
Cardioprotective Effect against Ischemia–Reperfusion Injury of PAK-200, a Dihydropyridine Analog with an Inhibitory Effect on Cl− but Not Ca2+ Current
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
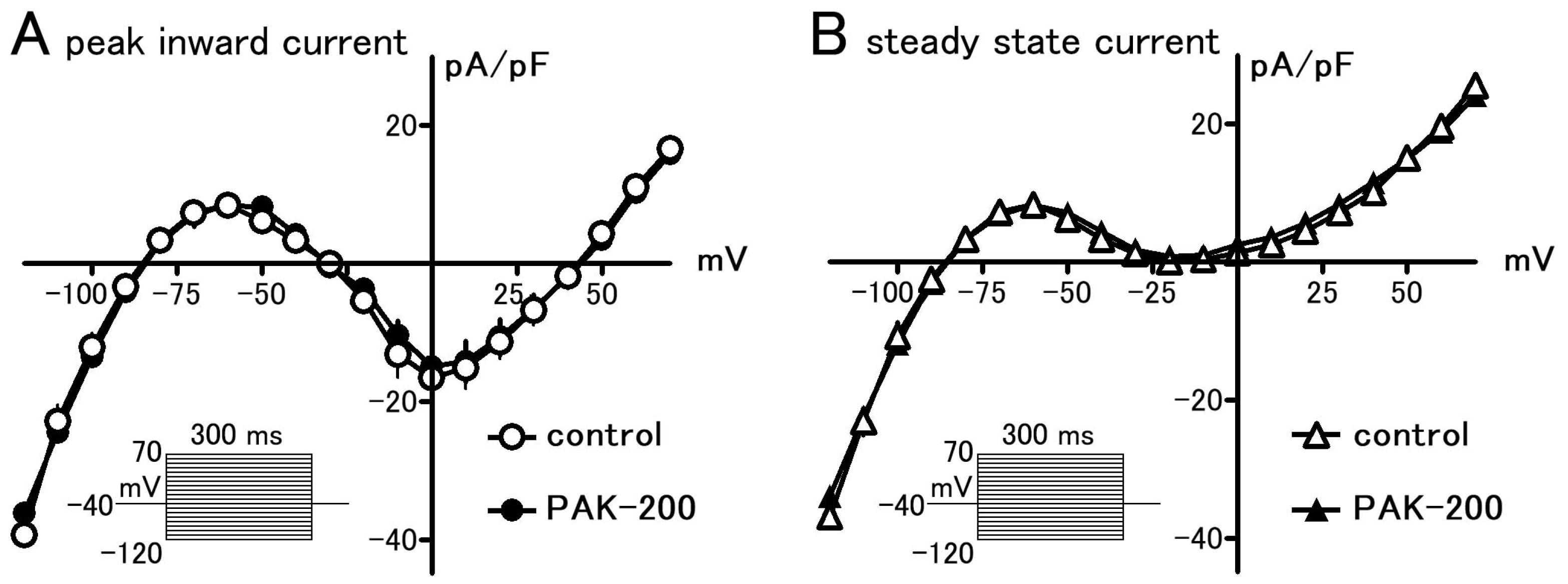
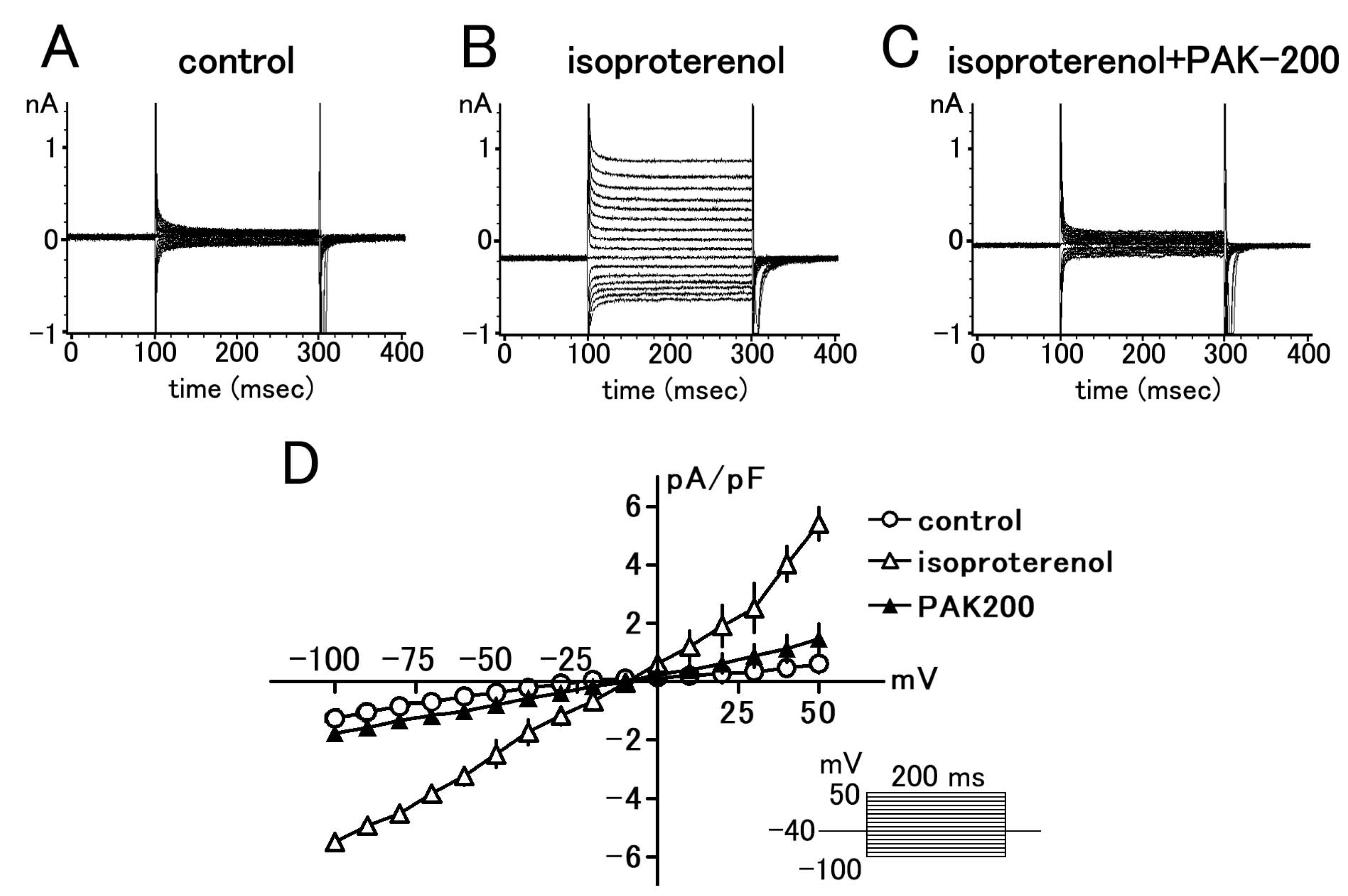
3.1. Effect of PAK-200 on the Peak Inward, Steady-State, and Cl− Currents
3.2. Effect of PAK-200 on the Spontaneous Beating of the Right Atria
3.3. Mechanical Parameters during Ischemia–Reperfusion and the Effect of PAK-200
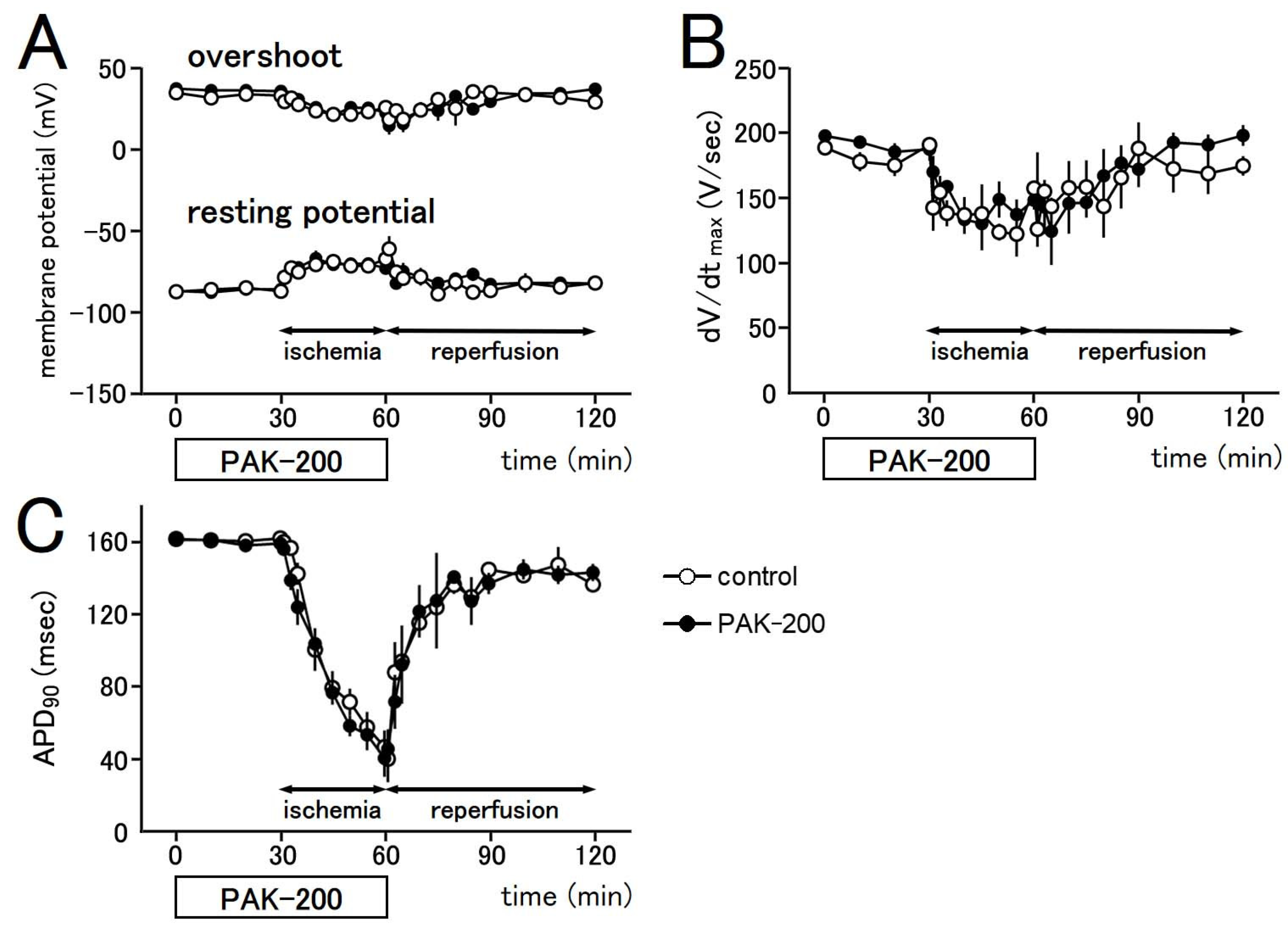
3.4. Electrophysiological Parameters during Ischemia–Reperfusion and the Effect of PAK-200
3.5. Arrhythmic Contractions after Reperfusion and the Effect of PAK-200
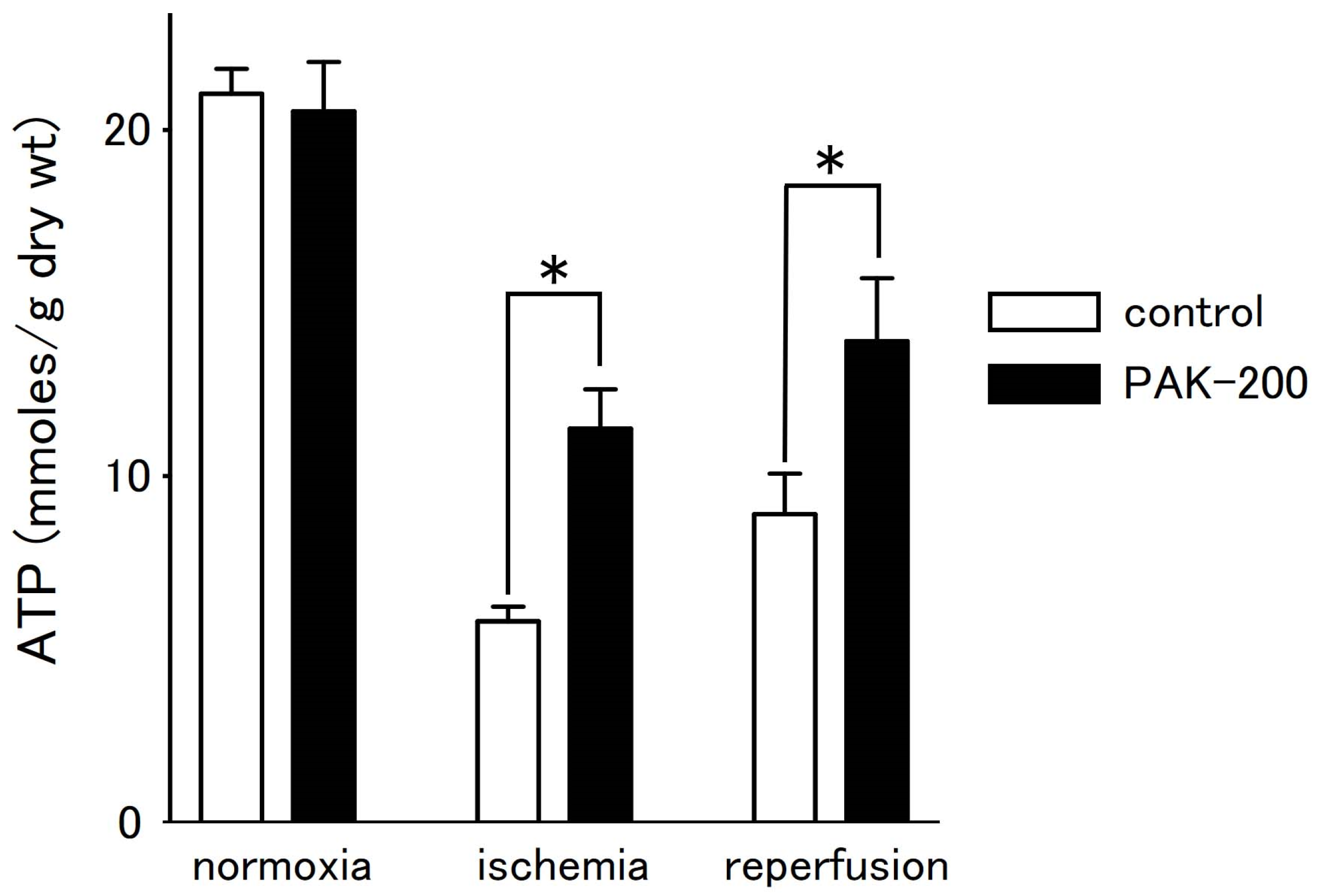
3.6. Tissue ATP Level during Ischemia–Reperfusion and the Effect of PAK-200
3.7. Effect of PAK-200 on Mitochondrial Membrane Potential
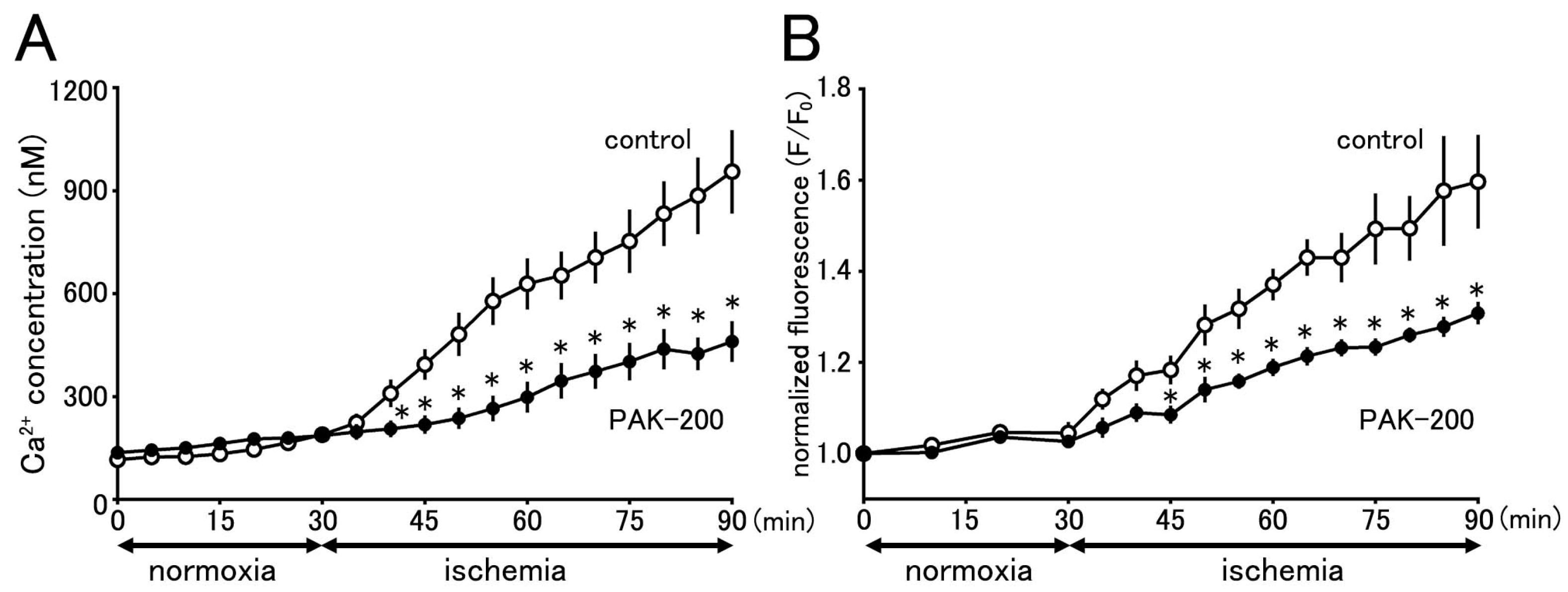
3.8. Cytoplasmic Ca2+ Concentration during Ischemia and the Effect of PAK-200
3.9. Effect of PAK-200 on the Mitochondrial Ca2+ Concentration
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Steenbergen, C.; Murphy, E.; Watts, J.A.; London, R.E. Correlation between cytosolic free calcium, contracture, ATP, and irreversible ischemic injury in perfused rat heart. Circ. Res. 1990, 66, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Tani, M. Mechanisms of Ca2+ overload in reperfused ischemic myocardium. Annu. Rev. Physiol. 1990, 52, 543–559. [Google Scholar] [CrossRef] [PubMed]
- Nayler, W.G.; Ferrari, R.; Williams, A. Protective effect of pretreatment with verapamil, nifedipine and propranolol on mitochondrial function in the ischemic and reperfused myocardium. Am. J. Cardiol. 1980, 46, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Cole, W.C.; McPherson, C.D.; Sontag, D. ATP-regulated K+ channels protect the myocardium against ischemia/reperfusion damage. Circ. Res. 1991, 69, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Okazaki, K.; Shigenobu, K. Cardioprotective effects of NIP-121, a novel ATP-sensitive potassium channel opener, during ischemia and reperfusion in coronary perfused guinea pig myocardium. J. Cardiovasc. Pharmacol. 1996, 27, 695–701. [Google Scholar] [CrossRef]
- Tanaka, H.; Matsui, S.; Shigenobu, K. Cardioprotective effects of beraprost sodium against experimental ischemia and reperfusion as compared with propranolol and diltiazem. Res. Commun. Mol. Pathol. Pharmacol. 1998, 99, 321–327. [Google Scholar]
- Hume, J.R.; Duan, D.; Collier, M.L.; Yamazaki, J.; Horowitz, B. Anion transport in heart. Physiol. Rev. 2000, 80, 31–81. [Google Scholar] [CrossRef]
- Duan, D. Phenomics of cardiac chloride channels: The systematic study of chloride channel function in the heart. J. Physiol. 2009, 587, 2163–2177. [Google Scholar] [CrossRef]
- Adkins, G.B.; Curtis, M.J. Potential role of cardiac chloride channels and transporters as novel therapeutic targets. Pharmacol. Ther. 2015, 145, 67–75. [Google Scholar] [CrossRef]
- Harvey, R.D.; Hume, J.R. Autonomic regulation of a chloride current in heart. Science 1989, 244, 983–985. [Google Scholar] [CrossRef] [PubMed]
- Levesque, P.C.; Hart, P.J.; Hume, J.R.; Kenyon, J.L.; Horowitz, B. Expression of cystic fibrosis transmembrane regulator Cl− channels in heart. Circ. Res. 1992, 71, 1002–1007. [Google Scholar] [CrossRef]
- Ridley, P.D.; Curtis, M.J. Anion manipulation: A new antiarrhythmic approach. Action of substitution of chloride with nitrate on ischemia- and reperfusion-induced ventricular fibrillation and contractile function. Circ. Res. 1992, 70, 617–632. [Google Scholar] [CrossRef] [PubMed]
- Curtis, M.J.; Garlick, P.B.; Ridley, P.D. Anion manipulation, a novel antiarrhythmic approach: Mechanism of action. J. Mol. Cell Cardiol. 1993, 25, 417–436. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.F.; Liu, J.; Nishi, K. Effects of stilbene derivatives SITS and DIDS on development of intracellular acidosis during ischemia in isolated guinea pig ventricular papillary muscle in vitro. Jpn. J. Pharmacol. 1996, 72, 161–174. [Google Scholar] [CrossRef]
- Tanaka, H.; Matsui, S.; Kawanishi, T.; Shigenobu, K. Use of chloride blockers: A novel approach for cardioprotection against ischemia-reperfusion damage. J. Pharmacol. Exp. Ther. 1996, 278, 854–861. [Google Scholar]
- Bryant, S.H.; Morales-Aguilera, A. Chloride conductance in normal and myotonic muscle fibres and the action of monocarboxylic aromatic acids. J. Physiol. 1971, 219, 367–383. [Google Scholar] [CrossRef]
- Payne, J.N.; Lawes, I.N.; Proctor, G.B.; Horobin, R.W. Variation between different samples of SITS with respect to axonal transport and toxicity. Neurosci. Lett. 1983, 42, 229–234. [Google Scholar] [CrossRef]
- Shudo, N.; Fujii, R.; Matsumoto, T.; Mizoguchi, T.; Seto, K.; Sakoda, R.; Akiyama, S. Potentiation of the vincristine effect on P388 mouse leukemia cells by a newly synthesized dihydropyridine analogue, PAK-200. Jpn. J. Cancer Res. 1992, 83, 1011–1017. [Google Scholar] [CrossRef]
- Niwa, K.; Yamada, K.; Furukawa, T.; Shudo, N.; Seto, K.; Matsumoto, T.; Takao, S.; Akiyama, S.; Shimazu, H. Effect of a dihydropyridine analogue, 2-[benzyl(phenyl)amino]ethyl 1,4-dihydro-2,6-dimethyl-5-(5,5-dimethyl-2-oxo-1,3,2-dioxaphosphorinan-2-yl)-1-(2-morpholinoethyl)-4-(3-nitrophenyl)-3-pyridinecarboxylate on reversing in vivo resistance of tumor cells to adriamycin. Cancer Res. 1992, 52, 3655–3660. [Google Scholar]
- Vanhoefer, U.; Muller, M.R.; Hilger, R.A.; Lindtner, B.; Klaassen, U.; Schleucher, N.; Rustum, Y.M.; Seeber, S.; Harstrick, A. Reversal of MDR1-associated resistance to topotecan by PAK-200S, a new dihydropyridine analogue, in human cancer cell lines. Br. J. Cancer 1999, 81, 1304–1310. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.F. ABC transporters: From microorganisms to man. Annu. Rev. Cell Biol. 1992, 8, 67–113. [Google Scholar] [CrossRef]
- Isenberg, G.; Klockner, U. Calcium tolerant ventricular myocytes prepared by preincubation in a “KB medium”. Pflugers Arch. 1982, 395, 6–18. [Google Scholar] [CrossRef] [PubMed]
- McDonald, T.F.; MacLeod, D.P. Anoxia-recovery cycle in ventricular muscle: Action potential duration, contractility and ATP content. Pflugers Arch. 1971, 325, 305–322. [Google Scholar] [CrossRef] [PubMed]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [CrossRef]
- Ishida, H.; Hirota, Y.; Genka, C.; Nakazawa, H.; Nakaya, H.; Sato, T. Opening of mitochondrial KATP channels attenuates the ouabain-induced calcium overload in mitochondria. Circ. Res. 2001, 89, 856–858. [Google Scholar] [CrossRef] [PubMed]
- Satoh, H. Sino-atrial nodal cells of mammalian hearts: Ionic currents and gene expression of pacemaker ionic channels. J. Smooth Muscle Res. 2003, 39, 175–193. [Google Scholar] [CrossRef]
- Sarai, N.; Matsuoka, S.; Kuratomi, S.; Ono, K.; Noma, A. Role of individual ionic current systems in the SA node hypothesized by a model study. Jpn. J. Physiol. 2003, 53, 125–134. [Google Scholar] [CrossRef]
- Tanaka, H.; Sekine, T.; Terada, M.; Kobayashi, Y.; Shigenobu, K. Cardioprotective effect against ischemia-reperfusion injury of AHC-52, a dihydropyridine compound with inhibitory effect on Cl− but not Ca2+ current. Naunyn Schmiedebergs Arch. Pharmacol. 1997, 356, 853–855. [Google Scholar] [CrossRef]
- Steenbergen, C.; Deleeuw, G.; Rich, T.; Williamson, J.R. Effects of acidosis and ischemia on contractility and intracellular pH of rat heart. Circ. Res. 1977, 41, 849–858. [Google Scholar] [CrossRef]
- Kubler, W.; Katz, A.M. Mechanism of early “pump” failure of the ischemic heart: Possible role of adenosine triphosphate depletion and inorganic phosphate accumulation. Am. J. Cardiol. 1977, 40, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Perlman, M.; London, R.E.; Steenbergen, C. Amiloride delays the ischemia-induced rise in cytosolic free calcium. Circ. Res. 1991, 68, 1250–1258. [Google Scholar] [CrossRef] [PubMed]
- Allshire, A.; Piper, H.M.; Cuthbertson, K.S.; Cobbold, P.H. Cytosolic free Ca2+ in single rat heart cells during anoxia and reoxygenation. Biochem. J. 1987, 244, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Steenbergen, C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 2008, 88, 581–609. [Google Scholar] [CrossRef]
- Schramm, M.; Klieber, H.G.; Daut, J. The energy expenditure of actomyosin-ATPase, Ca2+-ATPase and Na+, K+-ATPase in guinea-pig cardiac ventricular muscle. J. Physiol. 1994, 481, 647–662. [Google Scholar] [CrossRef] [PubMed]
- Lue, W.M.; Boyden, P.A. Abnormal electrical properties of myocytes from chronically infarcted canine heart. Alterations in Vmax and the transient outward current. Circulation 1992, 85, 1175–1188. [Google Scholar] [CrossRef]
- Patterson, E.; Scherlag, B.J.; Lazzara, R. Rapid inward current in ischemically-injured subepicardial myocytes bordering myocardial infarction. J. Cardiovasc. Electrophysiol. 1993, 4, 9–22. [Google Scholar] [CrossRef]
- Ye, L.; Zhu, W.; Backx, P.H.; Cortez, M.A.; Wu, J.; Chow, Y.; McKerlie, C.; Wang, A.; Tsui, L.; Gross, G.J.; et al. Arrhythmia and sudden death associated with elevated cardiac chloride channel activity. J. Cell Mol. Med. 2011, 15, 2307–2316. [Google Scholar] [CrossRef]
- Mitchell, P.; Moyle, J. Chemiosmotic hypothesis of oxidative phosphorylation. Nature 1967, 213, 137–139. [Google Scholar] [CrossRef]
- Ruiz-Meana, M.; Garcia-Dorado, D.; Pina, P.; Inserte, J.; Agullo, L.; Soler-Soler, J. Cariporide preserves mitochondrial proton gradient and delays ATP depletion in cardiomyocytes during ischemic conditions. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, 999. [Google Scholar] [CrossRef]
- De Stefani, D.; Raffaello, A.; Terado, E.; Szabò, I.; Rizzuto, R. A 40 kDa protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Namekata, I.; Hamaguchi, S.; Iida-Tanaka, N.; Kusakabe, T.; Kato, K.; Kawanishi, T.; Tanaka, H. Fluorescence analysis of the mitochondrial effect of a plasmalemmal Na+/Ca2+ exchanger inhibitor, SEA0400, in permeabilized H9c2 Cardiomyocytes. Biol. Pharm. Bull. 2017, 40, 1551–1555. [Google Scholar] [CrossRef]
- Matsuoka, S.; Jo, H.; Sarai, N.; Noma, A. An in silico study of energy metabolism in cardiac excitation-contraction coupling. Jpn. J. Physiol. 2004, 54, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Maack, C.; O’Rourke, B. Excitation-contraction coupling and mitochondrial energetics. Basic. Res. Cardiol. 2007, 102, 369–392. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.S.B.; Boyman, L.; Lederer, W.J. Mitochondrial calcium and the regulation of metabolism in the heart. J. Mol. Cell Cardiol. 2015, 78, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Duron, H.E.; Chaudhuri, D. Beyond the TCA cycle: New insights into mitochondrial calcium regulation of oxidative phosphorylation. Biochem. Soc. Trans. 2023, 51, 1661–1673. [Google Scholar] [CrossRef] [PubMed]
- Ramachandra, C.J.A.; Hernandez-Resendiz, S.; Crespo-Avilan, G.E.; Lin, Y.; Hausenloy, D.J. Mitochondria in acute myocardial infarction and cardioprotection. EBioMedicine 2020, 57, 102884. [Google Scholar] [CrossRef]
- Namekata, I.; Hamaguchi, S.; Tanaka, H. Pharmacological discrimination of plasmalemmal and mitochondrial sodium-calcium exchanger in cardiomyocyte-derived H9c2 cells. Biol. Pharm. Bull. 2015, 38, 147–150. [Google Scholar] [CrossRef]
- Sander, P.; Gudermann, T.; Schredelseker, J. A Calcium Guard in the Outer Membrane: Is VDAC a Regulated Gatekeeper of Mitochondrial Calcium Uptake? Int. J. Mol. Sci. 2021, 22, 946. [Google Scholar] [CrossRef]
- Sekine, T.; Kawanishi, T.; Tanaka, H.; Shigenbu, K. Cardioprotective effects and fluorescence profiles of AHC-93, a novel dihydropyridine compound with Cl− current blocking activity. Res. Comm. Pharmacol. Toxicol. 1998, 3, 10007368768. [Google Scholar]
- Budriesi, R.; Ioan, P.; Leoni, A.; Pedemonte, N.; Locatelli, A.; Micucci, M.; Chiarini, A.; Galietta, L.J.V. Cystic fibrosis: A new target for 4-Imidazo[2,1-b]thiazole-1,4-dihydropyridines. J. Med. Chem. 2011, 54, 3885–3894. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Namekata, I.; Tamura, M.; Kase, J.; Hamaguchi, S.; Tanaka, H. Cardioprotective Effect against Ischemia–Reperfusion Injury of PAK-200, a Dihydropyridine Analog with an Inhibitory Effect on Cl− but Not Ca2+ Current. Biomolecules 2023, 13, 1719. https://doi.org/10.3390/biom13121719
Namekata I, Tamura M, Kase J, Hamaguchi S, Tanaka H. Cardioprotective Effect against Ischemia–Reperfusion Injury of PAK-200, a Dihydropyridine Analog with an Inhibitory Effect on Cl− but Not Ca2+ Current. Biomolecules. 2023; 13(12):1719. https://doi.org/10.3390/biom13121719
Chicago/Turabian StyleNamekata, Iyuki, Miku Tamura, Jyunya Kase, Shogo Hamaguchi, and Hikaru Tanaka. 2023. "Cardioprotective Effect against Ischemia–Reperfusion Injury of PAK-200, a Dihydropyridine Analog with an Inhibitory Effect on Cl− but Not Ca2+ Current" Biomolecules 13, no. 12: 1719. https://doi.org/10.3390/biom13121719