche-Monar, C.; Ordoñez, J.E.; Chang, O.; Gonzales-Zubiate, F.A. gRNA Design: How Its Evolution Impacted on CRISPR/Cas9 Systems Refinement. Biomolecules 2023, 13, 1698. https://doi.org/10.3390/biom13121698
Motoche-Monar C, Ordoñez JE, Chang O, Gonzales-Zubiate FA. gRNA Design: How Its Evolution Impacted on CRISPR/Cas9 Systems Refinement. Biomolecules. 2023; 13(12):1698. https://doi.org/10.3390/biom13121698
Chicago/Turabian StyleMotoche-Monar, Cristofer, Julián E. Ordoñez, Oscar Chang, and Fernando A. Gonzales-Zubiate. 2023. "gRNA Design: How Its Evolution Impacted on CRISPR/Cas9 Systems Refinement" Biomolecules 13, no. 12: 1698. https://doi.org/10.3390/biom13121698