
Inherited Retinal Degeneration: Towards the Development of a Combination Therapy Targeting Histone Deacetylase, Poly (ADP-Ribose) Polymerase, and Calpain
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Retinal Explant Culture Procedure
2.3. Retinal Drug Treatments
2.4. Detection of Cell Death
2.5. HDAC In Situ Activity Assay
2.6. Calpain In Situ Activity Assay
2.7. Calpain-2 Immunostaining
2.8. PARP Activity Assay
2.9. Western Blot
2.10. Image and Data Analysis
3. Results
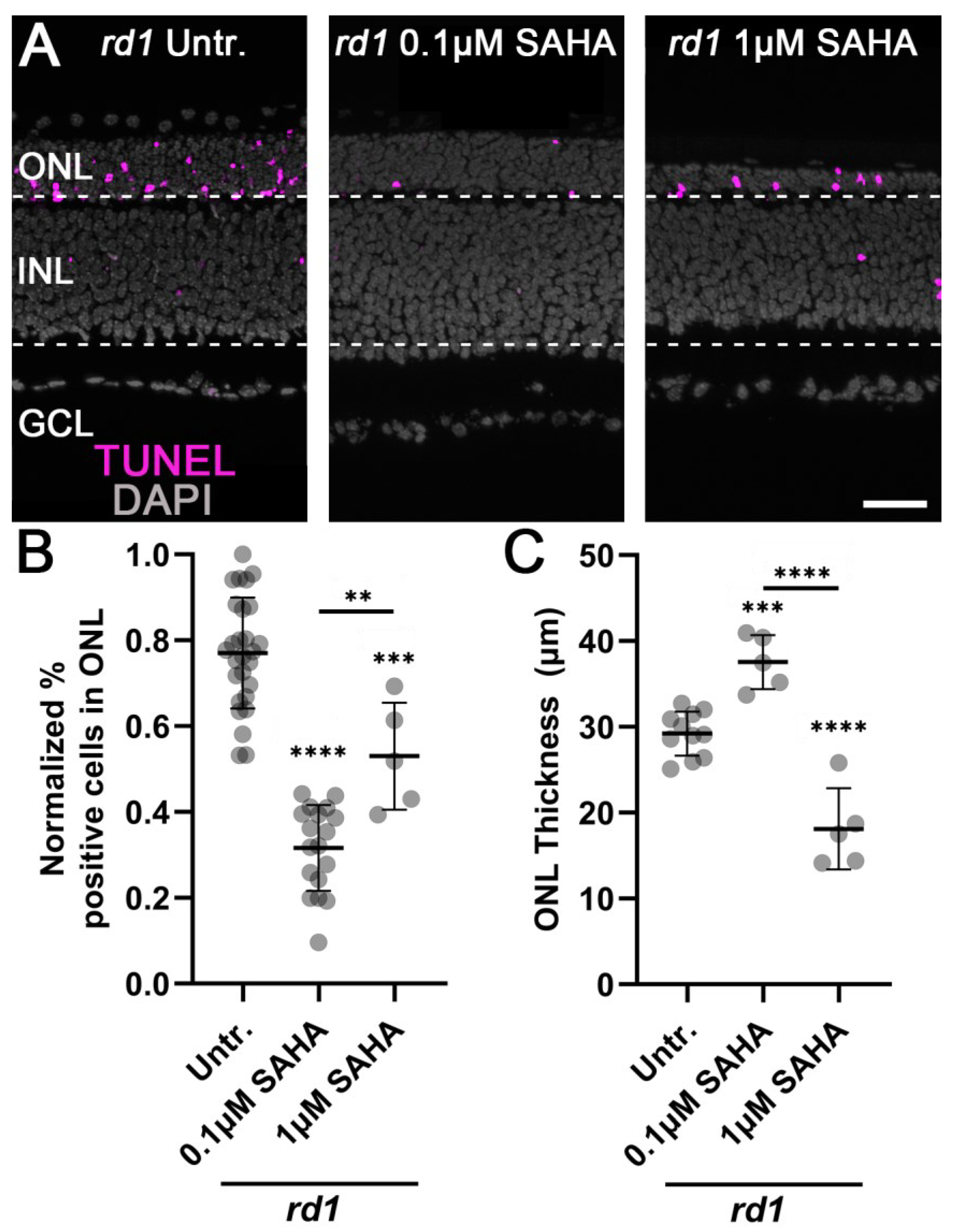
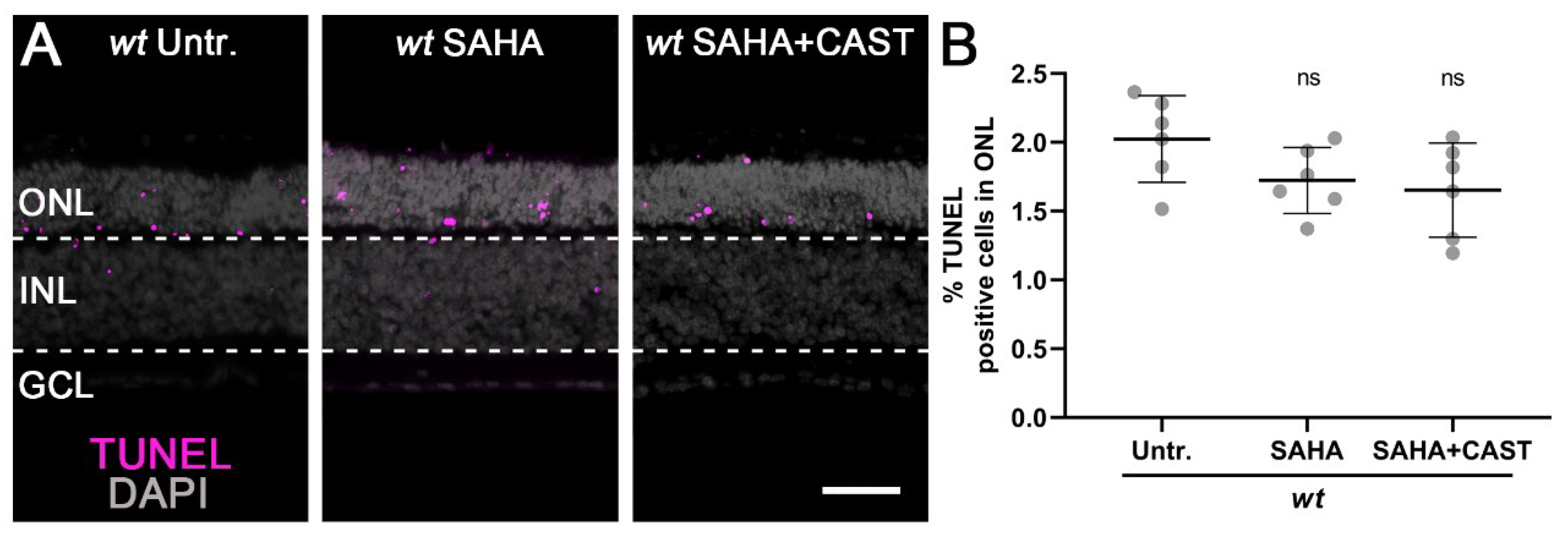
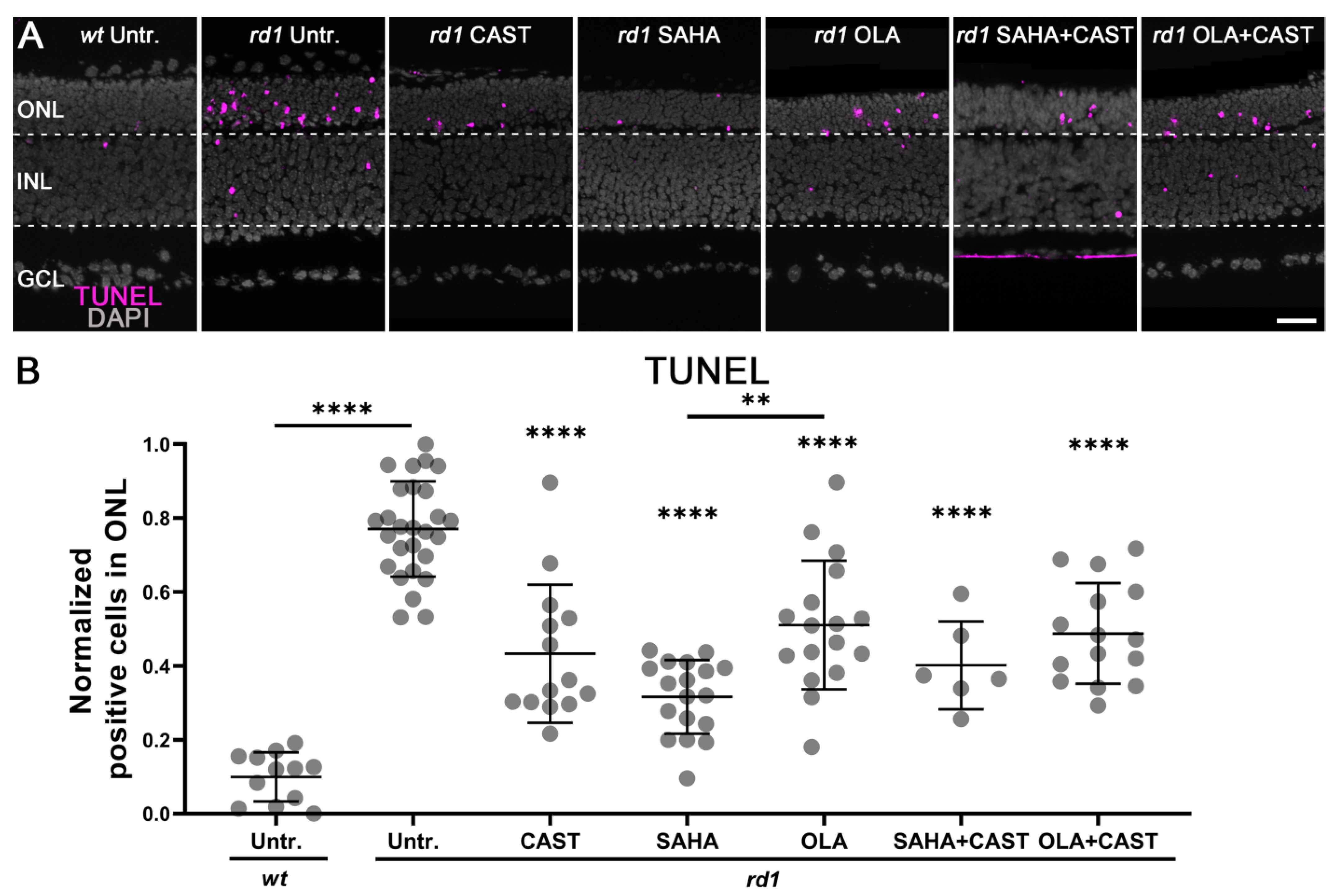
3.1. Monotherapy and Combination Therapy Both Delay rd1 Photoreceptor Degeneration
3.2. Monotherapy and Combination Therapy Reduce Calpain Activity
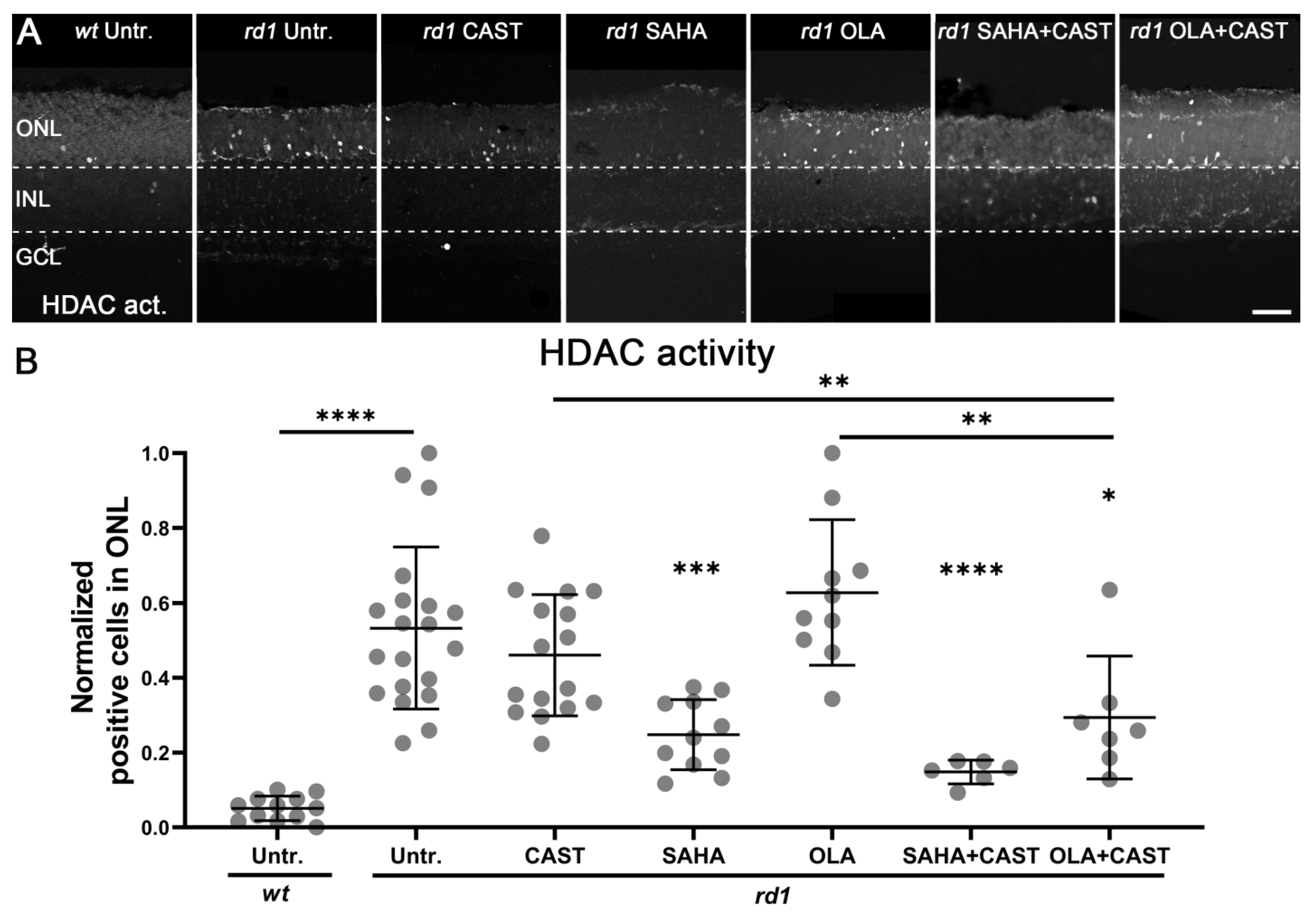
3.3. HDAC Activity Is Not Affected by PARP or Calpain Inhibition
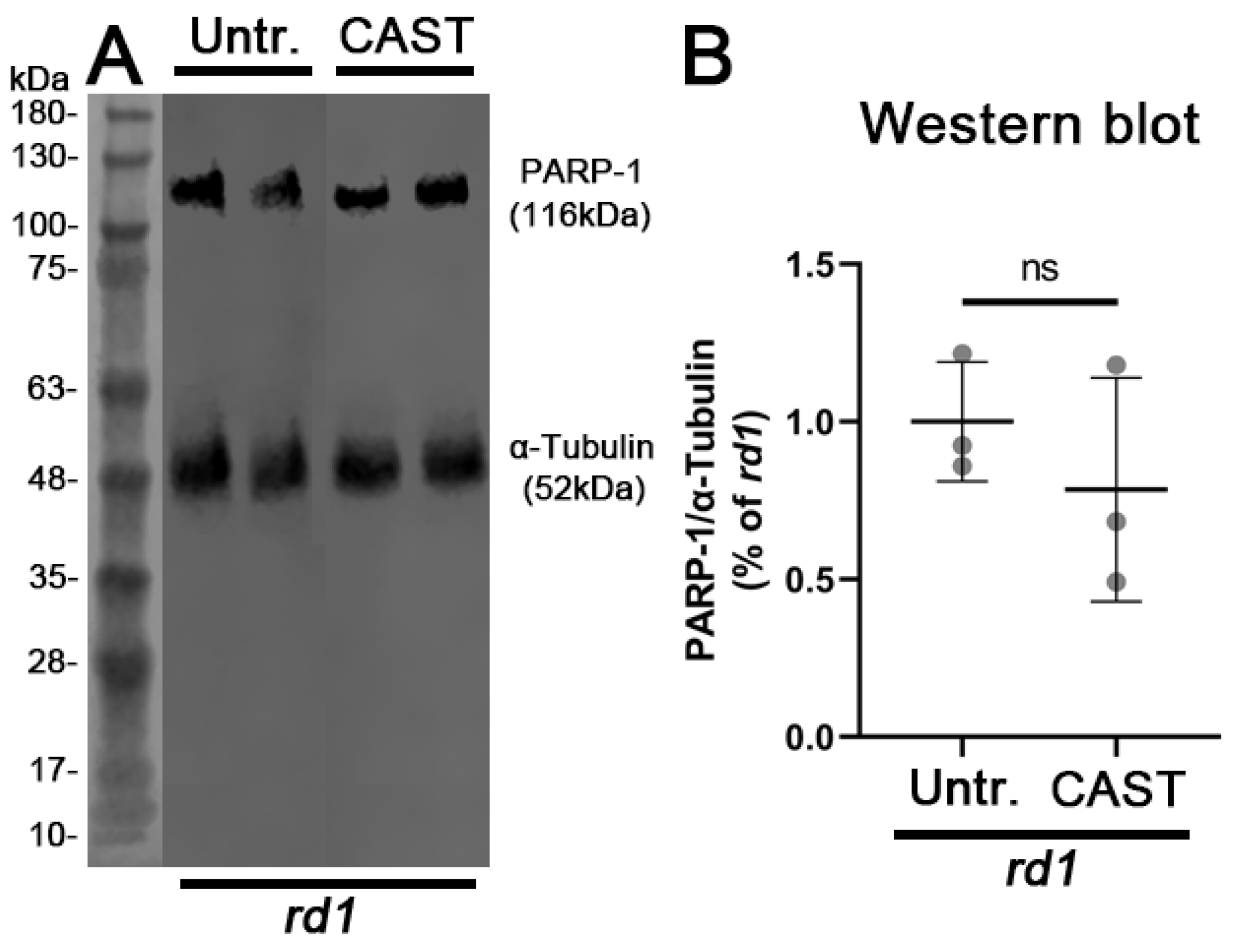
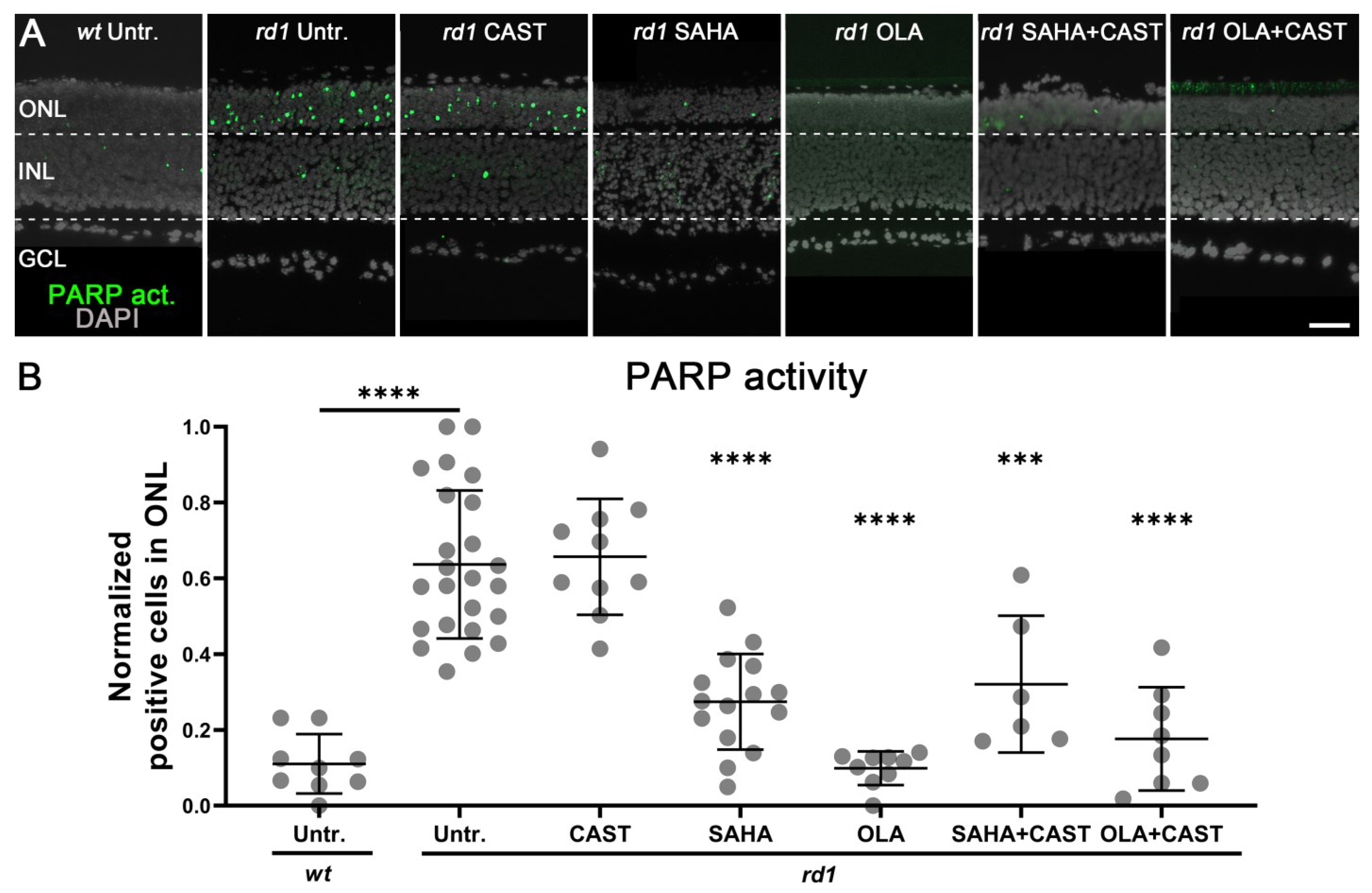
3.4. PARP Activation Depends on HDAC but Not on Calpain Activity
4. Discussion
4.1. Histone Deacetylases and Photoreceptor Degeneration
4.2. PARP as a Therapeutic Target in cGMP-Dependent Cell Death
4.3. Calpains as a Therapeutic Target in IRD
4.4. Molecular Interactions between HDAC, PARP, and Calpain
4.5. Towards the Identification of the HDAC, PARP, and Calpain Isoforms Responsible for Photoreceptor Degeneration
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Duncan, J.L.; Pierce, E.A.; Laster, A.M.; Daiger, S.P.; Birch, D.G.; Ash, J.D.; Iannaccone, A.; Flannery, J.G.; Sahel, J.A.; Zack, D.J.; et al. Inherited Retinal Degenerations: Current Landscape and Knowledge Gaps. Transl. Vis. Sci. Technol. 2018, 7, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertelsen, M.; Jensen, H.; Bregnhoj, J.F.; Rosenberg, T. Prevalence of generalized retinal dystrophy in Denmark. Ophthalmic Epidemiol. 2014, 21, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Cross, N.; Van Steen, C.; Zegaoui, Y.; Satherley, A.; Angelillo, L. Retinitis Pigmentosa: Burden of Disease and Current Unmet Needs. Clin. Ophthalmol. 2022, 16, 1993–2010. [Google Scholar] [CrossRef] [PubMed]
- Sahel, J.A.; Marazova, K.; Audo, I. Clinical characteristics and current therapies for inherited retinal degenerations. Cold Spring Harb. Perspect. Med. 2014, 5, a017111. [Google Scholar] [CrossRef]
- Arango-Gonzalez, B.; Trifunović, D.; Sahaboglu, A.; Kranz, K.; Michalakis, S.; Farinelli, P.; Koch, S.; Koch, F.; Cottet, S.; Janssen-Bienhold, U.; et al. Identification of a common non-apoptotic cell death mechanism in hereditary retinal degeneration. PLoS ONE 2014, 9, e112142. [Google Scholar] [CrossRef]
- Yan, J.; Chen, Y.; Zhu, Y.; Paquet-Durand, F. Programmed Non-Apoptotic Cell Death in Hereditary Retinal Degeneration: Crosstalk between cGMP-Dependent Pathways and PARthanatos? Int. J. Mol. Sci. 2021, 22, 10567. [Google Scholar] [CrossRef]
- Das, S.; Chen, Y.; Yan, J.; Christensen, G.; Belhadj, S.; Tolone, A.; Paquet-Durand, F. The role of cGMP-signalling and calcium-signalling in photoreceptor cell death: Perspectives for therapy development. Pflug. Arch. 2021, 473, 1411–1421. [Google Scholar] [CrossRef]
- Power, M.; Das, S.; Schutze, K.; Marigo, V.; Ekstrom, P.; Paquet-Durand, F. Cellular mechanisms of hereditary photoreceptor degeneration—Focus on cGMP. Prog. Retin. Eye Res. 2020, 74, 100772. [Google Scholar] [CrossRef]
- Keeler, C.E. The Inheritance of a Retinal Abnormality in White Mice. Proc. Natl. Acad. Sci. USA 1924, 10, 329–333. [Google Scholar] [CrossRef] [Green Version]
- Bowes, C.; Li, T.; Danciger, M.; Baxter, L.; Applebury, M.; Farber, D. Retinal degeneration in the rd mouse is caused by a defect in the beta subunit of rod cGMP-phosphodiesterase. Nature 1990, 347, 677–680. [Google Scholar] [CrossRef]
- Farber, D.B.; Lolley, R.N. Cyclic guanosine monophosphate: Elevation in degenerating photoreceptor cells of the C3H mouse retina. Science 1974, 186, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Paquet-Durand, F.; Hauck, S.M.; Van Veen, T.; Ueffing, M.; Ekström, P. PKG activity causes photoreceptor cell death in two retinitis pigmentosa models. J. Neurochem. 2009, 108, 796–810. [Google Scholar] [CrossRef] [PubMed]
- Sancho-Pelluz, J.; Alavi, M.V.; Sahaboglu, A.; Kustermann, S.; Farinelli, P.; Azadi, S.; van Veen, T.; Romero, F.J.; Paquet-Durand, F.; Ekström, P. Excessive HDAC activation is critical for neurodegeneration in the rd1 mouse. Cell Death Dis. 2010, 1, e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paquet-Durand, F.; Silva, J.; Talukdar, T.; Johnson, L.E.; Azadi, S.; Van Veen, T.; Ueffing, M.; Hauck, S.M.; Ekström, P.A. Excessive activation of poly(ADP-ribose) polymerase contributes to inherited photoreceptor degeneration in the retinal degeneration 1 mouse. J. Neurosci. 2007, 27, 10311–10319. [Google Scholar] [CrossRef]
- Paquet-Durand, F.; Azadi, S.; Hauck, S.M.; Ueffing, M.; Van Veen, T.; Ekström, P. Calpain is activated in degenerating photoreceptors in the rd1 mouse. J. Neurochem. 2006, 96, 802–814. [Google Scholar] [CrossRef]
- Jaliffa, C.; Ameqrane, I.; Dansault, A.; Leemput, J.; Vieira, V.; Lacassagne, E.; Provost, A.; Bigot, K.; Masson, C.; Menasche, M.; et al. Sirt1 involvement in rd10 mouse retinal degeneration. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3562–3572. [Google Scholar] [CrossRef] [Green Version]
- Trifunović, D.; Arango-Gonzalez, B.; Comitato, A.; Barth, M.; Del Amo, E.M.; Kulkarni, M.; Sahaboglu, A.; Hauck, S.M.; Urtti, A.; Arsenijevic, Y.; et al. HDAC inhibition in the cpfl1 mouse protects degenerating cone photoreceptors in vivo. Hum. Mol. Genet. 2016, 25, 4462–4472. [Google Scholar] [CrossRef] [Green Version]
- Sundaramurthi, H.; Roche, S.L.; Grice, G.L.; Moran, A.; Dillion, E.T.; Campiani, G.; Nathan, J.A.; Kennedy, B.N. Selective Histone Deacetylase 6 Inhibitors Restore Cone Photoreceptor Vision or Outer Segment Morphology in Zebrafish and Mouse Models of Retinal Blindness. Front. Cell Dev. Biol. 2020, 8, 689. [Google Scholar] [CrossRef]
- Samardzija, M.; Corna, A.; Gomez-Sintes, R.; Jarboui, M.A.; Armento, A.; Roger, J.E.; Petridou, E.; Haq, W.; Paquet-Durand, F.; Zrenner, E.; et al. HDAC inhibition ameliorates cone survival in retinitis pigmentosa mice. Cell Death Differ. 2021, 28, 1317–1332. [Google Scholar] [CrossRef]
- Ko, H.L.; Ren, E.C. Functional Aspects of PARP1 in DNA Repair and Transcription. Biomolecules 2012, 2, 524–548. [Google Scholar] [CrossRef] [Green Version]
- Morales, J.; Li, L.; Fattah, F.J.; Dong, Y.; Bey, E.A.; Patel, M.; Gao, J.; Boothman, D.A. Review of poly (ADP-ribose) polymerase (PARP) mechanisms of action and rationale for targeting in cancer and other diseases. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 15–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, P. Biology of Poly(ADP-Ribose) Polymerases: The Factotums of Cell Maintenance. Mol. Cell 2015, 58, 947–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- David, K.K.; Andrabi, S.A.; Dawson, T.M.; Dawson, V.L. Parthanatos, a messenger of death. Front. Biosci. 2009, 14, 1116–1128. [Google Scholar] [CrossRef] [Green Version]
- Sahaboglu, A.; Miranda, M.; Canjuga, D.; Avci-Adali, M.; Savytska, N.; Secer, E.; Feria-Pliego, J.; Kayık, G.; Durdagi, S. Drug repurposing studies of PARP inhibitors as a new therapy for inherited retinal degeneration. Cell. Mol. Life Sci. 2020, 77, 2199–2216. [Google Scholar] [CrossRef]
- Yan, J.; Gunter, A.; Das, S.; Muhlfriedel, R.; Michalakis, S.; Jiao, K.; Seeliger, M.W.; Paquet-Durand, F. Inherited Retinal Degeneration: PARP-Dependent Activation of Calpain Requires CNG Channel Activity. Biomolecules 2022, 12, 455. [Google Scholar] [CrossRef] [PubMed]
- Sahaboglu, A.; Barth, M.; Secer, E.; Amo, E.M.; Urtti, A.; Arsenijevic, Y.; Zrenner, E.; Paquet-Durand, F. Olaparib significantly delays photoreceptor loss in a model for hereditary retinal degeneration. Sci. Rep. 2016, 6, 39537. [Google Scholar] [CrossRef] [Green Version]
- Potz, B.A.; Abid, M.R.; Sellke, F.W. Role of Calpain in Pathogenesis of Human Disease Processes. J. Nat. Sci. 2016, 2, e218. [Google Scholar]
- Michalakis, S.; Becirovic, E.; Biel, M. Retinal Cyclic Nucleotide-Gated Channels: From Pathophysiology to Therapy. Int. J. Mol. Sci. 2018, 19, 749. [Google Scholar] [CrossRef] [Green Version]
- Paquet-Durand, F.; Beck, S.; Michalakis, S.; Goldmann, T.; Huber, G.; Mühlfriedel, R.; Trifunović, D.; Fischer, M.D.; Fahl, E.; Duetsch, G.; et al. A key role for cyclic nucleotide gated (CNG) channels in cGMP-related retinitis pigmentosa. Hum. Mol. Genet. 2011, 20, 941–947. [Google Scholar] [CrossRef] [Green Version]
- Kaur, J.; Mencl, S.; Sahaboglu, A.; Farinelli, P.; Van Veen, T.; Zrenner, E.; Ekstrom, P.; Paquet-Durand, F.; Arango-Gonzalez, B. Calpain and PARP activation during photoreceptor cell death in P23H and S334ter rhodopsin mutant rats. PLoS ONE 2011, 6, e22181. [Google Scholar] [CrossRef] [Green Version]
- Paquet-Durand, F.; Johnson, L.; Ekström, P. Calpain activity in retinal degeneration. J. Neurosci. Res. 2007, 85, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hrzenjak, A.; Moinfar, F.; Kremser, M.L.; Strohmeier, B.; Petru, E.; Zatloukal, K.; Denk, H. Histone deacetylase inhibitor vorinostat suppresses the growth of uterine sarcomas in vitro and in vivo. Mol. Cancer 2010, 9, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menear, K.; Adcock, C.; Boulter, R.; Cockcroft, X.; Copsey, L.; Cranston, A.; Dillon, K.; Drzewiecki, J.; Garman, S.; Gomez, S.; et al. 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin-1-one: A novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1. J. Med. Chem. 2008, 51, 6581–6591. [Google Scholar] [CrossRef] [PubMed]
- Fiorino, F.; Gil-Parrado, S.; Assfalg-Machleidt, I.; Machleidt, W.; Moroder, L. A new cell-permeable calpain inhibitor. J. Pept. Sci. 2007, 13, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Belhadj, S.; Tolone, A.; Christensen, G.; Das, S.; Chen, Y.; Paquet-Durand, F. Long-Term, Serum-Free Cultivation of Organotypic Mouse Retina Explants with Intact Retinal Pigment Epithelium. J. Vis. Exp. 2020, 165, e61868. [Google Scholar] [CrossRef]
- Goldman, A.; Ursitti, J.; Mozdzanowski, J.; Speicher, D. Electroblotting from Polyacrylamide Gels. Curr. Protoc. Protein Sci. 2015, 82, 10.7.1–10.7.6. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, Y.; Bi, X.; Baudry, M. Calpain-1 and Calpain-2 in the Brain: New Evidence for a Critical Role of Calpain-2 in Neuronal Death. Cells 2020, 9, 2698. [Google Scholar] [CrossRef]
- Ekstrom, P.A.; Ueffing, M.; Zrenner, E.; Paquet-Durand, F. Novel in situ activity assays for the quantitative molecular analysis of neurodegenerative processes in the retina. Curr. Med. Chem. 2014, 21, 3478–3493. [Google Scholar] [CrossRef]
- Power, M.J.; Rogerson, L.E.; Schubert, T.; Berens, P.; Euler, T.; Paquet-Durand, F. Systematic spatiotemporal mapping reveals divergent cell death pathways in three mouse models of hereditary retinal degeneration. J. Comp. Neurol. 2020, 528, 1113–1139. [Google Scholar] [CrossRef] [Green Version]
- Buki, K.G.; Bauer, P.I.; Kun, E. Isolation and identification of a proteinase from calf thymus that cleaves poly (ADP-ribose) polymerase and histone H1. Biochim. Biophys. Acta BBA Protein Struct. Mol. Enzymol. 1997, 1338, 100–106. [Google Scholar]
- Chen, B.; Cepko, C.L. HDAC4 regulates neuronal survival in normal and diseased retinas. Science 2009, 323, 256–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trifunović, D.; Petridou, E.; Comitato, A.; Marigo, V.; Ueffing, M.; Paquet-Durand, F. Primary Rod and Cone Degeneration Is Prevented by HDAC Inhibition. Adv. Exp. Med. Biol. 2018, 1074, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Vent-Schmidt, R.Y.J.; Wen, R.H.; Zong, Z.; Chiu, C.N.; Tam, B.M.; May, C.G.; Moritz, O.L. Opposing Effects of Valproic Acid Treatment Mediated by Histone Deacetylase Inhibitor Activity in Four Transgenic X. laevis Models of Retinitis Pigmentosa. J. Neurosci. 2017, 37, 1039–1054. [Google Scholar] [CrossRef] [PubMed]
- Mitton, K.P.; Guzman, A.E.; Deshpande, M.; Byrd, D.; DeLooff, C.; Mkoyan, K.; Zlojutro, P.; Wallace, A.; Metcalf, B.; Laux, K.; et al. Different effects of valproic acid on photoreceptor loss in Rd1 and Rd10 retinal degeneration mice. Mol. Vis. 2014, 20, 1527–1544. [Google Scholar]
- Zhang, Q.; Wang, S.; Chen, J.; Yu, Z. Histone Deacetylases (HDACs) Guided Novel Therapies for T-cell lymphomas. Int. J. Med. Sci. 2019, 16, 424–442. [Google Scholar] [CrossRef] [Green Version]
- Wawruszak, A.; Borkiewicz, L.; Okon, E.; Kukula-Koch, W.; Afshan, S.; Halasa, M. Vorinostat (SAHA) and Breast Cancer: An Overview. Cancers 2021, 13, 4700. [Google Scholar] [CrossRef]
- Himawan, E.; Ekström, P.; Buzgo, M.; Gaillard, P.; Stefánsson, E.; Marigo, V.; Loftsson, T.; Paquet-Durand, F. Drug delivery to retinal photoreceptors. Drug Discov. Today 2019, 24, 1637–1643. [Google Scholar] [CrossRef]
- Curtin, N.J.; Szabo, C. Poly(ADP-ribose) polymerase inhibition: Past, present and future. Nat. Rev. Drugs Discov. 2020, 19, 711–736. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Sahaboglu, A.; Sharif, A.; Feng, L.; Secer, E.; Zrenner, E.; Paquet-Durand, F. Temporal progression of PARP activity in the Prph2 mutant rd2 mouse: Neuroprotective effects of the PARP inhibitor PJ34. PLoS ONE 2017, 12, e0181374. [Google Scholar] [CrossRef] [PubMed]
- Sahaboglu, A.; Tanimoto, N.; Kaur, J.; Sancho-Pelluz, J.; Huber, G.; Fahl, E.; Arango-Gonzalez, B.; Zrenner, E.; Ekström, P.; Löwenheim, H. PARP1 gene knock-out increases resistance to retinal degeneration without affecting retinal function. PLoS ONE 2010, 5, e15495. [Google Scholar]
- Jiao, K.; Sahaboglu, A.; Zrenner, E.; Ueffing, M.; Ekstrom, P.A.; Paquet-Durand, F. Efficacy of PARP inhibition in Pde6a mutant mouse models for retinitis pigmentosa depends on the quality and composition of individual human mutations. Cell Death Discov. 2016, 2, 16040. [Google Scholar] [CrossRef] [PubMed]
- Vosler, P.S.; Sun, D.; Wang, S.; Gao, Y.; Kintner, D.B.; Signore, A.P.; Cao, G.; Chen, J. Calcium dysregulation induces apoptosis-inducing factor release: Cross-talk between PARP-1- and calpain-signaling pathways. Exp. Neurol. 2009, 218, 213–220. [Google Scholar] [CrossRef] [Green Version]
- Storr, S.J.; Carragher, N.O.; Frame, M.C.; Parr, T.; Martin, S.G. The calpain system and cancer. Nat. Rev. Cancer 2011, 11, 364–374. [Google Scholar] [CrossRef]
- Metwally, E.; Zhao, G.; Zhang, Y.Q. The calcium-dependent protease calpain in neuronal remodeling and neurodegeneration. Trends Neurosci. 2021, 44, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.L.; Mattson, M.P. Caspase and calpain substrates: Roles in synaptic plasticity and cell death. J. Neurosci. Res. 1999, 58, 167–190. [Google Scholar]
- Baudry, M.; Bi, X. Calpain-1 and Calpain-2: The Yin and Yang of Synaptic Plasticity and Neurodegeneration. Trends Neurosci. 2016, 39, 235–245. [Google Scholar] [CrossRef] [Green Version]
- Baudry, M. Calpain-1 and Calpain-2 in the Brain: Dr. Jekill and Mr Hyde? Curr. Neuropharmacol. 2019, 17, 823–829. [Google Scholar] [CrossRef]
- Misztak, P.; Sowa-Kucma, M.; Szewczyk, B.; Nowak, G. Vorinostat (SAHA) May Exert Its Antidepressant-Like Effects Through the Modulation of Oxidative Stress Pathways. Neurotox. Res. 2021, 39, 170–181. [Google Scholar] [CrossRef]
- Bjelland, S.; Seeberg, E. Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutat. Res. 2003, 531, 37–80. [Google Scholar] [CrossRef] [PubMed]
- Kryston, T.B.; Georgiev, A.B.; Pissis, P.; Georgakilas, A.G. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat. Res. 2011, 711, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Woodhouse, B.C.; Dianova, I.I.; Parsons, J.L.; Dianov, G.L. Poly(ADP-ribose) polymerase-1 modulates DNA repair capacity and prevents formation of DNA double strand breaks. DNA Repair 2008, 7, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Eustermann, S.; Wu, W.F.; Langelier, M.F.; Yang, J.C.; Easton, L.E.; Riccio, A.A.; Pascal, J.M.; Neuhaus, D. Structural Basis of Detection and Signaling of DNA Single-Strand Breaks by Human PARP-1. Mol. Cell 2015, 60, 742–754. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Himawan, E.; Belhadj, S.; Perez Garcia, R.O.; Paquet Durand, F.; Schipper, N.; Buzgo, M.; Simaite, A.; Marigo, V. Efficient Delivery of Hydrophilic Small Molecules to Retinal Cell Lines Using Gel Core-Containing Solid Lipid Nanoparticles. Pharmaceutics 2021, 14, 74. [Google Scholar] [CrossRef]
- Hegedus, L.; Padanyi, R.; Molnar, J.; Paszty, K.; Varga, K.; Kenessey, I.; Sarkozy, E.; Wolf, M.; Grusch, M.; Hegyi, Z.; et al. Histone Deacetylase Inhibitor Treatment Increases the Expression of the Plasma Membrane Ca(2+) Pump PMCA4b and Inhibits the Migration of Melanoma Cells Independent of ERK. Front. Oncol. 2017, 7, 95. [Google Scholar] [CrossRef] [Green Version]
- Dell’Orco, D.; Koch, K.W.; Rispoli, G. Where vision begins. Pflug. Arch. 2021, 473, 1333–1337. [Google Scholar] [CrossRef]
- Van Hasselt, J.G.C.; Iyengar, R. Systems Pharmacology: Defining the Interactions of Drug Combinations. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 21–40. [Google Scholar] [CrossRef] [Green Version]
- Jia, H.; Morris, C.D.; Williams, R.M.; Loring, J.F.; Thomas, E.A. HDAC inhibition imparts beneficial transgenerational effects in Huntington’s disease mice via altered DNA and histone methylation. Proc. Natl. Acad. Sci. USA 2015, 112, E56–E64. [Google Scholar] [CrossRef] [Green Version]
- Tolić, A.; Ravichandran, M.; Rajić, J.; Đorđević, M.; Đorđević, M.; Dinić, S.; Grdović, N.; Jovanović, J.A.; Mihailović, M.; Nestorović, N. TET-mediated DNA hydroxymethylation is negatively influenced by the PARP-dependent PARylation. Epigenetics Chromatin 2022, 15, 11. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Y. Regulation of TET protein stability by calpains. Cell Rep. 2014, 6, 278–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.; Xu, W.; Li, Y.; Wei, C.; Hu, Y.; Hu, Z.; Paquet-Durand, F.; Jiao, K. Inhibition of the MAPK/c-Jun-EGR1 Pathway Decreases Photoreceptor Cell Death in the rd1 Mouse Model for Inherited Retinal Degeneration. Int. J. Mol. Sci. 2022, 23, 14600. [Google Scholar] [CrossRef] [PubMed]
- El-Brolosy, M.A.; Stainier, D.Y. Genetic compensation: A phenomenon in search of mechanisms. PLoS Genet. 2017, 13, e1006780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Awady, R.; Saleh, E.; Hamoudi, R.; Ramadan, W.S.; Mazitschek, R.; Nael, M.A.; Elokely, K.M.; Abou-Gharbia, M.; Childers, W.E.; Srinivasulu, V. Discovery of novel class of histone deacetylase inhibitors as potential anticancer agents. Bioorganic Med. Chem. 2021, 42, 116251. [Google Scholar] [CrossRef] [PubMed]
- Richon, V.M.; Emiliani, S.; Verdin, E.; Webb, Y.; Breslow, R.; Rifkind, R.A.; Marks, P.A. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc. Natl. Acad. Sci. USA 1998, 95, 3003–3007. [Google Scholar] [CrossRef] [Green Version]
- Pandey, U.B.; Nie, Z.; Batlevi, Y.; McCray, B.A.; Ritson, G.P.; Nedelsky, N.B.; Schwartz, S.L.; DiProspero, N.A.; Knight, M.A.; Schuldiner, O. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007, 447, 860–864. [Google Scholar] [CrossRef]
- Kamaletdinova, T.; Fanaei-Kahrani, Z.; Wang, Z.-Q. The enigmatic function of PARP1: From PARylation activity to PAR readers. Cells 2019, 8, 1625. [Google Scholar] [CrossRef] [Green Version]
- Maki, M.; Bagci, H.; Hamaguchi, K.; Ueda, M.; Murachi, T.; Hatanaka, M. Inhibition of calpain by a synthetic oligopeptide corresponding to an exon of the human calpastatin gene. J. Biol. Chem. 1989, 264, 18866–18869. [Google Scholar] [CrossRef]
- Ono, Y.; Saido, T.C.; Sorimachi, H. Calpain research for drug discovery: Challenges and potential. Nat. Rev. Drug Discov. 2016, 15, 854–876. [Google Scholar] [CrossRef]
- De Tullio, R.; Averna, M.; Pedrazzi, M.; Sparatore, B.; Salamino, F.; Pontremoli, S.; Melloni, E. Differential regulation of the calpain–calpastatin complex by the L-domain of calpastatin. Biochim. Biophys. Acta BBA Mol. Cell Res. 2014, 1843, 2583–2591. [Google Scholar] [CrossRef] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, Y.; Yan, J.; Yang, M.; Xu, W.; Hu, Z.; Paquet-Durand, F.; Jiao, K. Inherited Retinal Degeneration: Towards the Development of a Combination Therapy Targeting Histone Deacetylase, Poly (ADP-Ribose) Polymerase, and Calpain. Biomolecules 2023, 13, 581. https://doi.org/10.3390/biom13040581
Dong Y, Yan J, Yang M, Xu W, Hu Z, Paquet-Durand F, Jiao K. Inherited Retinal Degeneration: Towards the Development of a Combination Therapy Targeting Histone Deacetylase, Poly (ADP-Ribose) Polymerase, and Calpain. Biomolecules. 2023; 13(4):581. https://doi.org/10.3390/biom13040581
Chicago/Turabian StyleDong, Yujie, Jie Yan, Ming Yang, Wenrong Xu, Zhulin Hu, François Paquet-Durand, and Kangwei Jiao. 2023. "Inherited Retinal Degeneration: Towards the Development of a Combination Therapy Targeting Histone Deacetylase, Poly (ADP-Ribose) Polymerase, and Calpain" Biomolecules 13, no. 4: 581. https://doi.org/10.3390/biom13040581