

Functional Loss of Terminal Complement Complex Protects Rabbits from Injury-Induced Osteoarthritis on Structural and Cellular Level

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Model and Genetic Characterization
2.2. CH50 Assay and TCC Staining on Erythrocytes
2.3. Western Blot Analysis
2.4. ACLT Model
2.5. Quantification of PGE2 in the Synovial Fluid
2.6. μCT Analysis
2.7. Histopathological Assessment and TUNEL Staining
2.8. Statistical Analyses
3. Results
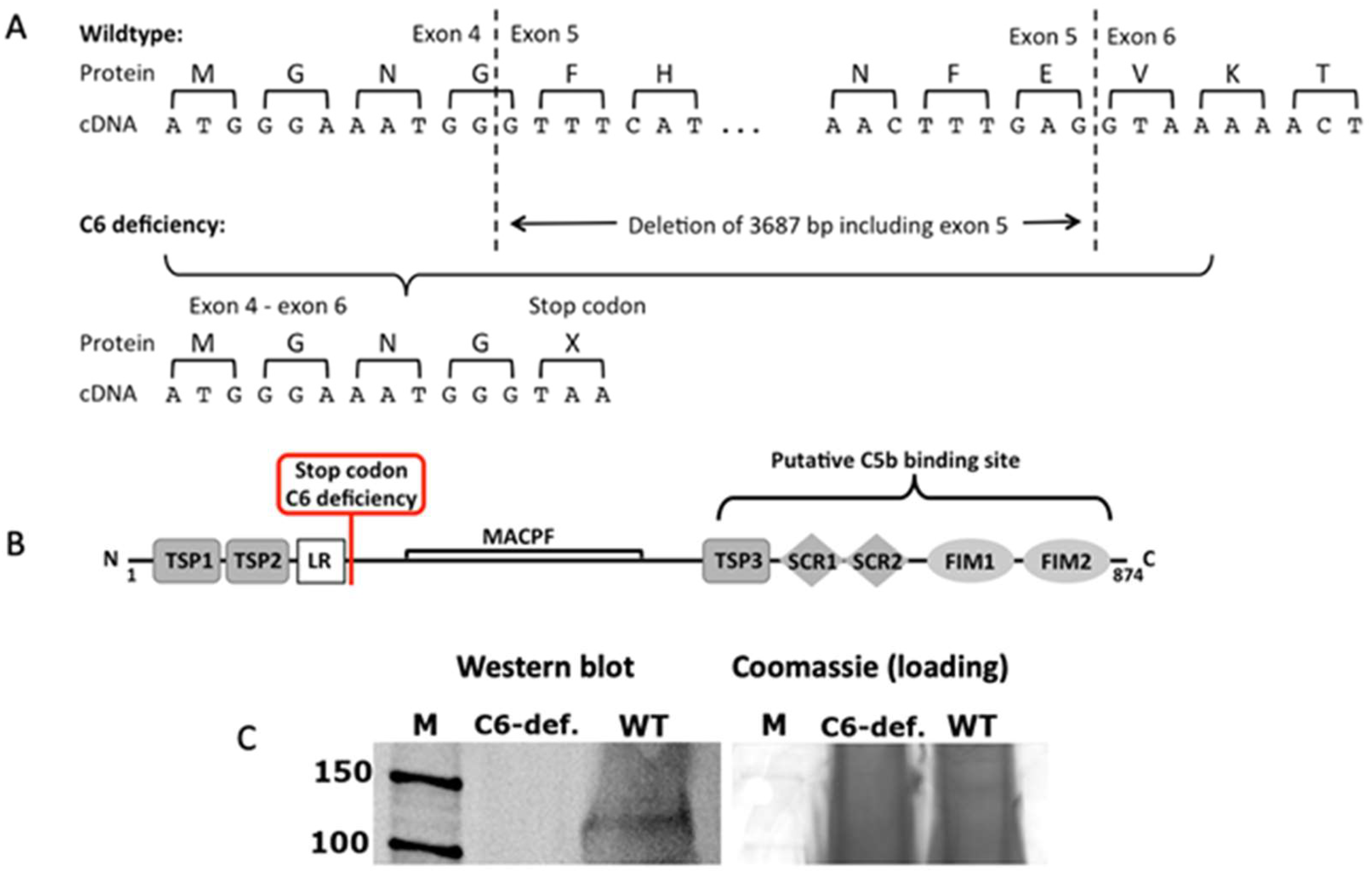
3.1. Inability to Form Functional TCC in C6-Deficient Rabbits Results from the Loss of the C5b Binding Site in C6
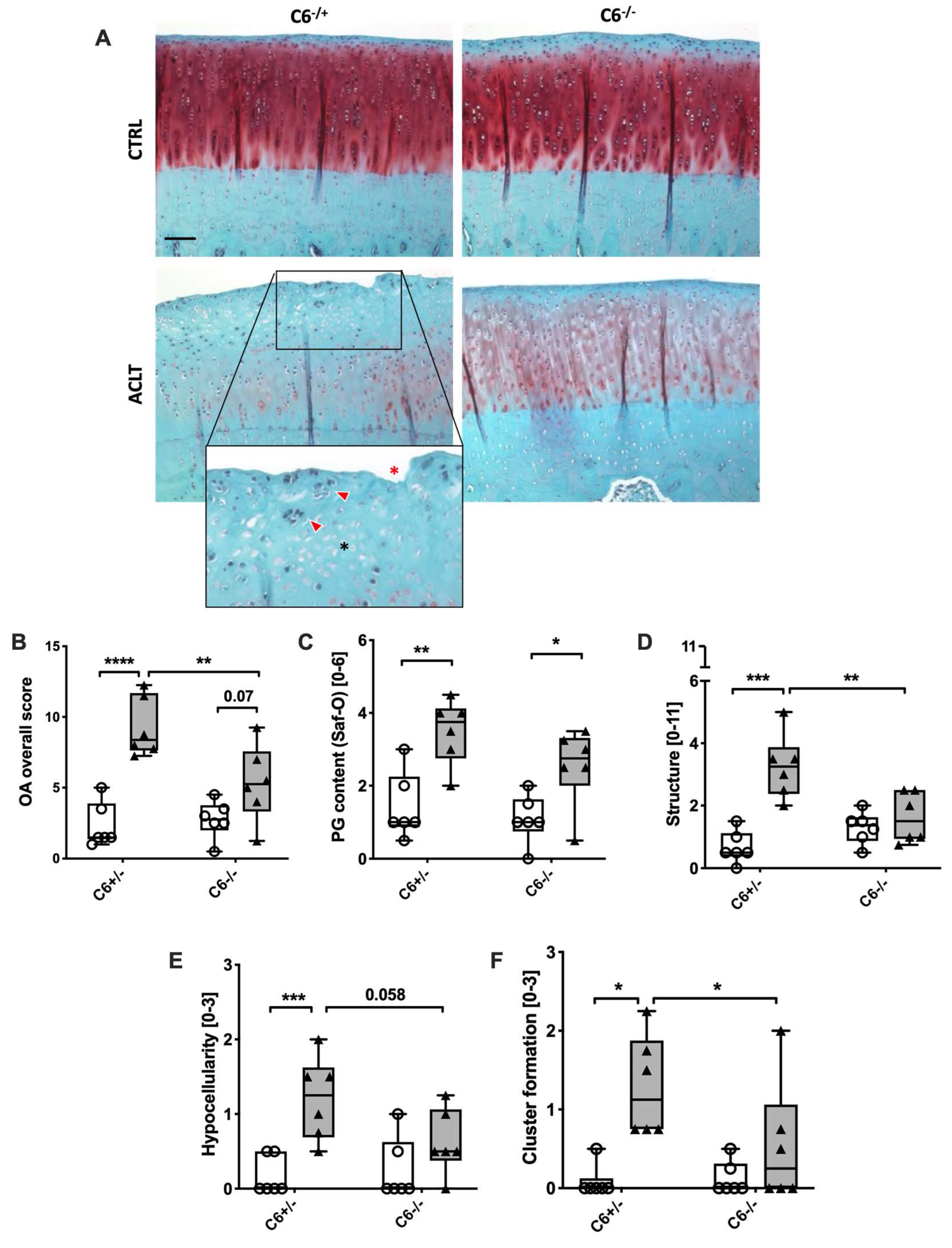
3.2. C6-Deficiency Attenuates Structural Cartilage Damage and Cluster Formation after ACLT Surgery
3.3. C6-Deficiency Reduces Osteophyte Formation but Has No Significant Effect on Synovial Inflammation after ACLT Surgery
3.4. ACLT-Related Alteration of the Subchondral Bone Is More Pronounced in C6-Deficient Rabbits
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huber-Lang, M.; Ignatius, A.; Brenner, R.E. Role of Complement on Broken Surfaces After Trauma. Adv. Exp. Med. Biol. 2015, 865, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Tegla, C.A.; Cudrici, C.; Patel, S.; Trippe, R., III; Rus, V.; Niculescu, F.; Rus, H. Membrane attack by complement: The assembly and biology of terminal complement complexes. Immunol. Res. 2011, 51, 45–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Rozelle, A.L.; Lepus, C.M.; Scanzello, C.R.; Song, J.J.; Larsen, D.M.; Crish, J.F.; Bebek, G.; Ritter, S.Y.; Lindstrom, T.M.; et al. Identification of a central role for complement in osteoarthritis. Nat. Med. 2011, 17, 1674–1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riegger, J.; Huber-Lang, M.; Brenner, R.E. Crucial role of the terminal complement complex in chondrocyte death and hypertrophy after cartilage trauma. Osteoarthr. Cartil. 2020, 28, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Carpanini, S.M.; Torvell, M.; Bevan, R.J.; Byrne, R.A.J.; Daskoulidou, N.; Saito, T.; Saido, T.C.; Taylor, P.R.; Hughes, T.R.; Zelek, W.M.; et al. Terminal complement pathway activation drives synaptic loss in Alzheimer's disease models. Acta Neuropathol. Com. 2022, 10, 99. [Google Scholar] [CrossRef]
- Struglics, A.; Okroj, M.; Sward, P.; Frobell, R.; Saxne, T.; Lohmander, L.S.; Blom, A.M. The complement system is activated in synovial fluid from subjects with knee injury and from patients with osteoarthritis. Arthritis Res. 2016, 18, 223. [Google Scholar] [CrossRef] [Green Version]
- Tatomir, A.; Rao, G.; Boodhoo, D.; Vlaicu, S.I.; Beltrand, A.; Anselmo, F.; Rus, V.; Rus, H. Histone Deacetylase SIRT1 Mediates C5b-9-Induced Cell Cycle in Oligodendrocytes. Front. Immunol. 2020, 11, 619. [Google Scholar] [CrossRef]
- Teixeira, G.Q.; Yong, Z.; Goncalves, R.M.; Kuhn, A.; Riegger, J.; Brisby, H.; Barreto Henriksson, H.; Ruf, M.; Nerlich, A.; Mauer, U.M.; et al. Terminal complement complex formation is associated with intervertebral disc degeneration. Eur. Spine J. 2021, 30, 217–226. [Google Scholar] [CrossRef]
- Soane, L.; Cho, H.J.; Niculescu, F.; Rus, H.; Shin, M.L. C5b-9 terminal complement complex protects oligodendrocytes from death by regulating bad through phosphatidylinositol 3-kinase/Akt pathway. J. Immunol. 2001, 167, 2305–2311. [Google Scholar] [CrossRef] [Green Version]
- Modinger, Y.; Rapp, A.E.; Vikman, A.; Ren, Z.; Fischer, V.; Bergdolt, S.; Haffner-Luntzer, M.; Song, W.C.; Lambris, J.D.; Huber-Lang, M.; et al. Reduced Terminal Complement Complex Formation in Mice Manifests in Low Bone Mass and Impaired Fracture Healing. Am. J. Pathol. 2019, 189, 147–161. [Google Scholar] [CrossRef]
- Rother, K.; Rother, U.; Muller-Eberhard, H.J.; Nilsson, J.R. Deficiency of the sixth component of complement in rabbits with an inherited complement defect. J. Exp. Med. 1966, 124, 773–785. [Google Scholar] [CrossRef]
- Joos, H.; Leucht, F.; Riegger, J.; Hogrefe, C.; Fiedler, J.; Durselen, L.; Reichel, H.; Ignatius, A.; Brenner, R.E. Differential Interactive Effects of Cartilage Traumatization and Blood Exposure In Vitro and In Vivo. Am. J. Sport. Med. 2015, 43, 2822–2832. [Google Scholar] [CrossRef]
- Wurzner, R.; Mewar, D.; Fernie, B.A.; Hobart, M.J.; Lachmann, P.J. Importance of the third thrombospondin repeat of C6 for terminal complement complex assembly. Immunology 1995, 85, 214–219. [Google Scholar]
- Ojanen, S.P.; Finnila, M.A.J.; Makela, J.T.A.; Saarela, K.; Happonen, E.; Herzog, W.; Saarakkala, S.; Korhonen, R.K. Anterior cruciate ligament transection of rabbits alters composition, structure and biomechanics of articular cartilage and chondrocyte deformation 2 weeks post-surgery in a site-specific manner. J. Biomech. 2020, 98, 109450. [Google Scholar] [CrossRef]
- Florea, C.; Malo, M.K.; Rautiainen, J.; Makela, J.T.; Fick, J.M.; Nieminen, M.T.; Jurvelin, J.S.; Davidescu, A.; Korhonen, R.K. Alterations in subchondral bone plate, trabecular bone and articular cartilage properties of rabbit femoral condyles at 4 weeks after anterior cruciate ligament transection. Osteoarthr. Cartil. 2015, 23, 414–422. [Google Scholar] [CrossRef] [Green Version]
- Arokoski, M.E.; Tiitu, V.; Jurvelin, J.S.; Korhonen, R.K.; Fick, J.M. Topographical investigation of changes in depth-wise proteoglycan distribution in rabbit femoral articular cartilage at 4 weeks after transection of the anterior cruciate ligament. J. Orthop. Res. 2015, 33, 1278–1286. [Google Scholar] [CrossRef] [Green Version]
- Hsia, A.W.; Anderson, M.J.; Heffner, M.A.; Lagmay, E.P.; Zavodovskaya, R.; Christiansen, B.A. Osteophyte formation after ACL rupture in mice is associated with joint restabilization and loss of range of motion. J. Orthop. Res. 2017, 35, 466–473. [Google Scholar] [CrossRef] [Green Version]
- Laverty, S.; Girard, C.A.; Williams, J.M.; Hunziker, E.B.; Pritzker, K.P. The OARSI histopathology initiative—Ecommendations for histological assessments of osteoarthritis in the rabbit. Osteoarthr. Cartil. 2010, 18 (Suppl. 3), S53–S65. [Google Scholar] [CrossRef] [Green Version]
- Boulocher, C.B.; Viguier, E.R.; Cararo Rda, R.; Fau, D.J.; Arnault, F.; Collard, F.; Maitre, P.A.; Roualdes, O.; Duclos, M.E.; Vignon, E.P.; et al. Radiographic assessment of the femorotibial joint of the CCLT rabbit experimental model of osteoarthritis. BMC Med. Imaging 2010, 10, 3. [Google Scholar] [CrossRef] [Green Version]
- Möhler, V. Einfluss der C6-Komplement-Defizienz auf die Pathogenese der Arthrose im Kaninchenmodell nach Kreuzbandresektion. Ph.D. Thesis, University of Ulm, Ulm, Germany, 2016. [Google Scholar]
- Tramontini, N.; Huber, C.; Liu-Bryan, R.; Terkeltaub, R.A.; Kilgore, K.S. Central role of complement membrane attack complex in monosodium urate crystal-induced neutrophilic rabbit knee synovitis. Arthritis Rheum. 2004, 50, 2633–2639. [Google Scholar] [CrossRef]
- Tramontini, N.L.; Kuipers, P.J.; Huber, C.M.; Murphy, K.; Naylor, K.B.; Broady, A.J.; Kilgore, K.S. Modulation of leukocyte recruitment and IL-8 expression by the membrane attack complex of complement (C5b-9) in a rabbit model of antigen-induced arthritis. Inflammation 2002, 26, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Schmiedt, W.; Kinscherf, R.; Deigner, H.P.; Kamencic, H.; Nauen, O.; Kilo, J.; Oelert, H.; Metz, J.; Bhakdi, S. Complement C6 deficiency protects against diet-induced atherosclerosis in rabbits. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1790–1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, W.; Schafer, H.J.; Bhakdi, S.; Klask, R.; Hansen, S.; Schaarschmidt, S.; Schofer, J.; Hugo, F.; Hamdoch, T.; Mathey, D. Influence of the terminal complement-complex on reperfusion injury, no-reflow and arrhythmias: A comparison between C6-competent and C6-deficient rabbits. Cardiovasc. Res. 1996, 32, 294–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhole, D.; Stahl, G.L. Molecular basis for complement component 6 (C6) deficiency in rats and mice. Immunobiology 2004, 209, 559–568. [Google Scholar] [CrossRef]
- Kotimaa, J.; Klar-Mohammad, N.; Gueler, F.; Schilders, G.; Jansen, A.; Rutjes, H.; Daha, M.R.; van Kooten, C. Sex matters: Systemic complement activity of female C57BL/6J and BALB/cJ mice is limited by serum terminal pathway components. Mol. Immunol. 2016, 76, 13–21. [Google Scholar] [CrossRef]
- Tedesco, F.; Lachmann, P.J. The quantitation of C6 in rabbit and human sera. Clin. Exp. Immunol. 1971, 9, 359–370. [Google Scholar]
- Riegger, J.; Palm, H.G.; Brenner, R.E. The Functional Role of Chondrogenic Stem/Progenito R Cells: Novel Evidence for Immunomodulatory Properties and Regenerative Potential after Cartilage Injury. Eur. Cells Mater. 2018, 36, 110–127. [Google Scholar] [CrossRef]
- Riegger, J.; Brenner, R.E. Pathomechanisms of Posttraumatic Osteoarthritis: Chondrocyte Behavior and Fate in a Precarious Environment. Int. J. Mol. Sci. 2020, 21, 1560. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Wu, Z.; Ying, S.; Liu, L.; Zhao, C.; Yao, C.; Zhang, Z.; Luo, C.; Wang, W.; Zhao, D.; et al. Sublytic C5b-9 induces glomerular mesangial cell proliferation via ERK1/2-dependent SOX9 phosphorylation and acetylation by enhancing Cyclin D1 in rat Thy-1 nephritis. Exp. Mol. Med. 2021, 53, 572–590. [Google Scholar] [CrossRef]
- Fosbrink, M.; Niculescu, F.; Rus, V.; Shin, M.L.; Rus, H. C5b-9-induced endothelial cell proliferation and migration are dependent on Akt inactivation of forkhead transcription factor FOXO1. J. Biol. Chem. 2006, 281, 19009–19018. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Jin, Y.; Zhang, S.; Ding, Y.; Huo, K.; Yang, J.; Zhao, L.; Nian, B.; Zhong, T.P.; Lu, W.; et al. PGE2 activates EP4 in subchondral bone osteoclasts to regulate osteoarthritis. Bone Res. 2022, 10, 27. [Google Scholar] [CrossRef]
- Hardy, M.M.; Seibert, K.; Manning, P.T.; Currie, M.G.; Woerner, B.M.; Edwards, D.; Koki, A.; Tripp, C.S. Cyclooxygenase 2-dependent prostaglandin E2 modulates cartilage proteoglycan degradation in human osteoarthritis explants. Arthritis Rheum. 2002, 46, 1789–1803. [Google Scholar] [CrossRef]
- Huang, L.W.; Riihioja, I.; Tanska, P.; Ojanen, S.; Palosaari, S.; Kroger, H.; Saarakkala, S.J.; Herzog, W.; Korhonen, R.K.; Finnila, M.A.J. Early changes in osteochondral tissues in a rabbit model of post-traumatic osteoarthritis. J. Orthop. Res. 2021, 39, 2556–2567. [Google Scholar] [CrossRef]
- Wang, S.X.; Laverty, S.; Dumitriu, M.; Plaas, A.; Grynpas, M.D. The effects of glucosamine hydrochloride on subchondral bone changes in an animal model of osteoarthritis. Arthritis Rheum. 2007, 56, 1537–1548. [Google Scholar] [CrossRef]
- Li, G.Y.; Yin, J.M.; Gao, J.J.; Cheng, T.S.; Pavlos, N.J.; Zhang, C.Q.; Zheng, M.H. Subchondral bone in osteoarthritis: Insight into risk factors and microstructural changes. Arthritis Res. 2013, 15, 223. [Google Scholar] [CrossRef] [Green Version]
- Arakawa, K.; Takahata, K.; Enomoto, S.; Oka, Y.; Ozone, K.; Nakagaki, S.; Murata, K.; Kanemura, N.; Kokubun, T. The difference in joint instability affects the onset of cartilage degeneration or subchondral bone changes. Osteoarthr. Cartil. 2022, 30, 451–460. [Google Scholar] [CrossRef]
- Riegger, J.; Leucht, F.; Palm, H.G.; Ignatius, A.; Brenner, R.E. Initial Harm Reduction by N-Acetylcysteine Alleviates Cartilage Degeneration after Blunt Single-Impact Cartilage Trauma in Vivo. Int. J. Mol. Sci. 2019, 20, 2916. [Google Scholar] [CrossRef] [Green Version]
- Riegger, J.; Joos, H.; Palm, H.G.; Friemert, B.; Reichel, H.; Ignatius, A.; Brenner, R.E. Antioxidative therapy in an ex vivo human cartilage trauma-model: Attenuation of trauma-induced cell loss and ECM-destructive enzymes by N-acetyl cysteine. Osteoarthr. Cartil. 2016, 24, 2171–2180. [Google Scholar] [CrossRef] [PubMed]
- Riegger, J.; Brenner, R.E. Evidence of necroptosis in osteoarthritic disease: Investigation of blunt mechanical impact as possible trigger in regulated necrosis. Cell Death Dis. 2019, 10, 683. [Google Scholar] [CrossRef] [Green Version]
- Riegger, J.; Zimmermann, M.; Joos, H.; Kappe, T.; Brenner, R.E. Hypothermia Promotes Cell-Protective and Chondroprotective Effects After Blunt Cartilage Trauma. Am. J. Sport Med. 2018, 46, 420–430. [Google Scholar] [CrossRef]
- Hornum, L.; Hansen, A.J.; Tornehave, D.; Fjording, M.S.; Colmenero, P.; Watjen, I.F.; Nielsen, N.H.S.; Bliddal, H.; Bartels, E.M. C5a and C5aR are elevated in joints of rheumatoid and psoriatic arthritis patients, and C5aR blockade attenuates leukocyte migration to synovial fluid. PLoS ONE 2017, 12, e0189017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banda, N.K.; Hyatt, S.; Antonioli, A.H.; White, J.T.; Glogowska, M.; Takahashi, K.; Merkel, T.J.; Stahl, G.L.; Mueller-Ortiz, S.; Wetsel, R.; et al. Role of C3a Receptors, C5a Receptors, and Complement Protein C6 Deficiency in Collagen Antibody-Induced Arthritis in Mice. J. Immunol. 2012, 188, 1469–1478. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riegger, J.; Joos, H.; Möhler, V.; Leucht, F.; Rading, K.; Kubisch, C.; Ignatius, A.; Huber-Lang, M.; Brenner, R.E. Functional Loss of Terminal Complement Complex Protects Rabbits from Injury-Induced Osteoarthritis on Structural and Cellular Level. Biomolecules 2023, 13, 216. https://doi.org/10.3390/biom13020216
Riegger J, Joos H, Möhler V, Leucht F, Rading K, Kubisch C, Ignatius A, Huber-Lang M, Brenner RE. Functional Loss of Terminal Complement Complex Protects Rabbits from Injury-Induced Osteoarthritis on Structural and Cellular Level. Biomolecules. 2023; 13(2):216. https://doi.org/10.3390/biom13020216
Chicago/Turabian StyleRiegger, Jana, Helga Joos, Valentin Möhler, Frank Leucht, Katrin Rading, Christian Kubisch, Anita Ignatius, Markus Huber-Lang, and Rolf E. Brenner. 2023. "Functional Loss of Terminal Complement Complex Protects Rabbits from Injury-Induced Osteoarthritis on Structural and Cellular Level" Biomolecules 13, no. 2: 216. https://doi.org/10.3390/biom13020216
APA StyleRiegger, J., Joos, H., Möhler, V., Leucht, F., Rading, K., Kubisch, C., Ignatius, A., Huber-Lang, M., & Brenner, R. E. (2023). Functional Loss of Terminal Complement Complex Protects Rabbits from Injury-Induced Osteoarthritis on Structural and Cellular Level. Biomolecules, 13(2), 216. https://doi.org/10.3390/biom13020216