

Combination of Experimental and Bioinformatic Approaches for Identification of Immunologically Relevant Protein–Peptide Interactions


 , and
, and
Abstract
1. Introduction
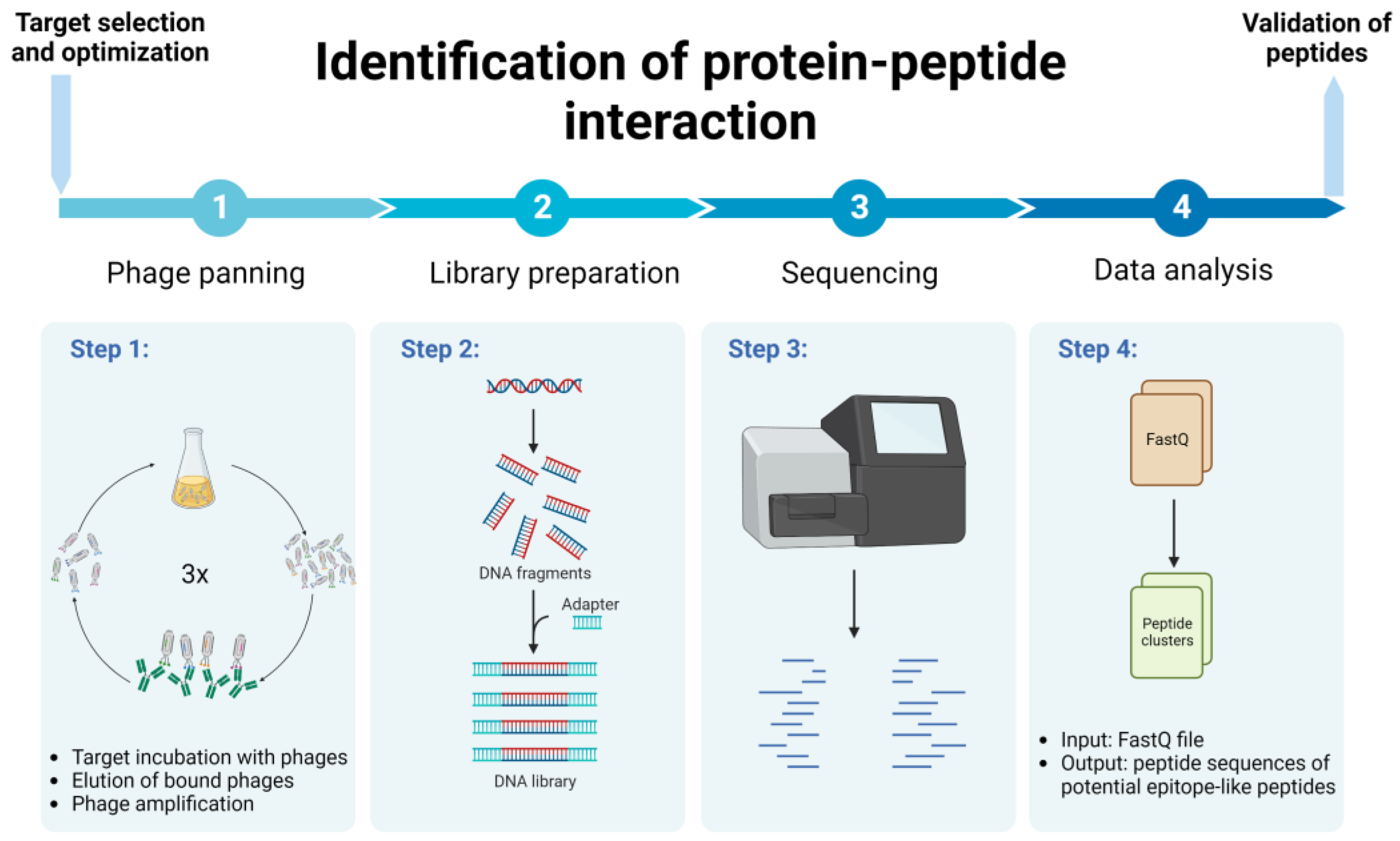
2. Materials and Methods
2.1. Phage Panning
2.2. NGS Data Analysis
2.3. Microarray Analysis
3. Results
3.1. Elimination of Non-Target Binders
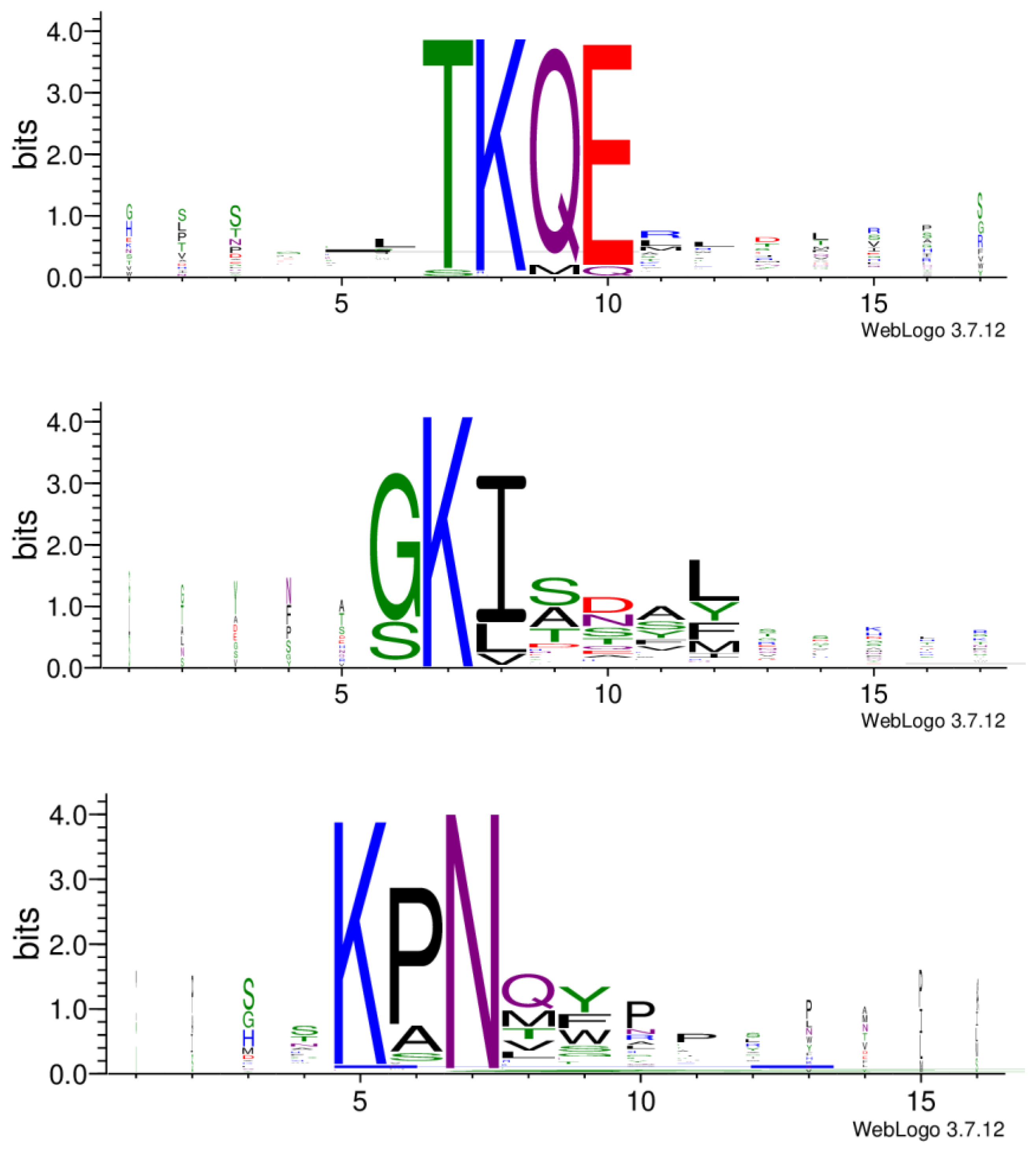
3.2. Amino Acid Motifs of Potential Conformational Epitopes
3.3. Identification of Three Peptide Clusters Indicates Three Potential Epitope Regions on the Allergen
3.4. Binding of IgG Antibodies to Epitope-like Peptides
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef]
- Vinogradov, A.A.; Yin, Y.; Suga, H. Macrocyclic Peptides as Drug Candidates: Recent Progress and Remaining Challenges. J. Am. Chem. Soc. 2019, 141, 4167–4181. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.C.-L.; Harris, J.L.; Khanna, K.K.; Hong, J.-H. A Comprehensive Review on Current Advances in Peptide Drug Development and Design. Int. J. Mol. Sci. 2019, 20, 2383. [Google Scholar] [CrossRef]
- Sathiamurthy, M.; Peters, B.; Bui, H.-H.; Sidney, J.; Mokili, J.; Wilson, S.S.; Fleri, W.; McGuinness, D.L.; Bourne, E.P.; Sette, A. An ontology for immune epitopes: Application to the design of a broad scope database of immune reactivities. Immunome Res. 2005, 1, 2. [Google Scholar] [CrossRef]
- Van Regenmortel, M.H. Mapping Epitope Structure and Activity: From One-Dimensional Prediction to Four-Dimensional Description of Antigenic Specificity. Methods 1996, 9, 465–472. [Google Scholar] [CrossRef]
- Pomés, A. Relevant B Cell Epitopes in Allergic Disease. Int. Arch. Allergy Immunol. 2009, 152, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Varshney, S.; Goldblum, R.M.; Kearney, C.; Watanabe, M.; Midoro-Horiuti, T. Major mountain cedar allergen, Jun a 1, contains conformational as well as linear IgE epitopes. Mol. Immunol. 2007, 44, 2781–2785. [Google Scholar] [CrossRef] [PubMed]
- King, T.P.; Spangfort, M.D. Structure and Biology of Stinging Insect Venom Allergens. Int. Arch. Allergy Immunol. 2000, 123, 99–106. [Google Scholar] [CrossRef]
- Ravn, U.; Gueneau, F.; Baerlocher, L.; Osteras, M.; Desmurs, M.; Malinge, P.; Magistrelli, G.; Farinelli, L.; Kosco-Vilbois, M.; Fischer, N. By-passing in vitro screening—Next generation sequencing technologies applied to antibody display and in silico candidate selection. Nucleic Acids Res. 2010, 38, e193. [Google Scholar] [CrossRef]
- Matochko, W.L.; Derda, R. Next-Generation Sequencing of Phage-Displayed Peptide Libraries. Pept. Libr. Methods Protoc. 2014, 1248, 249–266. [Google Scholar] [CrossRef]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef]
- Braun, R.; Schönberger, N.; Vinke, S.; Lederer, F.; Kalinowski, J.; Pollmann, K. Application of Next Generation Sequencing (NGS) in Phage Displayed Peptide Selection to Support the Identification of Arsenic-Binding Motifs. Viruses 2020, 12, 1360. [Google Scholar] [CrossRef]
- Frenzel, A.; Schirrmann, T.; Hust, M. Phage display-derived human antibodies in clinical development and therapy. Mabs 2016, 8, 1177–1194. [Google Scholar] [CrossRef] [PubMed]
- Luzar, J.; Molek, P.; Šilar, M.; Korosec, P.; Košnik, M.; Štrukelj, B.; Lunder, M. Identification and characterization of major cat allergen Fel d 1 mimotopes on filamentous phage carriers. Mol. Immunol. 2016, 71, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Zahirović, A.; Štrukelj, B.; Korošec, P.; Lunder, M. Epitope Mapping of Major Ragweed Allergen Amb a 1. Acta Chim. Slov. 2019, 66, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Zahirović, A.; Koren, A.; Kopač, P.; Štrukelj, B.; Korošec, P.; Lunder, M. Identification of bee venom Api m 1 IgE epitopes and characterization of corresponding mimotopes. J. Allergy Clin. Immunol. 2019, 143, 791–794. [Google Scholar] [CrossRef]
- New England Biolabs Inc. Ph.D. Phage Display Libraries, Instruction Manual; Version 1.0 9/09; New England Biolabs Inc.: Ipswich, MA, USA, 2009. [Google Scholar]
- Shave, S.; Mann, S.; Koszela, J.; Kerr, A.; Auer, M. PuLSE: Quality control and quantification of peptide sequences explored by phage display libraries. PLoS ONE 2018, 13, e0193332. [Google Scholar] [CrossRef] [PubMed]
- Boman, H.G. Antibacterial peptides: Basic facts and emerging concepts. J. Intern. Med. 2003, 254, 197–215. [Google Scholar] [CrossRef]
- He, B.; Chen, H.; Li, N.; Huang, J. SAROTUP: A suite of tools for finding potential target-unrelated peptides from phage display data. Int. J. Biol. Sci. 2019, 15, 1452–1459. [Google Scholar] [CrossRef]
- Grant, C.E.; Bailey, T.L. XSTREME: Comprehensive motif analysis of biological sequence datasets Introduction Abstract Results. Bioinformatics 2021, 9–11. [Google Scholar] [CrossRef]
- Krejci, A.; Hupp, T.R.; Lexa, M.; Vojtesek, B.; Muller, P. Hammock: A hidden Markov model-based peptide clustering algorithm to identify protein-interaction consensus motifs in large datasets. Bioinformatics 2015, 32, 9–16. [Google Scholar] [CrossRef]
- Mayrose, I.; Penn, O.; Erez, E.; Rubinstein, N.D.; Shlomi, T.; Freund, N.T.; Bublil, E.M.; Ruppin, E.; Sharan, R.; Gershoni, J.M.; et al. Pepitope: Epitope mapping from affinity-selected peptides. Bioinformatics 2007, 23, 3244–3246. [Google Scholar] [CrossRef]
- Menendez, A.; Scott, J.K. The nature of target-unrelated peptides recovered in the screening of phage-displayed random peptide libraries with antibodies. Anal. Biochem. 2005, 336, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Matochko, W.L.; Li, S.C.; Tang, S.K.; Derda, R. Prospective identification of parasitic sequences in phage display screens. Nucleic Acids Res. 2013, 42, 1784–1798. [Google Scholar] [CrossRef] [PubMed]
- Juds, C.; Schmidt, J.; Weller, M.G.; Lange, T.; Beck, U.; Conrad, T.; Börner, H.G. Combining Phage Display and Next-Generation Sequencing for Materials Sciences: A Case Study on Probing Polypropylene Surfaces. J. Am. Chem. Soc. 2020, 142, 10624–10628. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.; Brenner, S.E. WebLogo: A Sequence Logo Generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef]
- Frank, S.A. Immunology and Evolution of Infectious Disease; Princeton University Press: Princeton, NJ, USA, 2002. [Google Scholar]
- Vekris, A.; Pilalis, E.; Chatziioannou, A.; Petry, K.G. A Computational Pipeline for the Extraction of Actionable Biological Information From NGS-Phage Display Experiments. Front. Physiol. 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Brinton, L.T.; Bauknight, D.K.; Dasa, S.S.K.; Kelly, K.A. PHASTpep: Analysis Software for Discovery of Cell-Selective Peptides via Phage Display and Next-Generation Sequencing. PLoS ONE 2016, 11, e0155244. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nr. of All Unique Sequences | Nr. of Unique TUPs | % | |
|---|---|---|---|
| Ph.D.-7 | |||
| 7.1 | 207 | 55 | 25.6 |
| 7.2 | 195 | 36 | 18.5 |
| 7.3 | 202 | 27 | 13.4 |
| Ph.D.-12 | |||
| 12.1 | 209 | 62 | 29.7 |
| 12.2 | 201 | 34 | 16.9 |
| 12.3 | 184 | 16 | 8.7 |
| Ph.D.-C7C | |||
| C7.1C | 207 | 1 | 0.5 |
| C7.2C | 170 | 0 | 0 |
| C7.3C | 187 | 5 | 2.7 |
| Ph.D.-12 | Occ. | % | Boman Index | Ph.D.-7 | Occ. | % | Boman Index | Ph.D.-C7C | Occ. | % | Boman Index | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | DYHDPSLPTLRK | 111,085 | 63.30 | 3.237 | GKIFNTL | 18,843 | 16.05 | 0.143 | KPNQYPI | 13,587 | 10.04 | 1.154 |
| 2 | GKITQAMNVSQR | 4597 | 2.62 | 2.508 | GKIADLG | 8971 | 7.64 | 0.106 | KANQFPW | 8520 | 6.30 | 0.894 |
| 3 | STKQETYTDKHY | 4522 | 2.58 | 4.018 | NERALTL | 8018 | 6.83 | 2.756 | SGKPNVW | 4130 | 3.05 | 0.636 |
| 4 | QVNGLGERSQQM | 3429 | 1.95 | 2.933 | YSLQSVI | 6743 | 5.74 | −0.2 | LDKPNRY | 3895 | 2.88 | 3.166 |
| 5 | RDYHPRDHTATW | 2546 | 1.45 | 4.812 | GYKDFSA | 4736 | 4.03 | 1.726 | KANMWPS | 3642 | 2.69 | 0.727 |
| 6 | TAKYLPMRPGPL | 2047 | 1.17 | 0.687 | FGHYHYA | 3130 | 2.67 | 0.553 | NKPNQYF | 3431 | 2.54 | 2.108 |
| 7 | GKIVDSLGQSSP | 1652 | 0.94 | 1.188 | TNAWVDG | 2006 | 1.71 | 1.259 | KPNQSNY | 3214 | 2.38 | 2.817 |
| 8 | VKPNLYPSNDPI | 1410 | 0.80 | 1.434 | SITPMPA | 1985 | 1.69 | −0.444 | SHKPNVF | 2588 | 1.91 | 1.186 |
| 9 | GKISDRIKFDDG | 1314 | 0.75 | 3.407 | TPARHIY | 1831 | 1.56 | 2.223 | KANKYPS | 2152 | 1.59 | 1.879 |
| 10 | MGTKQEHLGPIR | 1313 | 0.75 | 2.165 | YKANQFL | 1769 | 1.51 | 1.166 | KPNMYPL | 1997 | 1.48 | 0.278 |
| 11 | HMETKQEKQIIW | 1259 | 0.72 | 2.376 | AHKSNHY | 1597 | 1.36 | 3.320 | KPNVYPL | 1722 | 1.27 | 0.090 |
| 12 | AEMTKQESILQR | 1213 | 0.69 | 3.095 | GKIDYFI | 1428 | 1.22 | 0.093 | KANQERT | 1699 | 1.26 | 4.184 |
| 13 | NWTKQERWAVSA | 1034 | 0.59 | 2.759 | FHPNTYN | 1313 | 1.12 | 2.524 | TKQEGRT | 1697 | 1.25 | 3.829 |
| 14 | GNNPLHVHHDKR | 997 | 0.57 | 3.879 | SPSTHWK | 1211 | 1.03 | 2.464 | KANSFGS | 1632 | 1.21 | 1.189 |
| 15 | DPKPNSSDYWYF | 995 | 0.57 | 2.617 | LSNNNLR | 1173 | 1.00 | 4.057 | KPNQFPR | 1599 | 1.18 | 3.012 |
| 16 | LPAHTKQEMRYL | 921 | 0.52 | 2.182 | VKPNQYA | 1162 | 0.99 | 1.717 | KPNQYPT | 1528 | 1.13 | 1.987 |
| 17 | SKIETSLNSMTN | 909 | 0.52 | 2.399 | GKIDSYF | 949 | 0.81 | 1.281 | SKPNMYS | 1521 | 1.12 | 1.580 |
| 18 | ITKQEALDTQIR | 905 | 0.52 | 2.970 | SKPNVYW | 833 | 0.71 | 1.337 | KPNLYPY | 1511 | 1.12 | 0.554 |
| 19 | HTKSNQWYPFQM | 905 | 0.52 | 2.198 | GHKMNHY | 804 | 0.69 | 2.623 | KENQWPS | 1392 | 1.03 | 2.561 |
| 20 | GKIGQYFSEYAT | 799 | 0.46 | 1.047 | GKITSMY | 775 | 0.66 | 0.143 | KPNVFPS | 1378 | 1.02 | 0.668 |
| Sequence | Median Signal Intensity | Sequence | Median Signal Intensity | ||
|---|---|---|---|---|---|
| 1 | HTKQELL | 42,935.17 | 1 | CGFKPNMFYYPELC | 39,741.17 |
| 2 | YSSLKPNKYAVW | 32,754.17 | 2 | CYSSLKPNKYAVWC | 32,991.50 |
| 3 | GFKPNMFYYPEL | 26,880.67 | 3 | CDPKPNSSDYWYFC | 29,583.17 |
| 4 | KANQFPW | 25,764.67 | 4 | CKANQFPWC | 27,706.17 |
| 5 | GKIGSFLGGGHI | 23,977.83 | 5 | CGKIDSYFC | 26,333.83 |
| 6 | DPKPNSSDYWYF | 23,049.67 | 6 | CVKPNLYPSNDPIC | 24,513.67 |
| 7 | GKIDSYF | 21,866.83 | 7 | CGKIDSFIRVEHGC | 23,558.17 |
| 8 | IAHKPNQGWWIH | 20,982.33 | CTRL | CQQLNIPPC | 11,382.17 |
| 9 | GFAGKIASTFVD | 20,143.00 | CTRL | CAHRVQTAC | 4026.17 |
| 10 | GKIDYFI | 19,626.83 | CTRL | CNLLMSHAC | 3364.50 |
| 11 | QGKPNQWANYFL | 17,935.50 | |||
| 12 | HMETKQEKQIIW | 16,990.83 | |||
| 13 | GKIDSFIRVEHG | 16,587.00 | |||
| 14 | GKIGQYFSEYAT | 16,452.83 | |||
| 15 | GKISSVMAHGDW | 15,244.00 | |||
| 16 | STKQETYTDKHY | 15,017.33 | |||
| 17 | LLANTGKIQKYL | 14,572.50 | |||
| 18 | TKQELPY | 13,254.17 | |||
| CTRL | QQLNIPP | 6598.50 | |||
| CTRL | AHRVQTA | 3720.00 | |||
| CTRL | NLLMSHA | 3311.83 |
| #1 | #2 | #3 | #4 | #5 | #6 | |
|---|---|---|---|---|---|---|
| p value | <0.0001 | 0.0003 | <0.0001 | <0.0001 | <0.0001 | <0.0001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Debeljak, J.; Korošec, P.; Šelb, J.; Rijavec, M.; Košnik, M.; Lunder, M. Combination of Experimental and Bioinformatic Approaches for Identification of Immunologically Relevant Protein–Peptide Interactions. Biomolecules 2023, 13, 310. https://doi.org/10.3390/biom13020310
Debeljak J, Korošec P, Šelb J, Rijavec M, Košnik M, Lunder M. Combination of Experimental and Bioinformatic Approaches for Identification of Immunologically Relevant Protein–Peptide Interactions. Biomolecules. 2023; 13(2):310. https://doi.org/10.3390/biom13020310
Chicago/Turabian StyleDebeljak, Jerneja, Peter Korošec, Julij Šelb, Matija Rijavec, Mitja Košnik, and Mojca Lunder. 2023. "Combination of Experimental and Bioinformatic Approaches for Identification of Immunologically Relevant Protein–Peptide Interactions" Biomolecules 13, no. 2: 310. https://doi.org/10.3390/biom13020310
APA StyleDebeljak, J., Korošec, P., Šelb, J., Rijavec, M., Košnik, M., & Lunder, M. (2023). Combination of Experimental and Bioinformatic Approaches for Identification of Immunologically Relevant Protein–Peptide Interactions. Biomolecules, 13(2), 310. https://doi.org/10.3390/biom13020310