1. Introduction
A microbiome defines a community of microorganisms and their activities in a given environment. This term encompasses the microbial species themselves, but also the collection of molecules they produce such as metagenomes [
1,
2]. Microbiome studies can be useful to different fields such as medicine or environmental protection. For example, the human gut microbiome is being extensively analyzed to uncover how its composition is linked to various disorders [
3], while the ocean microbiome provides information on the potential impact of climate change on marine biodiversity [
4].
The metagenomic study of microbiomes has gained a lot of interest due to the progress made in DNA sequencing technology. While the history of DNA sequencing started a decade after many proteins were already sequenced and RNA sequencing was being apprehended [
5], it quickly evolved in the late 1970s when Sanger and Gilbert independently developed methods that allowed sequencing of 50 and 100 nucleotides, respectively [
6,
7]. The automation of Sanger’s technique combined with the desire to sequence large fragments of DNA brought various improvements that led to the development of efficient machines able to perform DNA sequencing in a particularly parallel fashion. Current high-throughput sequencing methods can produce billions of DNA fragments simultaneously during a single run. In addition, high-throughput sequencing technology offers speed and a decrease in cost per base, but also offers high sequencing depth that comes with better sensitivity. This provides the means to study uncultivable microorganisms and to detect low abundance microorganisms of a microbial community.
In a typical metagenomic study, the genetic material in all organisms contained in a given sample is fragmented, and the DNA fragments sequenced are identified as reads. Following sequencing, diverse bioinformatic tools are used to remove low-quality sequences and to assemble overlapping reads into contiguous DNA segments, also called contigs. Contigs are then arranged through scaffolding into longer segments to eventually reconstruct genomes present in the sample. This complex process of de novo sequence assembly is further challenged when dealing with short-read sequences and the high sequencing depth that is required to differentiate similar or repetitive sequences. The recent development of third-generation sequencing platforms enabled the determination of long-read sequences. With a length of 10–25 kb [
8] from the Pacific Biosciences (PacBio) and 10–100 kb from Oxford Nanopore Technologies (ONT) platforms, de novo assembly will be greatly facilitated and improved as it can already be seen [
9,
10].
A complementary approach to analyze metagenomic data and provide information on the composition of microbial communities is the taxonomic classification of reads. This method involves assigning a taxonomic group to every read with the goal of classifying as many sequences as possible and identifying species present in the sample. One strategy for taxonomic classification consists of comparing k-mer signatures in metagenomic reads to a database of categorized k-mers. One of the state-of-the-art tools for metagenomic classification is Kraken [
11], which relies on a database of k-mers with each k-mer associated with the lowest common ancestor of all genomes containing that specific k-mer. Kraken has been criticized for employing a memory-intensive algorithm [
12,
13,
14], prompting its designers to release Kraken2, which features a more memory-efficient data structure [
15]. An alternative method to efficiently store and query the database of k-mers is a modified implementation of the FM-index, employed by Centrifuge [
16]. Both Kraken2 and Centrifuge have been praised in the literature for providing high accuracy and rapid runtimes [
13,
17].
The rapid development of deep learning techniques has inspired new applications in the analysis of metagenomic data. Deep learning models rely on artificial neural networks designed based on the structure and function of neurons in the human brain [
18]. Complex deep learning models containing many layers have the ability to extract relevant features and find abstract patterns in data, allowing them to achieve high accuracy. For example, the desire to push forward the capacities of deep neural networks has led to the development of new techniques and architectures to classify images, which can reach an accuracy of 99.84% [
19] on the MNIST handwritten digit classification dataset.
The first study to consider deep learning algorithms in the classification of DNA sequences built a convolutional neural network (CNN) to classify 16S small subunit ribosomal RNA (rRNA) genes which are commonly used for the identification of bacteria [
20]. This study employed the bag of words technique to represent reads simulated from 16S rRNA reference sequences in terms of k-mer occurrences, thus obtaining sparse matrices as input vectors for their neural network. A k-mer size of 7 was used to restrict the storage and computational complexities that occur with sparse input vectors. Despite these limitations, they reported an accuracy of 91% across 100 bacterial genera on an artificial validation dataset. Another CNN method was proposed to classify short reads of 16S rRNA genes across 2768 genera, and achieved better sensitivity compared to Kraken2 at the genus level on 100 bp and 200 bp synthetic reads generated using 16S rRNA genes as templates [
21]. While the tools mentioned above would support a high classification rate with amplicon sequencing data that targets specific genetic regions such as the 16S rRNA genes, other software have been designed to analyze the entire genetic materials sequenced from a sample. One such method called GeNet shows the improvement of training a CNN model with long DNA sequences by recording better classification of long metagenomic reads from a mock community consisting of ten microbial species, with comparable performances with Kraken and Centrifuge at the species and genus levels [
22]. A more recent tool called DeepMicrobes targets 2505 bacterial species from the human gut and implements a bidirectional long short-term memory (LSTM) in addition to a self-attention mechanism [
23]. DeepMicrobes outperforms other traditional taxonomic classification tools at the genus level on mock communities, suggesting the potential of LSTM in metagenomic read classification. However, LSTM is significantly slower than CNN. Finally, a recent model called BERTax, based on the state-of-the-art model BERT for natural language processing, classifies DNA sequences at the superkingdom, phylum, and genus taxonomic levels and shows generalization on unknown data compared to other approaches mentioned previously [
24]. For a more in depth analysis of the deep learning techniques applied to taxonomic classification, we recommend a review published recently by [
25].
Here, we present DL-TODA, a deep learning model based on CNN that classifies short metagenomic reads from over 3000 bacterial species. Compared to the aforementioned tools, DL-TODA is trained with a modified version of the deep neural network AlexNet, a successful CNN in computer vision. A training dataset containing 250 bp reads simulated from all complete bacterial genomes available in the NCBI Reference Sequence database was used for training the DL-TODA model. This enabled the identification of bacteria originating from a wide range of free-living and host-associated habitats. DL-TODA classifies each read at the species level and supports the inference of higher-order taxa based on NCBI or GTDB taxonomy. A probability score is generated for each prediction, hence permitting the quality control of prediction results based on probability thresholds.
2. Materials and Methods
An overview of all steps involved in the training, validation, and testing of the DL-TODA model is presented in
Figure S1. Below, we provide detailed descriptions of the corresponding steps.
2.1. Bacterial Genome Selection
A total of 9859 complete bacterial genomes representing 3313 different species isolated from diverse free-living and host-associated environments were selected from the genome taxonomy database (GTDB) release 95 and the NCBI RefSeq database, downloaded on 7 March 2020. The genomes selected are not derived from metagenome or environmental samples and have a size equal to or above 500 kb. For each species, 70% of the genomes were randomly assigned for model training and the remaining 30% for model testing. In the cases of species with a single genome, the genome in question was automatically appointed to the training set. Additionally, all representative genomes from GTDB were automatically assigned for training. In total, we have 7405 and 2454 genomes assigned for training and testing, respectively.
Table 1 provides a summary of the number of taxa represented at species, genus, family, order, class and phylum levels in training, validation and testing sets, and for both GTDB and NCBI taxonomy. A smaller number of taxa are represented in the NCBI classification due to different assignments with the GTDB classification. For example, amongst the 537 genomes classified as
Escherichia coli by NCBI, 363, 93, 80 and 1 genomes are assigned by GTDB to
Escherichia flexneri,
Escherichia coli,
Escherichia dysenteriae, and
Escherichia coli_C, respectively. Additionally, 244 genomes lack specific assignments in at least one of the given taxonomic ranks in the NCBI taxonomy database. For example, genome GCA_000317655.1 is not assigned a class in the NCBI taxonomy but is assigned to the class of Cyanobacteria in the GTDB taxonomy.
2.2. Reads Simulation
Paired-end reads of 250 bp were simulated using ART Illumina read simulator (version 2.5.8) [
26]. A coverage of 7 and 3 was used for read simulations using training and testing genomes, respectively. A mean fragment length of 300 bp and a standard deviation of fragment length of 10 bp were chosen according to ART Illumina usage information. The built-in error profile of MiSeq v1 (MSv1) was used for simulation. The command for running ART Illumina is art_illumina -ss ‘MSv1’ -i <input fasta file> -d <reads prefix id> -na -s <standard deviation of fragment length> -m <mean fragment length> -l <read length> -f <fold coverage> -p -o <output file>.
2.3. Training, Validation and Testing Sets
Paired-end reads obtained from training genomes were randomly shuffled and split into 70% for training and 30% for validation. The forward and reverse reads from testing genomes were treated separately and classified independently. Identical reads between the training and testing data were identified by clustering the training and testing reads using Mmseqs2 easy-linclust (version 13.45111) with a minimum sequence identity of 1.0 and a fraction of aligned residues of 1.0. To avoid biases in testing, testing reads that are identical to the training reads were removed from the testing set.
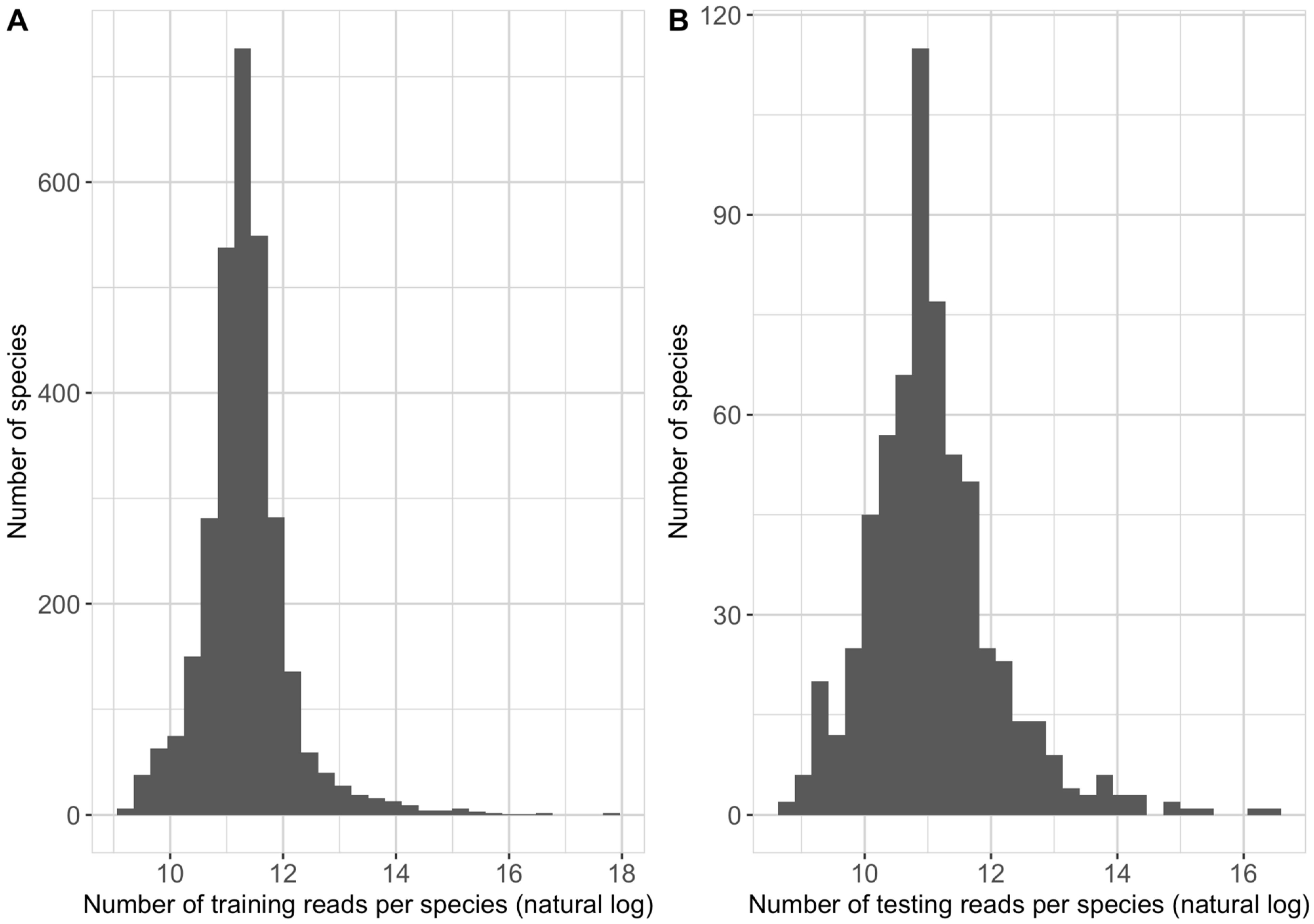
Table 2 summarizes the final number of reads included in the training, validation and testing sets of this study. The number of training reads allocated to each species in the NCBI taxonomy had a median of 80,067 and ranges between 10,359 and 56,838,380 (
Figure 1A). The number of testing reads allocated to each species in the NCBI taxonomy had a median of 56,839 and ranges between 6455 and 14,223,296 (
Figure 1B). The genome coverage represented in the training data was calculated for each species based on Equation (1), where the “number of training reads” are the number of reads assigned to a given species label in the training data, and the “average training genomes size” accounts for the average length of training genomes of the given species.
2.4. Deep Learning Neural Network
2.4.1. Reads Representation
DL-TODA represents each read as a vector of k-mers, using a sliding-window of size 12 across the 250 bp read sequence. Reads shorter than 250 bp were padded with 0s before representation of the k-mers. A vector of 239 integers was then used to represent each read based on a k-mer size of 12 and an indexed vocabulary of 12-mers (described in the section below). The read vectors were then stored in TensorflowRecord (TFRecord) files alongside labels corresponding to the species assignment (i.e., ground truth), and presented to the embedding layer.
2.4.2. K-mer Embedding
The DL-TODA model embeds each k-mer by choosing only the canonical form in a pair consisting of the k-mer and its reverse complement. The canonical k-mer corresponds to the k-mer that appears first, according to the alphabetical order. This strategy allows us to reduce the vocabulary learned by the neural network and therefore lower the complexity of the model. The number of all possible canonical 12-mers is 8,390,656, defined as
. The vocabulary of DL-TODA included all the canonical 12-mers and two additional digits, one accounting for unknown 12-mer with characters different from the four universal bases (i.e., A, T, G, C), and another for padded 0s to the right of sequences shorter than 250 bp. Following the vocabulary definition, each 12-mer was assigned an index between 0 and 8,390,657 in order to retrieve a vector of 60 real values from a list. These vectors were initiated in the Tensorflow embedding layer, with each real value drawn from the He Normal distribution [
27], and were updated during training.
2.4.3. DL-TODA Neural Network
The deep neural network architecture of DL-TODA is a modified version of AlexNet [
28] (
Figure 2) with a trainable embedding layer generating an (8,390,658 × 60) embedding matrix. The input layer of this neural network is a (239 × 60) matrix consisting of 239 rows of 12-mers embedded as 60 real value vectors (described above). The input data are then processed by five convolutional layers, two max pooling layers and three fully connected layers. The rectified linear unit (ReLU) activation function is applied throughout the neural network, except in the last layer, in which the softmax function transforms the output from the fully connected layer to a probability distribution over the 3313 species.
2.4.4. Loss Function and Probability Scores
The following cross entropy loss function (Equation (2)) was used to compute the difference between the species desired output (0 or 1) and the estimated probability of correct prediction for a given species for one example.
The estimated probability of every species is obtained by applying the softmax function [
29] to an output vector of 3313 real numbers.
2.5. Training and Testing
Data loading to the neural network was performed using the Nvidia Data Loading Library (DALI). Shuffling was carried out exclusively for the training and validation sets. Distributed training was executed by dispatching batches of 512 reads to four different GPUs (global batch size of 2048). Each GPU computed gradient updates independently; these were then averaged together and finally applied to the model. The accuracy and loss computed with the training and validation sets were monitored and saved throughout the training to create learning curves (
Figure 3). Additionally, the model was saved at the end of every epoch. Testing and applications to the oral and soil metagenomes were carried out similarly with a batch size per GPU of 512 reads distributed among four GPUs and using the trained model saved at epoch 14.
2.6. Evaluation of Model Performance
The performance of DL-TODA was assessed with the overall classification accuracy, defined in Equation (3), at different taxonomic ranks including species, genus, family, order, class and phylum.
At the species level, the number of correctly classified reads was directly obtained from the neural network. At higher taxonomic ranks, the number of correctly classified reads was calculated with the sum of all reads that were correctly assigned to the species within each taxon.
The percentage of classified vs. unclassified reads was also examined with the application of different thresholds on the predicted probability of species. The selection of threshold settings was guided by the overall distribution of probability scores among the correct or incorrect classification in the testing dataset (
Figure 4). The eqgamma function of the R package EnvStats (version 2.7.0) was used for identifying confidence intervals based on a gamma distribution for the elimination of incorrect predictions. The precision (Equation (4)), recall (Equation (5)) and F1-score (Equation (6)) were obtained for each species. The macro and micro average of each metric (Equations (7)–(12)) were computed to provide a comparison of the performance between DL-TODA, Kraken2 and Centrifuge. The number of true positives (TP), false positives (FP) and false negatives (FN) per species required to compute precision, recall and F1-score were obtained based on the generation of a confusion matrix.
2.7. Comparison with Kraken2 and Centrifuge
We evaluated the performance of DL-TODA in comparison with Kraken2 version 2.0.8 and Centrifuge version 1.0.3. For both programs, an index was built with the training genomes as references to classify the simulated reads in the testing set using the default settings. Given that both Kraken2 and Centrifuge classify reads to the NCBI taxonomy database, we used the NCBI taxonomy for analyzing the results from DL-TODA. Centrifuge provides multiple possible predictions per pair of reads or unpaired reads. Here, the top hit was systematically used as the predicted taxon.
2.8. Classification of Metagenomic Data
The functionality of DL-TODA was determined by classifying metagenomes obtained from sampling two distinct environments, human oral cavity and cropland soil. The human oral cavity datasets were identified following [
30]. The cropland soil datasets (NCBI accessions: ERR5004682, ERR5003895, ERR5003204, ERR5001925 and ERR4995171) were identified from the National Microbiome Data Collaborative (NMCD) data portal [
31], using “soil” as the keyword for ecosystem type and “cropland ecosystem” as the keywords for broad-scale environmental context. The metagenomic reads were retrieved using the SRA Toolkit from NCBI, converted to TFRecords and classified by DL-TODA with a probability score threshold above 0.5 (i.e., reads with probability scores below or equal to 0.5 were counted as unclassified). The relative abundance of each taxon was measured by dividing the number of reads classified to that taxon by the total number of reads in the metagenome (Equation (13)). The DL-TODA classification was compared with Kraken2 and Centrifuge classifications of the same metagenomes, using the training genomes as references.
2.9. Computational Requirements
The DL-TODA model was trained and tested on a compute node with 768 GB of High Performance DDR4 2666 MHz ECC system memory, 48 Intel Xeon Cascade Lake Scalable Cloud Ready Processor Cores/2.2 GHz processors and four Nvidia A100/40 GB HBM2 Memory GPUs. Kraken2 and Centrifuge were run on a compute node with 24 Intel(R) Xeon(R) CPU E5-4607 0/2.20 GHz processors and 512 GB of memory. The deep learning model was implemented with TensorFlow as a Python3 script, Horovod was used to distribute training across multiple GPUs and the Nvidia DALI was used to load the TFRecord files.
4. Discussion
Taxonomic classification of billions of short sequencing reads is an important step in the analysis of metagenomic data, shedding light into the function and diversity of microbiomes. Such analysis can be performed by several existing programs but still has room for improvement. K-mer based approaches, such as Kraken2 and Centrifuge, are the most common strategies to classify metagenomic data. While both Kraken2 and Centrifuge rely on the construction of reference databases, the use of a deep learning model in DL-TODA permits the extraction of features during model construction, hence circumventing the requirement of a reference database.
An accuracy similar to higher classification was achieved by DL-TODA compared to Kraken2 and Centrifuge on an independent test set of over a hundred million simulated metagenomic reads (
Figure 6). A look at the precision, recall and F1-score (
Table 3) further demonstrated the better performance of DL-TODA, as it carried a higher micro average on all three metrics compared to Kraken2 and Centrifuge. However, lower macro than micro averages were observed in DL-TODA, indicating potential differences in how well it recognizes different species. In contrast, Kraken2 and Centrifuge appeared to perform more equally across species, as their macro average metrics are comparable to the corresponding micro average metrics.
One possible reason why DL-TODA may have performed poorly on some species may be the lack of sufficient training data. This is supported by the positive correlations between depth of training genome coverage and minimum precisions observed (
Figure S2). For example, when the coverage is greater than 55 (
), the precision values are consistently higher than 0.75, suggesting that a higher and potentially more diverse set of training data may lead to an enhanced performance of DL-TODA. However, we note that some species, despite having a low number of training reads, reached high precisions in DL-TODA predictions. This may indicate that the DL-TODA model is efficient at extracting traits from these species for label classification. While reaching high performances on a majority of the species tested (
Figure 5), DL-TODA seems to assign low probability scores to reads from a few species, resulting in low precisions approaching zero for the prediction of these species, especially when a probability threshold is used. Given the variability in the classification of different species, the probability threshold may be individually adjusted for each species to optimize the performance of DL-TODA. A careful selection of the probability threshold may require more benchmarking efforts to maximize the prediction accuracy while minimizing the fraction of unclassified data; this may be a topic of future research using diverse test cases. Future studies that seek to reveal the correlations between different genomic features (e.g., GC content, tetranucleotide frequency, distribution of mobile genetic elements, etc.) and the outcomes of read classifications can also help guide the further advancement of DL-TODA models and enhance their precision across all species.
The application of DL-TODA to the human oral and cropland soil metagenomes supports a general consensus on the prediction of top-ranking taxa, but distinct predictions on the relative abundance of different taxonomic groups compared to Kraken2 and Centrifuge (
Figure 7). In the human oral metagenomes, DL-TODA identified a higher proportion of Clostridia, which is known to be abundant and diverse in the human oral microbiome [
33] compared to Centrifuge and Kraken2. Likewise, in the cropland soil metagenomes, a higher proportion of Clostridia and Coriobacteriia was identified by DL-TODA compared to Centrifuge or Kraken2. The abundance of Clostridia and Coriobacteriia, as predicted by DL-TODA, aligns well with prior studies of diverse agricultural related soil types [
34,
35,
36]. Due to the lack of ground truth data, it is difficult to fully assess the accuracy of different tools on the metagenomes. However, the Centrifuge and Kraken2 predictions seem to be highly skewed towards assigning large proportions to a small number of taxa. For example, the class Actinomycetia was assigned the highest proportions by both Centrifuge and Kraken2 in both the oral and soil metagenomes, suggesting the potential biases of Centrifuge and Kraken2 towards classifying certain taxa. In contrast, the prediction of DL-TODA is less biased towards a single taxon, and it predicted different rankings of the dominant taxa between the human oral cavity and cropland soil, two highly distinct environments. The total number of reads classified remains low across all three tools, and the percentage of classified reads varies among the two environments tested (
Table 4). Large differences were observed with Kraken2, which classified over 20% of reads in the oral metagenome but only around 5% of reads in the soil metagenome. Centrifuge seems to have classified the highest proportion of reads among all three tools in both the oral and soil metagenomes. Considering that Centrifuge assigns the same taxa to paired reads, similar strategies may be employed by DL-TODA to leverage the read pairs for enhancing the number of classified reads. It is noted that the DL-TODA predictions were based on a probability threshold higher than 0.5 which was uniformly applied to all taxa. Based on discussions in the above paragraph, further optimization of the probability threshold, together with the introduction of more training data, especially for some underrepresented species, will likely further enhance the number of classified reads in the metagenomes.
Overall, DL-TODA is a new deep learning-based model for the taxonomic classification of metagenomic reads. The model showed a high accuracy in classifying synthetic reads and demonstrated the potential of recognizing a wide range of taxonomic groups from diverse environments. Besides DL-TODA, several other deep learning models have recently been created for the classification of metagenomic data, showing varied accuracy and generalizability, usually at the genus or higher taxonomic levels [
23,
24]. DL-TODA is distinct from these deep learning-based read classification tools. It uses a convolutional neural network designed based on the architecture of AlexNet and classifies metagenomic reads at the species level. DL-TODA has the ability to classify over 3000 bacterial species, covering all the phyla represented in the current GTDB and NCBI databases. An additional advantage of DL-TODA is the possibility to resume training with new data without needing to reanalyze the previous training sets. This allows the model to be efficiently updated with newly discovered genomes. DL-TODA also supports the calibration of classification results based on a probability score associated with each taxonomic assignment. The implementation of DL-TODA is designed to support high efficiency in processing high volumes of metagenomic data. By making use of Horovod, DL-TODA distributes the training and testing tasks across multiple GPUs in parallel, faster than with the data distribution strategy provided by TensorFlow. This feature, in addition to loading data directly to the GPU memory using the Nvidia DALI library, creates an efficient pipeline for dealing with large datasets. Future developments will include investigating solutions to reduce the size and number of parameters in DL-TODA to further accelerate the training and testing processes. Given the rapid growth of deep learning applications in metagenomic data analysis, future benchmarking studies would provide useful guidelines for the application of different deep learning tools and will likely nurture the engagement of a broader scientific community.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}