
Distinct Roles of SELENOF in Different Human Cancers


Abstract
:1. Introduction
2. SELENOF’s Discovery, Structure, Cellular Localization and Expression
3. SELENOF’s Putative Roles in Redox Protein Quality Control and Metabolism
4. SELENOF Activities in Different Types of Cancers
5. Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Schrauzer, G.N.; White, D.A.; Schneider, C.J. Cancer mortality correlation studies-III: Statistical association with dietary selenium intakes. Bioinorg. Chem. 1977, 7, 23–34. [Google Scholar] [CrossRef]
- Schrauzer, G.N.; White, D.A.; Schneider, C.J. Cancer mortality correlation studies-IV: Associations with dietary intakes and blood levels of certain trace elements, notably Se-antagonists. Bioinorg. Chem. 1977, 7, 35–56. [Google Scholar] [CrossRef] [PubMed]
- el-Bayoumy, K.; Upadhyaya, P.; Chae, Y.H.; Sohn, O.S.; Rao, C.V.; Fiala, E.; Reddy, B.S. Chemoprevention of cancer by organoselenium compounds. J. Cell. Biochem. 1995, 22, 92–100. [Google Scholar] [CrossRef] [PubMed]
- El-Bayoumy, K.; Sinha, R. Mechanisms of mammary cancer chemoprevention by organoselenium compounds. Mutat. Res. 2004, 551, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Ip, C. Selenium inhibition of chemical carcinogenesis. Fed. Proc. 1985, 44, 2573–2578. [Google Scholar] [PubMed]
- Brigelius-Flohe, R. Selenium compounds and selenoproteins in cancer. Chem. Biodivers. 2008, 5, 389–395. [Google Scholar] [CrossRef]
- Fleming, J.; Ghose, A.; Harrison, P.R. Molecular mechanisms of cancer prevention by selenium compounds. Nutr. Cancer 2001, 40, 42–49. [Google Scholar] [CrossRef]
- Nadiminty, N.; Gao, A.C. Mechanisms of selenium chemoprevention and therapy in prostate cancer. Mol. Nutr. Food Res. 2008, 52, 1247–1260. [Google Scholar] [CrossRef]
- Fernandes, A.P.; Gandin, V. Selenium compounds as therapeutic agents in cancer. Biochim. Biophys. Acta 2015, 1850, 1642–1660. [Google Scholar] [CrossRef]
- Klein, E.A.; Thompson, I.M.; Lippman, S.M.; Goodman, P.J.; Albanes, D.; Taylor, P.R.; Coltman, C. SELECT: The next prostate cancer prevention trial. Selenum and Vitamin E Cancer Prevention Trial. J. Urol. 2001, 166, 1311–1315. [Google Scholar] [CrossRef]
- Klein, E.A.; Thompson, I.M., Jr.; Tangen, C.M.; Crowley, J.J.; Lucia, M.S.; Goodman, P.J.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; et al. Vitamin E and the risk of prostate cancer: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2011, 306, 1549–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristal, A.R.; Darke, A.K.; Morris, J.S.; Tangen, C.M.; Goodman, P.J.; Thompson, I.M.; Meyskens, F.L., Jr.; Goodman, G.E.; Minasian, L.M.; Parnes, H.L.; et al. Baseline selenium status and effects of selenium and vitamin e supplementation on prostate cancer risk. J. Natl. Cancer Inst. 2014, 106, djt456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinceti, M.; Filippini, T.; Del Giovane, C.; Dennert, G.; Zwahlen, M.; Brinkman, M.; Zeegers, M.P.; Horneber, M.; D’Amico, R.; Crespi, C.M. Selenium for preventing cancer. Cochrane Database Syst. Rev. 2018, 1, CD005195. [Google Scholar] [CrossRef]
- Flowers, B.; Poles, A.; Kastrati, I. Selenium and breast cancer—An update of clinical and epidemiological data. Arch. Biochem. Biophys. 2022, 732, 109465. [Google Scholar] [CrossRef]
- Gladyshev, V.N.; Hatfield, D.L. Analysis of selenocysteine-containing proteins. Curr. Protoc. Protein Sci. 2001, 20, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Gladyshev, V.N.; Kryukov, G.V. Evolution of selenocysteine-containing proteins: Significance of identification and functional characterization of selenoproteins. Biofactors 2001, 14, 87–92. [Google Scholar] [CrossRef]
- Kryukov, G.V.; Castellano, S.; Novoselov, S.V.; Lobanov, A.V.; Zehtab, O.; Guigo, R.; Gladyshev, V.N. Characterization of mammalian selenoproteomes. Science 2003, 300, 1439–1443. [Google Scholar] [CrossRef] [Green Version]
- Lobanov, A.V.; Hatfield, D.L.; Gladyshev, V.N. Eukaryotic selenoproteins and selenoproteomes. Biochim. Biophys. Acta 2009, 1790, 1424–1428. [Google Scholar] [CrossRef] [Green Version]
- Hondal, R.J.; Marino, S.M.; Gladyshev, V.N. Selenocysteine in thiol/disulfide-like exchange reactions. Antioxid. Redox Signal. 2013, 18, 1675–1689. [Google Scholar] [CrossRef] [Green Version]
- Hatfield, D.L.; Gladyshev, V.N. How selenium has altered our understanding of the genetic code. Mol. Cell. Biol. 2002, 22, 3565–3576. [Google Scholar] [CrossRef] [Green Version]
- Berry, M.J.; Banu, L.; Chen, Y.; Mandel, S.J.; Kiefer, J.D.; Harney, J.W.; Larsen, P.R. Recognition of UGA as a selenocysteine codon in Type I deiodinase requires sequences in the 3’ untranslated region. Nature 1991, 353, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Labunskyy, V.M.; Hatfield, D.L.; Gladyshev, V.N. Selenoproteins: Molecular pathways and physiological roles. Physiol. Rev. 2014, 94, 739–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tujebajeva, R.; Copeland, P.; Xu, X.-M.; Carlson, B.; Harney, J.; Driscoll, D.; Hatfield, D.; Berry, M. Decoding apparatus for eukaryotic selenocysteine insertion. EMBO Rep. 2000, 1, 158–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Copeland, P.R. Regulation of gene expression by stop codon recoding: Selenocysteine. Gene 2003, 312, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Zhuo, P.; Diamond, A.M. Molecular mechanisms by which selenoproteins affect cancer risk and progression. Biochim. Biophys. Acta 2009, 115, 227–242. [Google Scholar] [CrossRef] [Green Version]
- Meplan, C.; Hesketh, J. Selenium and cancer: A story that should not be forgotten-insights from genomics. Cancer Treat. Res. 2014, 159, 145–166. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Lubos, E.; Yang, Y.; Galbraith, J.D.; Kelly, N.; Zhang, Y.Y.; Leopold, J.A.; Loscalzo, J. Glutathione peroxidase-1 regulates mitochondrial function to modulate redox-dependent cellular responses. J. Biol. Chem. 2009, 284, 11913–11921. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, P.; Abo El-Magd, N.F.; Hesketh, J.; Bermano, G. The Role of rs713041 Glutathione Peroxidase 4 (GPX4) Single Nucleotide Polymorphism on Disease Susceptibility in Humans: A Systematic Review and Meta-Analysis. Int. J. Mol. Sci. 2022, 23, 15762. [Google Scholar] [CrossRef]
- Kadkol, S.; Diamond, A.M. The Interaction between Dietary Selenium Intake and Genetics in Determining Cancer Risk and Outcome. Nutrients 2020, 12, 2424. [Google Scholar] [CrossRef]
- Hu, J.; Zhou, G.W.; Wang, N.; Wang, Y.J. GPX1 Pro198Leu polymorphism and breast cancer risk: A meta-analysis. Breast Cancer Res. Treat. 2010, 124, 425–431. [Google Scholar] [CrossRef]
- Garberg, P.; Hogberg, J. Studies on Se incorporation in selenoproteins; effects of peroxisome proliferators and hydrogen peroxide generating system. Chem. Biol. Interact. 1992, 81, 291–306. [Google Scholar] [CrossRef] [PubMed]
- Kalcklosch, M.; Kyriakopoulos, A.; Hammel, C.; Behne, D. A new selenoprotein found in the glandular epithelial cells of the rat prostate. Biochem. Biophys. Res. Commun. 1995, 217, 162–170. [Google Scholar] [CrossRef]
- Gladyshev, V.N.; Jeang, K.-T.; Wootton, J.C.; Hatfield, D.L. A new human selenium-containing protein. Purification, characterization and cDNA sequence. J. Biol. Chem. 1998, 273, 8910–8915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumaraswamy, E.; Malykh, A.; Korotkov, K.V.; Kozyavkin, S.; Hu, Y.; Kwon, S.Y.; Moustafa, M.E.; Carlson, B.A.; Berry, M.J.; Lee, B.J.; et al. Structure-expression relationships of the 15-kDa selenoprotein gene. Possible role of the protein in cancer etiology. J. Biol. Chem. 2000, 275, 35540–35547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gladyshev, V.N.; Arner, E.S.; Berry, M.J.; Brigelius-Flohe, R.; Bruford, E.A.; Burk, R.F.; Carlson, B.A.; Castellano, S.; Chavatte, L.; Conrad, M.; et al. Selenoprotein Gene Nomenclature. J. Biol. Chem. 2016, 291, 24036–24040. [Google Scholar] [CrossRef] [Green Version]
- Nagai, H.; Negrini, M.; Carter, S.L.; Gillum, D.R.; Rosenberg, A.L.; Schwartz, G.F.; Croce, C.M. Detection and cloning of a common region of loss of heterozygosity at chromosome 1p in breast cancer. Cancer Res. 1995, 55, 1752–1757. [Google Scholar]
- Hu, Y.J.; Korotkov, K.V.; Mehta, R.; Hatfield, D.L.; Rotimi, C.N.; Luke, A.; Prewitt, T.E.; Cooper, R.S.; Stock, W.; Vokes, E.E.; et al. Distribution and functional consequences of nucleotide polymorphisms in the 3’-untranslated region of the human Sep15 gene. Cancer Res. 2001, 61, 2307–2310. [Google Scholar]
- Ekoue, D.N.; Ansong, E.; Liu, L.; Macias, V.; Deaton, R.; Lacher, C.; Picklo, M.; Nonn, L.; Gann, P.H.; Kajdacsy-Balla, A.; et al. Correlations of SELENOF and SELENOP genotypes with serum selenium levels and prostate cancer. Prostate 2018, 78, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Korotkov, K.V.; Kumaraswamy, E.; Zhou, Y.; Hatfield, D.L.; Gladyshev, V.N. Association between the 15-kDa selenoprotein and UDP-glucose:glycoprotein glucosyltransferase in the endoplasmic reticulum of mammalian cells. J. Biol. Chem. 2001, 276, 15330–15336. [Google Scholar] [CrossRef] [Green Version]
- Labunskyy, V.M.; Ferguson, A.D.; Fomenko, D.E.; Chelliah, Y.; Hatfield, D.L.; Gladyshev, V.N. A novel cysteine-rich domain of Sep15 mediates the interaction with UDP-glucose:glycoprotein glucosyltransferase. J. Biol. Chem. 2005, 280, 37839–37845. [Google Scholar] [CrossRef] [Green Version]
- Labunskyy, V.M.; Yoo, M.H.; Hatfield, D.L.; Gladyshev, V.N. Sep15, a thioredoxin-like selenoprotein, is involved in the unfolded protein response and differentially regulated by adaptive and acute ER stresses. Biochemistry 2009, 48, 8458–8465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunde, R.A.; Raines, A.M.; Barnes, K.M.; Evenson, J.K. Selenium status highly regulates selenoprotein mRNA levels for only a subset of the selenoproteins in the selenoproteome. Biosci. Rep. 2009, 29, 329–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, A.D.; Labunskyy, V.M.; Fomenko, D.E.; Arac, D.; Chelliah, Y.; Amezcua, C.A.; Rizo, J.; Gladyshev, V.N.; Deisenhofer, J. NMR structures of the selenoproteins Sep15 and SelM reveal redox activity of a new thioredoxin-like family. J. Biol. Chem. 2006, 281, 3536–3543. [Google Scholar] [CrossRef] [Green Version]
- Ren, B.; Huang, Y.; Zou, C.; Wu, Y.; Huang, Y.; Ni, J.; Tian, J. Transcriptional Regulation of Selenoprotein F by Heat Shock Factor 1 during Selenium Supplementation and Stress Response. Cells 2019, 8, 479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bang, J.; Huh, J.H.; Na, J.W.; Lu, Q.; Carlson, B.A.; Tobe, R.; Tsuji, P.A.; Gladyshev, V.N.; Hatfield, D.L.; Lee, B.J. Cell Proliferation and Motility Are Inhibited by G1 Phase Arrest in 15-kDa Selenoprotein-Deficient Chang Liver Cells. Mol. Cells 2015, 38, 457–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yim, S.H.; Everley, R.A.; Schildberg, F.A.; Lee, S.G.; Orsi, A.; Barbati, Z.R.; Karatepe, K.; Fomenko, D.E.; Tsuji, P.A.; Luo, H.R.; et al. Role of Selenof as a Gatekeeper of Secreted Disulfide-Rich Glycoproteins. Cell Rep. 2018, 23, 1387–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasaikina, M.V.; Fomenko, D.E.; Labunskyy, V.M.; Lachke, S.A.; Qiu, W.; Moncaster, J.A.; Zhang, J.; Wojnarowicz, M.W., Jr.; Natarajan, S.K.; Malinouski, M.; et al. Roles of the 15-kDa selenoprotein (Sep15) in redox homeostasis and cataract development revealed by the analysis of Sep 15 knockout mice. J. Biol. Chem. 2011, 286, 33203–33212. [Google Scholar] [CrossRef] [Green Version]
- Yin, N.; Zheng, X.; Zhou, J.; Liu, H.; Huang, K. Knockdown of 15-kDa selenoprotein (Sep15) increases hLE cells’ susceptibility to tunicamycin-induced apoptosis. J. Biol. Inorg. Chem. 2015, 20, 1307–1317. [Google Scholar] [CrossRef]
- Oakes, S.A. Endoplasmic Reticulum Stress Signaling in Cancer Cells. Am. J. Pathol. 2020, 190, 934–946. [Google Scholar] [CrossRef] [Green Version]
- Hong, L.K.; Kadkol, S.; Sverdlov, M.; Kastrati, I.; Elhodaky, M.; Deaton, R.; Sfanos, K.S.; Wang, H.; Liu, L.; Diamond, A.M. Loss of SELENOF Induces the Transformed Phenotype in Human Immortalized Prostate Epithelial Cells. Int. J. Mol. Sci. 2021, 22, 12040. [Google Scholar] [CrossRef]
- Tian, J.; Liu, J.; Li, J.; Zheng, J.; Chen, L.; Wang, Y.; Liu, Q.; Ni, J. The interaction of selenoprotein F (SELENOF) with retinol dehydrogenase 11 (RDH11) implied a role of SELENOF in vitamin A metabolism. Nutr. Metab. 2018, 15, 7. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Ren, B.; Wang, H.; Huang, R.; Zhou, J.; Liu, H.; Tian, J.; Huang, K. Hepatic proteomic analysis of selenoprotein F knockout mice by iTRAQ: An implication for the roles of selenoprotein F in metabolism and diseases. J. Proteom. 2020, 215, 103653. [Google Scholar] [CrossRef]
- Zheng, X.; Ren, B.; Li, X.; Yan, H.; Xie, Q.; Liu, H.; Zhou, J.; Tian, J.; Huang, K. Selenoprotein F knockout leads to glucose and lipid metabolism disorders in mice. J. Biol. Inorg. Chem. 2020, 25, 1009–1022. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, Y.; Zhou, J.; Liu, H. Selenoprotein F Knockout Caused Glucose Metabolism Disorder in Young Mice by Disrupting Redox Homeostasis. Antioxidants 2022, 11, 2105. [Google Scholar] [CrossRef] [PubMed]
- Labunskyy, V.M.; Lee, B.C.; Handy, D.E.; Loscalzo, J.; Hatfield, D.L.; Gladyshev, V.N. Both maximal expression of selenoproteins and selenoprotein deficiency can promote development of type 2 diabetes-like phenotype in mice. Antioxid. Redox Signal. 2011, 14, 2327–2336. [Google Scholar] [CrossRef]
- Zhou, J.; Huang, K.; Lei, X.G. Selenium and diabetes--evidence from animal studies. Free. Radic. Biol. Med. 2013, 65, 1548–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.G.; Zhao, T.; Xu, X.J.; Xu, Y.H.; Wei, X.L.; Jiang, M.; Luo, Z. Selenoprotein F (SELENOF)-mediated AKT1-FOXO3a-PYGL axis contributes to selenium supranutrition-induced glycogenolysis and lipogenesis. Biochim. Biophys. Acta Gene Regul. Mech. 2022, 1865, 194814. [Google Scholar] [CrossRef]
- Penney, K.L.; Schumacher, F.R.; Li, H.; Kraft, P.; Morris, J.S.; Kurth, T.; Mucci, L.A.; Hunter, D.J.; Kantoff, P.W.; Stampfer, M.J.; et al. A large prospective study of SEP15 genetic variation, interaction with plasma selenium levels, and prostate cancer risk and survival. Cancer Prev. Res. 2010, 3, 604–610. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef]
- Eidelman, E.; Twum-Ampofo, J.; Ansari, J.; Siddiqui, M.M. The Metabolic Phenotype of Prostate Cancer. Front. Oncol. 2017, 7, 131. [Google Scholar] [CrossRef] [Green Version]
- Bera, S.; Diamond, A.M. Role of SELENBP1 and SELENOF in prostate cancer bioenergetics. Arch. Biochem. Biophys. 2022, 732, 109451. [Google Scholar] [CrossRef]
- Zigrossi, A.; Hong, L.K.; Ekyalongo, R.C.; Cruz-Alvarez, C.; Gornick, E.; Diamond, A.M.; Kastrati, I. SELENOF is a new tumor suppressor in breast cancer. Oncogene 2022, 41, 1263–1268. [Google Scholar] [CrossRef]
- Apostolou, S.; Klein, J.O.; Mitsuuchi, Y.; Shetler, J.N.; Poulikakos, P.I.; Jhanwar, S.C.; Kruger, W.D.; Testa, J.R. Growth inhibition and induction of apoptosis in mesothelioma cells by selenium and dependence on selenoprotein SEP15 genotype. Oncogene 2004, 23, 5032–5040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irons, R.; Tsuji, P.A.; Carlson, B.A.; Ouyang, P.; Yoo, M.H.; Xu, X.M.; Hatfield, D.L.; Gladyshev, V.N.; Davis, C.D. Deficiency in the 15-kDa selenoprotein inhibits tumorigenicity and metastasis of colon cancer cells. Cancer Prev. Res. 2010, 3, 630–639. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, P.A.; Naranjo-Suarez, S.; Carlson, B.A.; Tobe, R.; Yoo, M.H.; Davis, C.D. Deficiency in the 15 kDa selenoprotein inhibits human colon cancer cell growth. Nutrients 2011, 3, 805–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuji, P.A.; Carlson, B.A.; Naranjo-Suarez, S.; Yoo, M.H.; Xu, X.M.; Fomenko, D.E.; Gladyshev, V.N.; Hatfield, D.L.; Davis, C.D. Knockout of the 15 kDa selenoprotein protects against chemically-induced aberrant crypt formation in mice. PLoS ONE 2012, 7, e50574. [Google Scholar] [CrossRef]
- Canter, J.A.; Ernst, S.E.; Peters, K.M.; Carlson, B.A.; Thielman, N.R.J.; Grysczyk, L.; Udofe, P.; Yu, Y.; Cao, L.; Davis, C.D.; et al. Selenium and the 15kDa Selenoprotein Impact Colorectal Tumorigenesis by Modulating Intestinal Barrier Integrity. Int. J. Mol. Sci. 2021, 22, 10651. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Risbridger, G.P.; Davis, I.D.; Birrell, S.N.; Tilley, W.D. Breast and prostate cancer: More similar than different. Nat. Rev. Cancer 2010, 10, 205–212. [Google Scholar] [CrossRef]
- Lin, J.H.; Giovannucci, E. Sex hormones and colorectal cancer: What have we learned so far? J. Natl. Cancer Inst. 2010, 102, 1746–1747. [Google Scholar] [CrossRef] [Green Version]
- Bates, G.J.; Fox, S.B.; Han, C.; Leek, R.D.; Garcia, J.F.; Harris, A.L.; Banham, A.H. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J. Clin. Oncol. 2006, 24, 5373–5380. [Google Scholar] [CrossRef] [PubMed]
- Salama, P.; Phillips, M.; Grieu, F.; Morris, M.; Zeps, N.; Joseph, D.; Platell, C.; Iacopetta, B. Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J. Clin. Oncol. 2009, 27, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Larionova, I.; Tuguzbaeva, G.; Ponomaryova, A.; Stakheyeva, M.; Cherdyntseva, N.; Pavlov, V.; Choinzonov, E.; Kzhyshkowska, J. Tumor-Associated Macrophages in Human Breast, Colorectal, Lung, Ovarian and Prostate Cancers. Front. Oncol. 2020, 10, 566511. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, P.A.; Carlson, B.A.; Yoo, M.H.; Naranjo-Suarez, S.; Xu, X.M.; He, Y.; Asaki, E.; Seifried, H.E.; Reinhold, W.C.; Davis, C.D.; et al. The 15kDa selenoprotein and thioredoxin reductase 1 promote colon cancer by different pathways. PLoS ONE 2015, 10, e0124487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
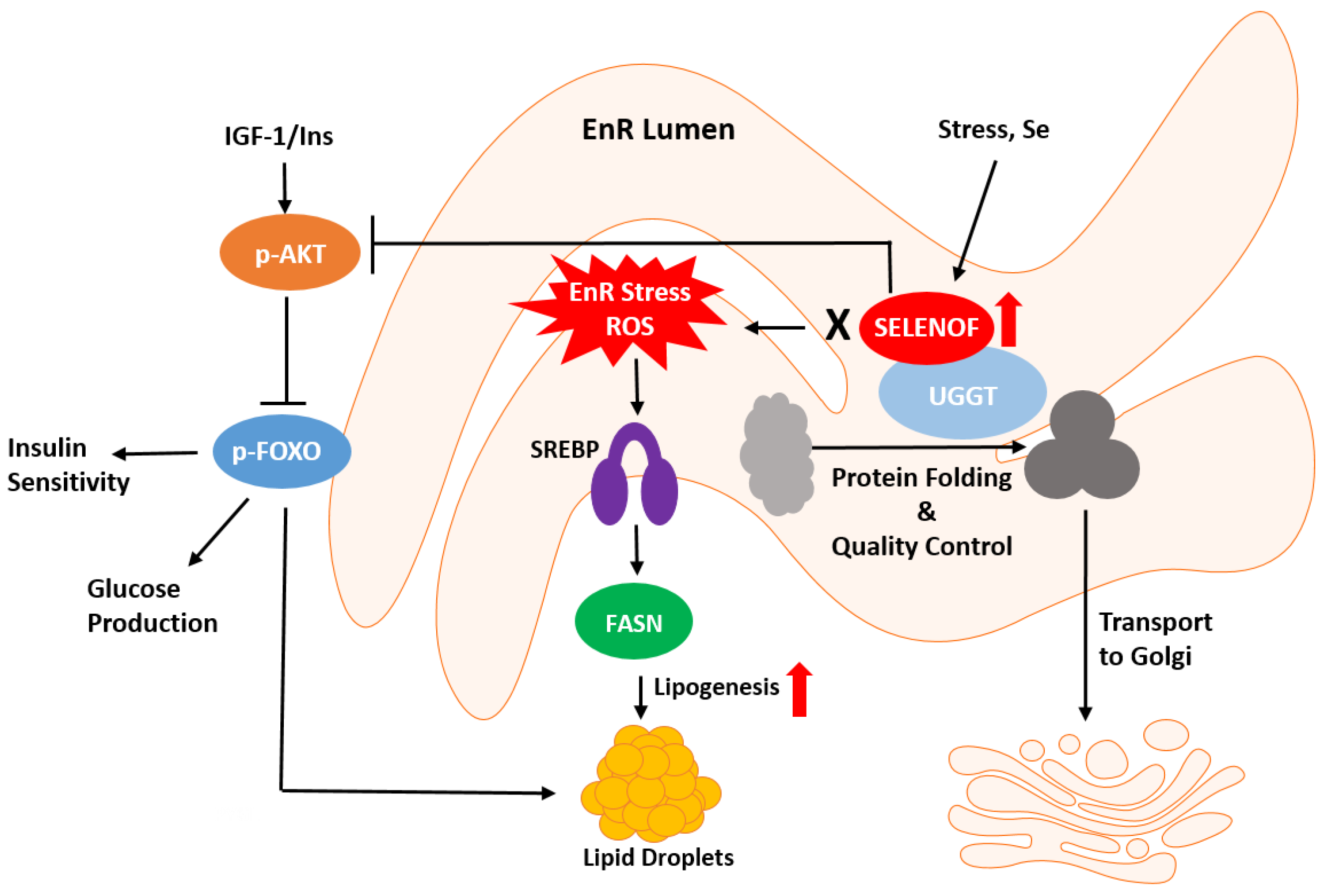

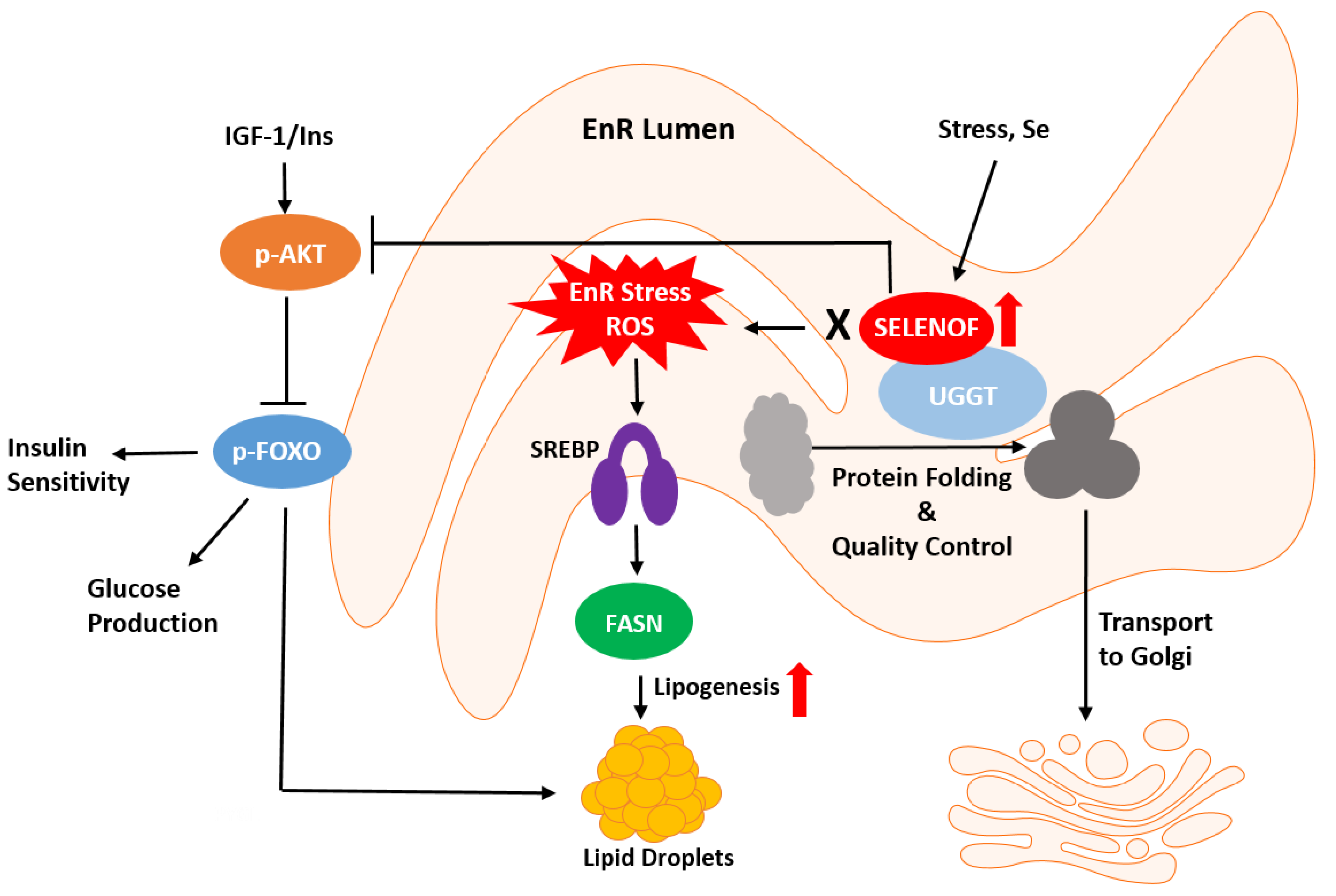{kind=link}
| Cancer | Model | Phenotype | Reference(s) |
|---|---|---|---|
| Prostate Cancer |
| ↓SELENOF correlates to increased cancer progression | [38,50,60] |
| Breast Cancer |
| ↓SELENOF correlates to increased proliferation and decreased cell death | [62] |
| Colorectal Cancer |
| ↓SELENOF correlates to decreased proliferation and aberrant crypt formation | [64,65,66,67] |
| Malignant Mesothelioma |
| Effectiveness of selenium supplementation dependent on polymorphism | [63] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flowers, B.; Bochnacka, O.; Poles, A.; Diamond, A.M.; Kastrati, I. Distinct Roles of SELENOF in Different Human Cancers. Biomolecules 2023, 13, 486. https://doi.org/10.3390/biom13030486
Flowers B, Bochnacka O, Poles A, Diamond AM, Kastrati I. Distinct Roles of SELENOF in Different Human Cancers. Biomolecules. 2023; 13(3):486. https://doi.org/10.3390/biom13030486
Chicago/Turabian StyleFlowers, Brenna, Oliwia Bochnacka, Allison Poles, Alan M. Diamond, and Irida Kastrati. 2023. "Distinct Roles of SELENOF in Different Human Cancers" Biomolecules 13, no. 3: 486. https://doi.org/10.3390/biom13030486
APA StyleFlowers, B., Bochnacka, O., Poles, A., Diamond, A. M., & Kastrati, I. (2023). Distinct Roles of SELENOF in Different Human Cancers. Biomolecules, 13(3), 486. https://doi.org/10.3390/biom13030486