
Hybrid Multitask Learning Reveals Sequence Features Driving Specificity in the CRISPR/Cas9 System


Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Assembly
2.1.1. Positive Off-Target Data Source
2.1.2. Negative Off-Target Data Source
2.1.3. sgRNA-Target Sequence Encoding
2.2. Mismatch and Bulge Propensity Analysis
2.3. Deep Learning Techniques, Model Evaluation, and Feature Importance
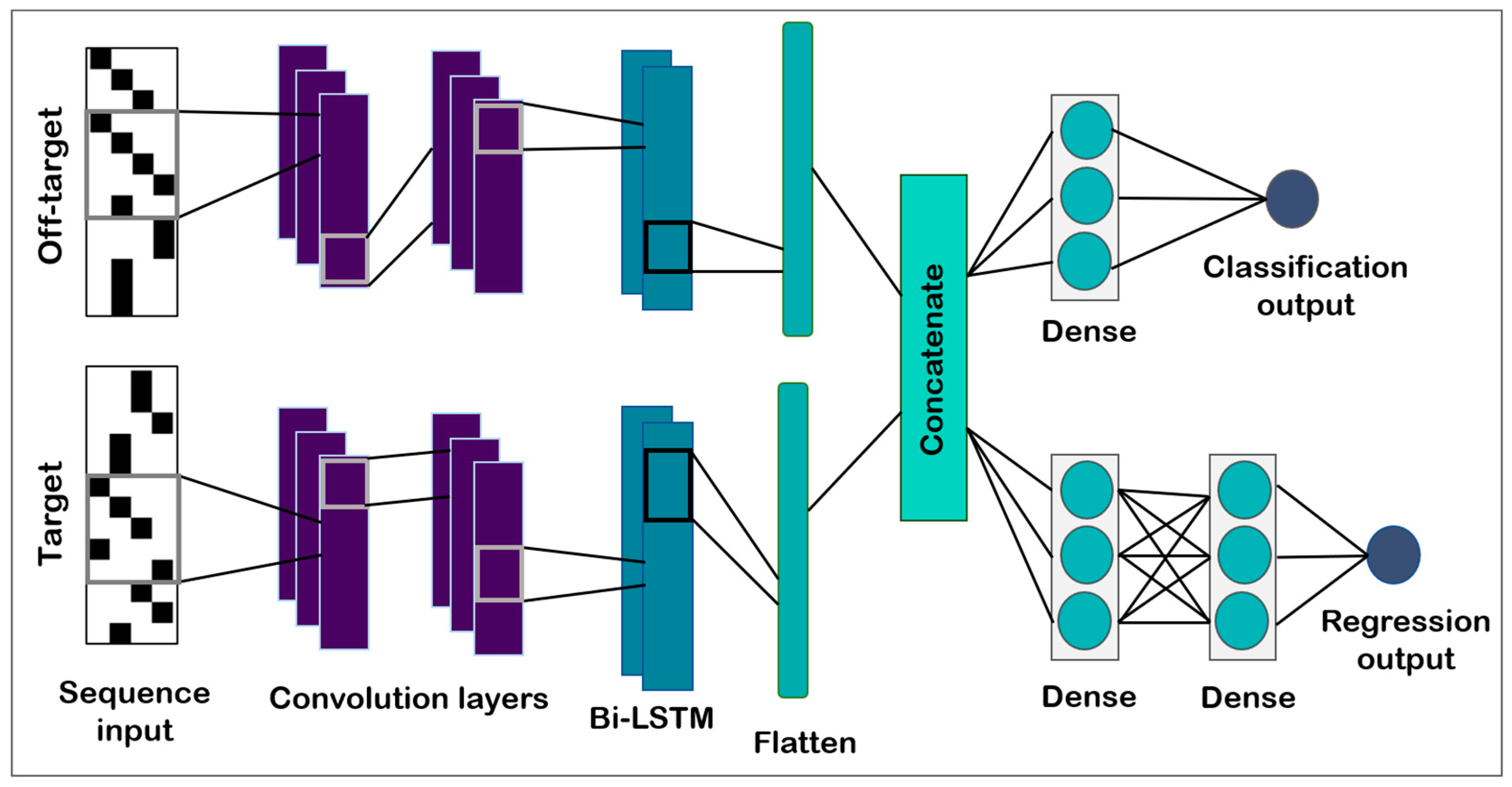
2.3.1. Model Architecture
2.3.2. Model Training
2.3.3. Evaluation Metrics
2.3.4. Identification of Important Sequence Features
3. Results
3.1. Data Assembly and Preparation
3.2. Analysis of Mismatch Occurrence and Nucleotide-Specific Mismatch Propensity
3.3. Analysis of Bulge Occurrence and Nucleotide-Specific Bulge Propensity
3.4. Predictive Performance of the CRISP–RCNN Model
3.5. Activation Maps
3.6. Average AMs’ Feature Importance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-Guided Human Genome Engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Wang, H.; Shivalila, C.S.; Cheng, A.W.; Shi, L.; Jaenisch, R. One-Step Generation of Mice Carrying Reporter and Conditional Alleles by CRISPR/Cas-Mediated Genome Engineering. Cell 2013, 154, 1370–1379. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, L.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [Green Version]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2014, 33, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic Screens in Human Cells Using the CRISPR-Cas9 System. Science 2014, 343, 80–84. [Google Scholar] [CrossRef] [Green Version]
- Doench, J.G.; Hartenian, E.; Graham, D.B.; Tothova, Z.; Hegde, M.; Smith, I.; Sullender, M.; Ebert, B.L.; Xavier, R.J.; Root, D.E. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat. Biotechnol. 2014, 32, 1262–1267. [Google Scholar] [CrossRef] [Green Version]
- Chuai, G.; Ma, H.; Yan, J.; Chen, M.; Hong, N.; Xue, D.; Zhou, C.; Zhu, C.; Chen, K.; Duan, B.; et al. DeepCRISPR: Optimized CRISPR guide RNA design by deep learning. Genome Biol. 2018, 19, 80. [Google Scholar] [CrossRef]
- Kim, H.K.; Kim, Y.; Lee, S.; Min, S.; Bae, J.Y.; Choi, J.W.; Park, J.; Jung, D.; Yoon, S.; Kim, H.H. Spcas9 activity prediction by deepspcas9, a deep learning–based model with high generalization performance. Sci. Adv. 2019, 5, eaax9249. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Cheng, X.; Liu, G.; Li, B.; Liu, X. Deep learning improves the ability of sgRNA off-target propensity prediction. BMC Bioinform. 2020, 21, 51. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Long, Y.; Yin, R.; Kwoh, C.K. DL-CRISPR: A Deep Learning Method for Off-Target Activity Prediction in CRISPR/Cas9 With Data Augmentation. IEEE Access 2020, 8, 76610–76617. [Google Scholar] [CrossRef]
- Zou, J.; Huss, M.; Abid, A.; Mohammadi, P.; Torkamani, A.; Telenti, A. A primer on deep learning in genomics. Nat. Genet. 2018, 51, 12–18. [Google Scholar] [CrossRef]
- Zhang, G.; Dai, Z.; Dai, X. C-RNNCrispr: Prediction of CRISPR/Cas9 sgRNA activity using convolutional and recurrent neural networks. Comput. Struct. Biotechnol. J. 2020, 18, 344–354. [Google Scholar] [CrossRef]
- Dhanjal, J.K.; Radhakrishnan, N.; Sundar, D. CRISPcut: A novel tool for designing optimal sgRNAs for CRISPR/Cas9 based experiments in human cells. Genomics 2018, 111, 560–566. [Google Scholar] [CrossRef]
- Smilkov, D.; Thorat, N.; Kim, B.; Viégas, F.; Wattenberg, M. Smoothgrad: Removing noise by adding noise. arXiv 2017, arXiv:1706.03825. [Google Scholar]
- Tsai, S.Q.; Nguyen, N.T.; Malagon-Lopez, J.; Topkar, V.V.; Aryee, J.M.-L.M.J.; Joung, S.Q.T.N.T.N.J.M.-L.V.V.T.M.J.A.J.K. CIRCLE-seq: A highly sensitive in vitro screen for genome-wide CRISPR–Cas9 nuclease off-targets. Nat. Methods 2017, 14, 607–614. [Google Scholar] [CrossRef] [Green Version]
- Chapman, B.; Chang, J. Biopython: Python tools for computational biology. ACM Sigbio Newsl. 2000, 20, 15–19. [Google Scholar] [CrossRef]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef] [Green Version]
- Koch, G.; Zemel, R.; Salakhutdinov, R. Siamese Neural Networks for One-Shot Image Recognition; W&CP: Lille, France, 2015. [Google Scholar]
- Salakhutdinov, R.; Tenenbaum, J.; Torralba, A. One-shot learning with a hierarchical nonparametric bayesian model. JMLR Workshop Conf. Proc. 2012, 2012, 195–206. [Google Scholar]
- Baxter, J. A Bayesian/Information Theoretic Model of Learning to Learn via Multiple Task Sampling. Mach. Learn. 1997, 28, 7–39. [Google Scholar] [CrossRef]
- LaValle, S.M.; Branicky, M.S.; Lindemann, S.R. On the Relationship between Classical Grid Search and Probabilistic Roadmaps. Int. J. Robot. Res. 2004, 23, 673–692. [Google Scholar] [CrossRef]
- Chollet, F. Keras, GitHub. Available online: https://github.com/fchollet/keras (accessed on 1 July 2021).
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Chicco, D.; Jurman, G. The advantages of the Matthews correlation coefficient (MCC) over F1 score and accuracy in binary classification evaluation. BMC Genom. 2020, 21, 6. [Google Scholar] [CrossRef] [Green Version]
- Cao, C.; Chicco, D.; Hoffman, M.M. The mcc-f1 curve: A performance evaluation technique for binary classification. arXiv 2020, arXiv:2006.11278. [Google Scholar]
- Simonyan, K.; Vedaldi, A.; Zisserman, A. Deep inside convolutional networks: Visualising image classification models and saliency maps. arXiv 2013, arXiv:1312.6034. [Google Scholar]
- Shrikumar, A.; Greenside, P.; Kundaje, A. Learning important features through propagating activation differences. In Proceedings of the 34th International Conference on Machine Learning, Sydney, Australia, 6–11 August 2017; Doina, P., Yee Whye, T., Eds.; PMLR: Sydney, Australia, 2017; Volume 70, pp. 3145–3153. [Google Scholar]
- Sundararajan, M.; Taly, A.; Yan, Q. Gradients of counterfactuals. arXiv 2016, arXiv:1611.02639. [Google Scholar]
- Semenova, E.; Jore, M.M.; Datsenko, K.A.; Semenova, A.; Westra, E.R.; Wanner, B.; van der Oost, J.; Brouns, S.J.J.; Severinov, K. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc. Natl. Acad. Sci. USA 2011, 108, 10098–10103. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ge, X.; Yang, F.; Zhang, L.; Zheng, J.; Tan, X.; Jin, Z.-B.; Qu, J.; Gu, F. Comparison of non-canonical PAMs for CRISPR/Cas9-mediated DNA cleavage in human cells. Sci. Rep. 2014, 4, 5405. [Google Scholar] [CrossRef] [Green Version]
- Cameron, P.; Settle, A.H.; Fuller, C.K.; Thompson, M.S.; Cigan, A.M.; Young, J.K.; May, A.P. SITE-Seq: A Genome-wide Method to Measure Cas9 Cleavage. Protoc. Exch. 2017. [Google Scholar] [CrossRef]
- Lin, J.; Wong, K.-C. Off-target predictions in CRISPR-Cas9 gene editing using deep learning. Bioinformatics 2018, 34, i656–i663. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Zhang, C.; Wang, B.; Li, B.; Wang, Q.; Liu, D.; Wang, H.; Zhou, Y.; Shi, L.; Lan, F.; et al. Optimized CRISPR guide RNA design for two high-fidelity Cas9 variants by deep learning. Nat. Commun. 2019, 10, 4284. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Khosla, A.; Lapedriza, A.; Oliva, A.A.T. Learning Deep Features for Discriminative Localization. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Las Vegas, NV, USA, 27–30 June 2016; IEEE: Las Vegas, NV, USA, 2016; pp. 2921–2929. [Google Scholar]
- Tang, L.; Yang, F.; He, X.; Xie, H.; Liu, X.; Fu, J.; Xi, H.; Lu, X.; Liu, C.; Song, Z.; et al. Efficient cleavage resolves PAM preferences of CRISPR-Cas in human cells. Cell Regen. 2019, 8, 44–50. [Google Scholar] [CrossRef]
- Mitchell, B.P.; Hsu, R.V.; Medrano, M.A.; Zewde, N.T.; Narkhede, Y.; Palermo, G. Spontaneous Embedding of DNA Mismatches Within the RNA:DNA Hybrid of CRISPR-Cas9. Front. Mol. Biosci. 2020, 7, 39. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | Target Nucleotide | Sequences with No Mismatches (%) | Most Substituted Nucleotide | Sequences with ‘Most Substituted’ Mismatch (%) |
|---|---|---|---|---|
| 3 | T | 78.57 | A | 12.37 |
| 4 | G | 94.26 | A | 3.79 |
| 5 | C | 85.58 | A | 6.19 |
| 6 | A | 87.41 | G | 7.65 |
| 7 | A | 90.93 | T | 4.25 |
| 8 | G | 86.66 | A | 9.31 |
| 9 | C | 83.09 | A | 7.25 |
| 11 | T | 92.83 | A | 4.09 |
| 12 | C | 94.77 | T | 3.01 |
| 13 | C | 95.46 | T | 3.93 |
| 14 | C | 96.32 | T | 2.29 |
| 15 | T | 89.28 | A | 7.89 |
| 16 | A | 95.85 | G | 3.65 |
| 18 | T | 83.38 | A | 8.35 |
| 19 | C | 94.14 | T | 3.74 |
| 20 | C | 88.82 | T | 4.93 |
| Position | Target Nucleotide | Sequences with No Mismatches (%) | Most Substituted Nucleotide | Sequences with ‘Most Substituted’ Mismatch (%) |
|---|---|---|---|---|
| 3 | T | 76.52 | A | 15.31 |
| 4 | G | 94.52 | A | 3.78 |
| 5 | C | 89.11 | T | 4.77 |
| 6 | A | 93.03 | G | 5.44 |
| 7 | C | 88.65 | T | 6.52 |
| 8 | G | 89.52 | A | 7.92 |
| 9 | C | 86.12 | A | 7.89 |
| 11 | T | 91.67 | A | 5.31 |
| 12 | A | 95.74 | G | 3.91 |
| 13 | C | 95.53 | T | 3.91 |
| 14 | C | 97.64 | T | 1.83 |
| 15 | C | 87.29 | A | 10.08 |
| 16 | A | 94.54 | G | 4.95 |
| 18 | T | 85.48 | A | 8.04 |
| 19 | C | 97.45 | T | 2.03 |
| 20 | C | 94.16 | T | 3.36 |
| Metric | Score |
|---|---|
| Precision | 0.91 |
| Recall | 0.87 |
| F1 score | 0.89 |
| MCC | 0.80 |
| Cohen’s Kappa | 0.77 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vora, D.S.; Yadav, S.; Sundar, D. Hybrid Multitask Learning Reveals Sequence Features Driving Specificity in the CRISPR/Cas9 System. Biomolecules 2023, 13, 641. https://doi.org/10.3390/biom13040641
Vora DS, Yadav S, Sundar D. Hybrid Multitask Learning Reveals Sequence Features Driving Specificity in the CRISPR/Cas9 System. Biomolecules. 2023; 13(4):641. https://doi.org/10.3390/biom13040641
Chicago/Turabian StyleVora, Dhvani Sandip, Shashank Yadav, and Durai Sundar. 2023. "Hybrid Multitask Learning Reveals Sequence Features Driving Specificity in the CRISPR/Cas9 System" Biomolecules 13, no. 4: 641. https://doi.org/10.3390/biom13040641