

Connecting Dots between Mitochondrial Dysfunction and Depression
Abstract
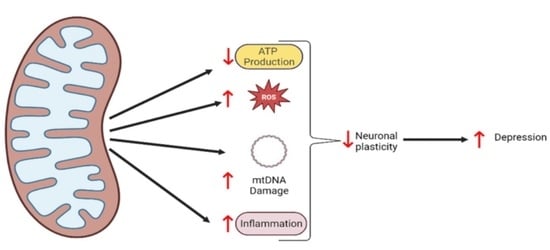
1. Introduction
2. Neurobiological Basis of Depression
3. Models of Depression
4. Mitochondrial Genetics and Depression
5. Mitochondrial Proteome in Depression
6. OXPHOS and ATP Production by Mitochondria in Depression
7. Oxidative Stress in Depression
8. Calcium Homeostasis and Depression
9. Inflammation and Mitochondria in Depression
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Roger, A.J.; Muñoz-Gómez, S.A.; Kamikawa, R. The Origin and Diversification of Mitochondria. Curr. Biol. CB 2017, 27, R1177–R1192. [Google Scholar] [CrossRef] [PubMed]
- Perkins, G.; Renken, C.; Martone, M.E.; Young, S.J.; Ellisman, M.; Frey, T. Electron Tomography of Neuronal Mitochondria: Three-Dimensional Structure and Organization of Cristae and Membrane Contacts. J. Struct. Biol. 1997, 119, 260–272. [Google Scholar] [CrossRef]
- Nagashima, S.; Tábara, L.-C.; Tilokani, L.; Paupe, V.; Anand, H.; Pogson, J.H.; Zunino, R.; McBride, H.M.; Prudent, J. Golgi-Derived PI(4)P-Containing Vesicles Drive Late Steps of Mitochondrial Division. Science 2020, 367, 1366–1371. [Google Scholar] [CrossRef] [PubMed]
- Collier, J.J.; Oláhová, M.; McWilliams, T.G.; Taylor, R.W. Mitochondrial Signalling and Homeostasis: From Cell Biology to Neurological Disease. Trends Neurosci. 2023, 46, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Pernas, L.; Scorrano, L. Mito-Morphosis: Mitochondrial Fusion, Fission, and Cristae Remodeling as Key Mediators of Cellular Function. Annu. Rev. Physiol. 2016, 78, 505–531. [Google Scholar] [CrossRef]
- Ge, Y.; Shi, X.; Boopathy, S.; McDonald, J.; Smith, A.W.; Chao, L.H. Two Forms of Opa1 Cooperate to Complete Fusion of the Mitochondrial Inner-Membrane. eLife 2020, 9, e50973. [Google Scholar] [CrossRef]
- Eura, Y.; Ishihara, N.; Yokota, S.; Mihara, K. Two Mitofusin Proteins, Mammalian Homologues of FZO, with Distinct Functions Are Both Required for Mitochondrial Fusion. J. Biochem. 2003, 134, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Farmer, T.; Naslavsky, N.; Caplan, S. Tying Trafficking to Fusion and Fission at the Mighty Mitochondria. Traffic Cph. Den. 2018, 19, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial Diseases. Nat. Rev. Dis. Primer 2016, 2, 16080. [Google Scholar] [CrossRef] [PubMed]
- Rb, H.; Ns, C. Mitochondrial Reactive Oxygen Species Regulate Cellular Signaling and Dictate Biological Outcomes. Trends Biochem. Sci. 2010, 35. [Google Scholar] [CrossRef]
- Angelova, P.R.; Abramov, A.Y. Functional Role of Mitochondrial Reactive Oxygen Species in Physiology. Free Radic. Biol. Med. 2016, 100, 81–85. [Google Scholar] [CrossRef]
- Cadenas, E.; Davies, K.J.A. Mitochondrial Free Radical Generation, Oxidative Stress, and Aging11This Article Is Dedicated to the Memory of Our Dear Friend, Colleague, and Mentor Lars Ernster (1920–1998), in Gratitude for All He Gave to Us. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Kamata, H.; Honda, S.-I.; Maeda, S.; Chang, L.; Hirata, H.; Karin, M. Reactive Oxygen Species Promote TNFalpha-Induced Death and Sustained JNK Activation by Inhibiting MAP Kinase Phosphatases. Cell 2005, 120, 649–661. [Google Scholar] [CrossRef] [PubMed]
- Izeradjene, K.; Douglas, L.; Tillman, D.M.; Delaney, A.B.; Houghton, J.A. Reactive Oxygen Species Regulate Caspase Activation in Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand-Resistant Human Colon Carcinoma Cell Lines. Cancer Res. 2005, 65, 7436–7445. [Google Scholar] [CrossRef]
- Rangaraju, V.; Lewis, T.L.; Hirabayashi, Y.; Bergami, M.; Motori, E.; Cartoni, R.; Kwon, S.-K.; Courchet, J. Pleiotropic Mitochondria: The Influence of Mitochondria on Neuronal Development and Disease. J. Neurosci. 2019, 39, 8200–8208. [Google Scholar] [CrossRef] [PubMed]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and Molecular Mechanisms of Mitochondrial Function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Cardanho-Ramos, C.; Morais, V.A. Mitochondrial Biogenesis in Neurons: How and Where. Int. J. Mol. Sci. 2021, 22, 13059. [Google Scholar] [CrossRef] [PubMed]
- Werth, J.; Thayer, S. Mitochondria Buffer Physiological Calcium Loads in Cultured Rat Dorsal Root Ganglion Neurons. J. Neurosci. 1994, 14, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Gleichmann, M.; Cheng, A. Mitochondria in Neuroplasticity and Neurological Disorders. Neuron 2008, 60, 748–766. [Google Scholar] [CrossRef]
- Gebara, E.; Zanoletti, O.; Ghosal, S.; Grosse, J.; Schneider, B.L.; Knott, G.; Astori, S.; Sandi, C. Mitofusin-2 in the Nucleus Accumbens Regulates Anxiety and Depression-like Behaviors Through Mitochondrial and Neuronal Actions. Biol. Psychiatry 2021, 89, 1033–1044. [Google Scholar] [CrossRef]
- Sr, S.; Mf, C. Mitochondrial Dysfunction and Oxidative Stress in Parkinson’s Disease. Prog. Neurobiol. 2013, 106, 17–32. [Google Scholar] [CrossRef]
- Ahmad, W.; Ijaz, B.; Shabbiri, K.; Ahmed, F.; Rehman, S. Oxidative Toxicity in Diabetes and Alzheimer’s Disease: Mechanisms behind ROS/RNS Generation. J. Biomed. Sci. 2017, 24, 76. [Google Scholar] [CrossRef] [PubMed]
- Bansal, Y.; Kuhad, A. Mitochondrial Dysfunction in Depression. Curr. Neuropharmacol. 2016, 14, 610–618. [Google Scholar] [CrossRef]
- Tripathi, A.; Scaini, G.; Barichello, T.; Quevedo, J.; Pillai, A. Mitophagy in Depression: Pathophysiology and Treatment Targets. Mitochondrion 2021, 61, 1–10. [Google Scholar] [CrossRef]
- Kimbrell, T.A.; Ketter, T.A.; George, M.S.; Little, J.T.; Benson, B.E.; Willis, M.W.; Herscovitch, P.; Post, R.M. Regional Cerebral Glucose Utilization in Patients with a Range of Severities of Unipolar Depression. Biol. Psychiatry 2002, 51, 237–252. [Google Scholar] [CrossRef]
- Drevets, W.C. Functional Anatomical Abnormalities in Limbic and Prefrontal Cortical Structures in Major Depression. In Progress in Brain Research; Cognition, emotion and autonomic responses: The integrative role of the prefrontal cortex and limbic structures; Elsevier: Amsterdam, The Netherlands, 2000; Volume 126, pp. 413–431. [Google Scholar]
- Mayberg, H.S.; Brannan, S.K.; Mahurin, R.K.; Jerabek, P.A.; Brickman, J.S.; Tekell, J.L.; Silva, J.A.; McGinnis, S.; Glass, T.G.; Martin, C.C.; et al. Cingulate Function in Depression: A Potential Predictor of Treatment Response. Neuroreport 1997, 8, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Cai, Y.; Xu, Y.; Dutt, A.; Shi, S.; Bramon, E. Cerebral Metabolism in Major Depressive Disorder: A Voxel-Based Meta-Analysis of Positron Emission Tomography Studies. BMC Psychiatry 2014, 14, 321. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhong, S.; Liao, X.; Chen, J.; He, T.; Lai, S.; Jia, Y. A Meta-Analysis of Oxidative Stress Markers in Depression. PLoS ONE 2015, 10, e0138904. [Google Scholar] [CrossRef]
- Allen, J.; Romay-Tallon, R.; Brymer, K.J.; Caruncho, H.J.; Kalynchuk, L.E. Mitochondria and Mood: Mitochondrial Dysfunction as a Key Player in the Manifestation of Depression. Front. Neurosci. 2018, 12. [Google Scholar] [CrossRef]
- Petschner, P.; Gonda, X.; Baksa, D.; Eszlari, N.; Trivaks, M.; Juhasz, G.; Bagdy, G. Genes Linking Mitochondrial Function, Cognitive Impairment and Depression Are Associated with Endophenotypes Serving Precision Medicine. Neuroscience 2018, 370, 207–217. [Google Scholar] [CrossRef]
- Kessler, R.C.; McGonagle, K.A.; Zhao, S.; Nelson, C.B.; Hughes, M.; Eshleman, S.; Wittchen, H.-U.; Kendler, K.S. Lifetime and 12-Month Prevalence of DSM-III-R Psychiatric Disorders in the United States: Results From the National Comorbidity Survey. Arch. Gen. Psychiatry 1994, 51, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Dadi, A.F.; Miller, E.R.; Bisetegn, T.A.; Mwanri, L. Global Burden of Antenatal Depression and Its Association with Adverse Birth Outcomes: An Umbrella Review. BMC Public Health 2020, 20, 173. [Google Scholar] [CrossRef]
- Zhu, S.; Zhao, L.; Fan, Y.; Lv, Q.; Wu, K.; Lang, X.; Li, Z.; Yi, Z.; Geng, D. Interaction between TNF-α and Oxidative Stress Status in First-Episode Drug-Naïve Schizophrenia. Psychoneuroendocrinology 2020, 114, 104595. [Google Scholar] [CrossRef]
- The Neurobiology of Depression. Available online: https://www.scientificamerican.com/article/the-neurobiology-of-depression/ (accessed on 22 December 2022).
- Li, Z.; Ruan, M.; Chen, J.; Fang, Y. Major Depressive Disorder: Advances in Neuroscience Research and Translational Applications. Neurosci. Bull. 2021, 37, 863–880. [Google Scholar] [CrossRef] [PubMed]
- Malki, K.; Keers, R.; Tosto, M.G.; Lourdusamy, A.; Carboni, L.; Domenici, E.; Uher, R.; McGuffin, P.; Schalkwyk, L.C. The Endogenous and Reactive Depression Subtypes Revisited: Integrative Animal and Human Studies Implicate Multiple Distinct Molecular Mechanisms Underlying Major Depressive Disorder. BMC Med. 2014, 12, 73. [Google Scholar] [CrossRef] [PubMed]
- Konttinen, H.; van Strien, T.; Männistö, S.; Jousilahti, P.; Haukkala, A. Depression, Emotional Eating and Long-Term Weight Changes: A Population-Based Prospective Study. Int. J. Behav. Nutr. Phys. Act. 2019, 16, 28. [Google Scholar] [CrossRef]
- Karabatsiakis, A.; Schönfeldt-Lecuona, C. Depression, Mitochondrial Bioenergetics, and Electroconvulsive Therapy: A New Approach towards Personalized Medicine in Psychiatric Treatment - a Short Review and Current Perspective. Transl. Psychiatry 2020, 10, 1–9. [Google Scholar] [CrossRef]
- Qiu, W.; Cai, X.; Zheng, C.; Qiu, S.; Ke, H.; Huang, Y. Update on the Relationship Between Depression and Neuroendocrine Metabolism. Front. Neurosci. 2021, 15. [Google Scholar] [CrossRef] [PubMed]
- Kessler, R.C.; Nelson, C.B.; McGonagle, K.A.; Liu, J.; Swartz, M.; Blazer, D.G. Comorbidity of DSM-III-R Major Depressive Disorder in the General Population: Results from the US National Comorbidity Survey. Br. J. Psychiatry. Suppl. 1996, 168, 17–30. [Google Scholar] [CrossRef]
- Kalia, M. Neurobiological Basis of Depression: An Update. Metabolism 2005, 54, 24–27. [Google Scholar] [CrossRef]
- Aboul-Fotouh, S. Chronic Treatment with Coenzyme Q10 Reverses Restraint Stress-Induced Anhedonia and Enhances Brain Mitochondrial Respiratory Chain and Creatine Kinase Activities in Rats. Behav. Pharmacol. 2013, 24, 552. [Google Scholar] [CrossRef]
- Maes, M.; Fišar, Z.; Medina, M.; Scapagnini, G.; Nowak, G.; Berk, M. New Drug Targets in Depression: Inflammatory, Cell-Mediated Immune, Oxidative and Nitrosative Stress, Mitochondrial, Antioxidant, and Neuroprogressive Pathways. And New Drug Candidates—Nrf2 Activators and GSK-3 Inhibitors. Inflammopharmacology 2012, 20, 127–150. [Google Scholar] [CrossRef]
- Iwamoto, K.; Bundo, M.; Kato, T. Altered Expression of Mitochondria-Related Genes in Postmortem Brains of Patients with Bipolar Disorder or Schizophrenia, as Revealed by Large-Scale DNA Microarray Analysis. Hum. Mol. Genet. 2005, 14, 241–253. [Google Scholar] [CrossRef]
- Uzbekov, M.G. Monoamine Oxidase as a Potential Biomarker of the Efficacy of Treatment of Mental Disorders. Biochem. Mosc. 2021, 86, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Caruso, G.; Benatti, C.; Blom, J.M.C.; Caraci, F.; Tascedda, F. The Many Faces of Mitochondrial Dysfunction in Depression: From Pathology to Treatment. Front. Pharmacol. 2019, 10, 995. [Google Scholar] [CrossRef] [PubMed]
- Thase, M.E. Preventing Relapse and Recurrence of Depression: A Brief Review of Therapeutic Options. CNS Spectr. 2006, 11, 12–21. [Google Scholar] [CrossRef]
- Trivedi, M.H.; Rush, A.J.; Wisniewski, S.R.; Nierenberg, A.A.; Warden, D.; Ritz, L.; Norquist, G.; Howland, R.H.; Lebowitz, B.; McGrath, P.J.; et al. Evaluation of Outcomes With Citalopram for Depression Using Measurement-Based Care in STAR*D: Implications for Clinical Practice. Am. J. Psychiatry 2006, 163, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, B.L.; van Praag, H.; Gage, F.H. Depression and the Birth and Death of Brain Cells: The Turnover of Neurons in the Hippocampus Might Help to Explain the Onset of and Recovery from Clinical Depression. Am. Sci. 2000, 88, 340–345. [Google Scholar] [CrossRef]
- Fenton, E.Y.; Fournier, N.M.; Lussier, A.L.; Romay-Tallon, R.; Caruncho, H.J.; Kalynchuk, L.E. Imipramine Protects against the Deleterious Effects of Chronic Corticosterone on Depression-like Behavior, Hippocampal Reelin Expression, and Neuronal Maturation. Prog. Neuropsychopharmacol. Biol. Psychiatry 2015, 60, 52–59. [Google Scholar] [CrossRef]
- Schoenfeld, T.J.; Cameron, H.A. Adult Neurogenesis and Mental Illness. Neuropsychopharmacology 2015, 40, 113–128. [Google Scholar] [CrossRef]
- Brummelte, S.; Galea, L.A.M. Chronic High Corticosterone Reduces Neurogenesis in the Dentate Gyrus of Adult Male and Female Rats. Neuroscience 2010, 168, 680–690. [Google Scholar] [CrossRef]
- Campbell, S.; Marriott, M.; Nahmias, C.; MacQueen, G.M. Lower Hippocampal Volume in Patients Suffering From Depression: A Meta-Analysis. Am. J. Psychiatry 2004, 161, 598–607. [Google Scholar] [CrossRef]
- Santarelli, L.; Saxe, M.; Gross, C.; Surget, A.; Battaglia, F.; Dulawa, S.; Weisstaub, N.; Lee, J.; Duman, R.; Arancio, O.; et al. Requirement of Hippocampal Neurogenesis for the Behavioral Effects of Antidepressants. Science 2003, 301, 805–809. [Google Scholar] [CrossRef]
- Lussier, A.L.; Caruncho, H.J.; Kalynchuk, L.E. Repeated Exposure to Corticosterone, but Not Restraint, Decreases the Number of Reelin-Positive Cells in the Adult Rat Hippocampus. Neurosci. Lett. 2009, 460, 170–174. [Google Scholar] [CrossRef] [PubMed]
- Pujadas, L.; Gruart, A.; Bosch, C.; Delgado, L.; Teixeira, C.M.; Rossi, D.; Lecea, L.d.; Martínez, A.; Delgado-García, J.M.; Soriano, E. Reelin Regulates Postnatal Neurogenesis and Enhances Spine Hypertrophy and Long-Term Potentiation. J. Neurosci. 2010, 30, 4636–4649. [Google Scholar] [CrossRef]
- Kitamura, T.; Saitoh, Y.; Takashima, N.; Murayama, A.; Niibori, Y.; Ageta, H.; Sekiguchi, M.; Sugiyama, H.; Inokuchi, K. Adult Neurogenesis Modulates the Hippocampus-Dependent Period of Associative Fear Memory. Cell 2009, 139, 814–827. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Jinnou, H.; Sawamoto, K.; Hitoshi, S. Adult Neurogenesis and Its Role in Brain Injury and Psychiatric Diseases. J. Neurochem. 2018, 147, 584–594. [Google Scholar] [CrossRef]
- Malberg, J.E. Implications of Adult Hippocampal Neurogenesis in Antidepressant Action. J. Psychiatry Neurosci. JPN 2004, 29, 196–205. [Google Scholar] [PubMed]
- Czéh, B.; Michaelis, T.; Watanabe, T.; Frahm, J.; de Biurrun, G.; van Kampen, M.; Bartolomucci, A.; Fuchs, E. Stress-Induced Changes in Cerebral Metabolites, Hippocampal Volume, and Cell Proliferation Are Prevented by Antidepressant Treatment with Tianeptine. Proc. Natl. Acad. Sci. USA 2001, 98, 12796–12801. [Google Scholar] [CrossRef]
- Perera, T.D.; Coplan, J.D.; Lisanby, S.H.; Lipira, C.M.; Arif, M.; Carpio, C.; Spitzer, G.; Santarelli, L.; Scharf, B.; Hen, R.; et al. Antidepressant-Induced Neurogenesis in the Hippocampus of Adult Nonhuman Primates. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 4894–4901. [Google Scholar] [CrossRef]
- Hitoshi, S.; Maruta, N.; Higashi, M.; Kumar, A.; Kato, N.; Ikenaka, K. Antidepressant Drugs Reverse the Loss of Adult Neural Stem Cells Following Chronic Stress. J. Neurosci. Res. 2007, 85, 3574–3585. [Google Scholar] [CrossRef]
- Duman, R.S.; Malberg, J.; Nakagawa, S. Regulation of Adult Neurogenesis by Psychotropic Drugs and Stress. J. Pharmacol. Exp. Ther. 2001, 299, 401–407. [Google Scholar] [PubMed]
- Boldrini, M.; Underwood, M.D.; Hen, R.; Rosoklija, G.B.; Dwork, A.J.; John Mann, J.; Arango, V. Antidepressants Increase Neural Progenitor Cells in the Human Hippocampus. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2009, 34, 2376–2389. [Google Scholar] [CrossRef]
- Reif, A.; Fritzen, S.; Finger, M.; Strobel, A.; Lauer, M.; Schmitt, A.; Lesch, K.-P. Neural Stem Cell Proliferation Is Decreased in Schizophrenia, but Not in Depression. Mol. Psychiatry 2006, 11, 514–522. [Google Scholar] [CrossRef]
- Kirby, D.M.; Rennie, K.J.; Smulders-Srinivasan, T.K.; Acin-Perez, R.; Whittington, M.; Enriquez, J.-A.; Trevelyan, A.J.; Turnbull, D.M.; Lightowlers, R.N. Transmitochondrial Embryonic Stem Cells Containing Pathogenic MtDNA Mutations Are Compromised in Neuronal Differentiation. Cell Prolif. 2009, 42, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Calingasan, N.Y.; Ho, D.J.; Wille, E.J.; Campagna, M.V.; Ruan, J.; Dumont, M.; Yang, L.; Shi, Q.; Gibson, G.E.; Beal, M.F. Influence of Mitochondrial Enzyme Deficiency on Adult Neurogenesis in Mouse Models of Neurodegenerative Diseases. Neuroscience 2008, 153, 986–996. [Google Scholar] [CrossRef] [PubMed]
- Baxter, K.K.; Uittenbogaard, M.; Yoon, J.; Chiaramello, A. The Neurogenic Basic Helix-Loop-Helix Transcription Factor NeuroD6 Concomitantly Increases Mitochondrial Mass and Regulates Cytoskeletal Organization in the Early Stages of Neuronal Differentiation. ASN Neuro 2009, 1, AN20090036. [Google Scholar] [CrossRef]
- Cuperfain, A.B.; Zhang, Z.L.; Kennedy, J.L.; Gonçalves, V.F. The Complex Interaction of Mitochondrial Genetics and Mitochondrial Pathways in Psychiatric Disease. Complex Psychiatry 2018, 4, 52–69. [Google Scholar] [CrossRef]
- Kasahara, T.; Kato, T. What Can Mitochondrial DNA Analysis Tell Us About Mood Disorders? Biol. Psychiatry 2018, 83, 731–738. [Google Scholar] [CrossRef]
- Bessa, J.M.; Ferreira, D.; Melo, I.; Marques, F.; Cerqueira, J.J.; Palha, J.A.; Almeida, O.F.X.; Sousa, N. The Mood-Improving Actions of Antidepressants Do Not Depend on Neurogenesis but Are Associated with Neuronal Remodeling. Mol. Psychiatry 2009, 14, 764–773. [Google Scholar] [CrossRef]
- David, D.J.; Samuels, B.A.; Rainer, Q.; Wang, J.-W.; Marsteller, D.; Mendez, I.; Drew, M.; Craig, D.A.; Guiard, B.P.; Guilloux, J.-P.; et al. Neurogenesis-Dependent and -Independent Effects of Fluoxetine in an Animal Model of Anxiety/Depression. Neuron 2009, 62, 479–493. [Google Scholar] [CrossRef]
- Surget, A.; Saxe, M.; Leman, S.; Ibarguen-Vargas, Y.; Chalon, S.; Griebel, G.; Hen, R.; Belzung, C. Drug-Dependent Requirement of Hippocampal Neurogenesis in a Model of Depression and of Antidepressant Reversal. Biol. Psychiatry 2008, 64, 293–301. [Google Scholar] [CrossRef]
- Lussier, A.L.; Lebedeva, K.; Fenton, E.Y.; Guskjolen, A.; Caruncho, H.J.; Kalynchuk, L.E. The Progressive Development of Depression-like Behavior in Corticosterone-Treated Rats Is Paralleled by Slowed Granule Cell Maturation and Decreased Reelin Expression in the Adult Dentate Gyrus. Neuropharmacology 2013, 71, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Sheline, Y.I.; Gado, M.H.; Kraemer, H.C. Untreated Depression and Hippocampal Volume Loss. Am. J. Psychiatry 2003, 160, 1516–1518. [Google Scholar] [CrossRef] [PubMed]
- Manji, H.K.; Drevets, W.C.; Charney, D.S. The Cellular Neurobiology of Depression. Nat. Med. 2001, 7, 541–547. [Google Scholar] [CrossRef]
- Zhang, F.; Peng, W.; Sweeney, J.A.; Jia, Z.; Gong, Q. Brain Structure Alterations in Depression: Psychoradiological Evidence. CNS Neurosci. Ther. 2018, 24, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Spellman, T.; Liston, C. Toward Circuit Mechanisms of Pathophysiology in Depression. Am. J. Psychiatry 2020, 177, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Pandya, M.; Altinay, M.; Malone, D.A.; Anand, A. Where in the Brain Is Depression? Curr. Psychiatry Rep. 2012, 14, 634–642. [Google Scholar] [CrossRef]
- Martins-de-Souza, D.; Guest, P.C.; Harris, L.W.; Vanattou-Saifoudine, N.; Webster, M.J.; Rahmoune, H.; Bahn, S. Identification of Proteomic Signatures Associated with Depression and Psychotic Depression in Post-Mortem Brains from Major Depression Patients. Transl. Psychiatry 2012, 2, e87. [Google Scholar] [CrossRef]
- Hardy, J.A.; Dodd, P.R. Metabolic and Functional Studies on Post-Mortem Human Brain. Neurochem. Int. 1983, 5, 253–266. [Google Scholar] [CrossRef]
- Fuchs, E.; Flügge, G. Experimental Animal Models for the Simulation of Depression and Anxiety. Dialogues Clin. Neurosci. 2006, 8, 323–333. [Google Scholar] [CrossRef]
- Czéh, B.; Simon, M. Benefits of Animal Models to Understand the Pathophysiology of Depressive Disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 106, 110049. [Google Scholar] [CrossRef] [PubMed]
- Vollmayr, B.; Mahlstedt, M.M.; Henn, F.A. Neurogenesis and Depression: What Animal Models Tell Us about the Link. Eur. Arch. Psychiatry Clin. Neurosci. 2007, 257, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Anisman, H.; Matheson, K. Stress, Depression, and Anhedonia: Caveats Concerning Animal Models. Neurosci. Biobehav. Rev. 2005, 29, 525–546. [Google Scholar] [CrossRef] [PubMed]
- Willner, P.; Mitchell, P.J. The Validity of Animal Models of Predisposition to Depression. Behav. Pharmacol. 2002, 13, 169–188. [Google Scholar] [CrossRef] [PubMed]
- Klinedinst, N.J.; Regenold, W.T. A Mitochondrial Bioenergetic Basis of Depression. J. Bioenerg. Biomembr. 2015, 47, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Nestler, E.J.; Hyman, S.E. Animal Models of Neuropsychiatric Disorders. Nat. Neurosci. 2010, 13, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Duman, C.H. Models of Depression. Vitam. Horm. 2010, 82, 1–21. [Google Scholar] [CrossRef]
- Mairesse, J.; Vercoutter-Edouart, A.S.; Marrocco, J.; Zuena, A.R.; Giovine, A.; Nicoletti, F.; Michalski, J.C.; Maccari, S.; Morley-Fletcher, S. Proteomic Characterization in the Hippocampus of Prenatally Stressed Rats. J. Proteomics 2012, 75, 1764–1770. [Google Scholar] [CrossRef]
- Marais, L.; Hattingh, S.M.; Stein, D.J.; Daniels, W.M.U. A Proteomic Analysis of the Ventral Hippocampus of Rats Subjected to Maternal Separation and Escitalopram Treatment. Metab. Brain Dis. 2009, 24, 569–586. [Google Scholar] [CrossRef]
- Marais, L.; van Rensburg, S.J.; van Zyl, J.M.; Stein, D.J.; Daniels, W.M.U. Maternal Separation of Rat Pups Increases the Risk of Developing Depressive-like Behavior after Subsequent Chronic Stress by Altering Corticosterone and Neurotrophin Levels in the Hippocampus. Neurosci. Res. 2008, 61, 106–112. [Google Scholar] [CrossRef]
- Piubelli, C.; Carboni, L.; Becchi, S.; Mathé, A.A.; Domenici, E. Regulation of Cytoskeleton Machinery, Neurogenesis and Energy Metabolism Pathways in a Rat Gene-Environment Model of Depression Revealed by Proteomic Analysis. Neuroscience 2011, 176, 349–380. [Google Scholar] [CrossRef]
- Yadid, G.; Nakash, R.; Deri, I.; Tamar, G.; Kinor, N.; Gispan, I.; Zangen, A. Elucidation of the Neurobiology of Depression: Insights from a Novel Genetic Animal Model. Prog. Neurobiol. 2000, 62, 353–378. [Google Scholar] [CrossRef] [PubMed]
- Malki, K.; Campbell, J.; Davies, M.; Keers, R.; Uher, R.; Ward, M.; Paya-Cano, J.; Aitchinson, K.J.; Binder, E.; Sluyter, F.; et al. Pharmacoproteomic Investigation into Antidepressant Response in Two Mouse Inbred Strains. Proteomics 2012, 12, 2355–2365. [Google Scholar] [CrossRef] [PubMed]
- Bisgaard, C.F.; Jayatissa, M.N.; Enghild, J.J.; Sanchéz, C.; Artemychyn, R.; Wiborg, O. Proteomic Investigation of the Ventral Rat Hippocampus Links DRP-2 to Escitalopram Treatment Resistance and SNAP to Stress Resilience in the Chronic Mild Stress Model of Depression. J. Mol. Neurosci. MN 2007, 32, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Bisgaard, C.F.; Bak, S.; Christensen, T.; Jensen, O.N.; Enghild, J.J.; Wiborg, O. Vesicular Signalling and Immune Modulation as Hedonic Fingerprints: Proteomic Profiling in the Chronic Mild Stress Depression Model. J. Psychopharmacol. Oxf. Engl. 2012, 26, 1569–1583. [Google Scholar] [CrossRef]
- Henningsen, K.; Palmfeldt, J.; Christiansen, S.; Baiges, I.; Bak, S.; Jensen, O.N.; Gregersen, N.; Wiborg, O. Candidate Hippocampal Biomarkers of Susceptibility and Resilience to Stress in a Rat Model of Depression. Mol. Cell. Proteomics MCP 2012, 11, M111.016428. [Google Scholar] [CrossRef]
- Kedracka-Krok, S.; Fic, E.; Jankowska, U.; Jaciuk, M.; Gruca, P.; Papp, M.; Kusmider, M.; Solich, J.; Debski, J.; Dadlez, M.; et al. Effect of Chronic Mild Stress and Imipramine on the Proteome of the Rat Dentate Gyrus. J. Neurochem. 2010, 113, 848–859. [Google Scholar] [CrossRef] [PubMed]
- Rezin, G.T.; Cardoso, M.R.; Gonçalves, C.L.; Scaini, G.; Fraga, D.B.; Riegel, R.E.; Comim, C.M.; Quevedo, J.; Streck, E.L. Inhibition of Mitochondrial Respiratory Chain in Brain of Rats Subjected to an Experimental Model of Depression. Neurochem. Int. 2008, 53, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Chai, Y.; Ding, J.-H.; Sun, X.-L.; Hu, G. Chronic Mild Stress Damages Mitochondrial Ultrastructure and Function in Mouse Brain. Neurosci. Lett. 2011, 488, 76–80. [Google Scholar] [CrossRef]
- Mu, J.; Xie, P.; Yang, Z.-S.; Yang, D.-L.; Lv, F.-J.; Luo, T.-Y.; Li, Y. Neurogenesis and Major Depression: Implications from Proteomic Analyses of Hippocampal Proteins in a Rat Depression Model. Neurosci. Lett. 2007, 416, 252–256. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, D.; Tang, G.; Zhou, C.; Cheng, K.; Zhou, J.; Wu, B.; Peng, Y.; Liu, C.; Zhan, Y.; et al. Proteomics Reveals Energy and Glutathione Metabolic Dysregulation in the Prefrontal Cortex of a Rat Model of Depression. Neuroscience 2013, 247, 191–200. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, N.; Hao, W.; Zhao, Q.; Ying, T.; Liu, S.; Li, Q.; Liang, Y.; Wang, T.; Dong, Y.; et al. Dynamic Proteomic Analysis of Protein Expression Profiles in Whole Brain of Balb/c Mice Subjected to Unpredictable Chronic Mild Stress: Implications for Depressive Disorders and Future Therapies. Neurochem. Int. 2011, 58, 904–913. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhou, C. Corticosterone Reduces Brain Mitochondrial Function and Expression of Mitofusin, BDNF in Depression-like Rodents Regardless of Exercise Preconditioning. Psychoneuroendocrinology 2012, 37, 1057–1070. [Google Scholar] [CrossRef]
- Carboni, L.; Piubelli, C.; Pozzato, C.; Astner, H.; Arban, R.; Righetti, P.G.; Hamdan, M.; Domenici, E. Proteomic Analysis of Rat Hippocampus after Repeated Psychosocial Stress. Neuroscience 2006, 137, 1237–1246. [Google Scholar] [CrossRef] [PubMed]
- Mallei, A.; Giambelli, R.; Gass, P.; Racagni, G.; Mathé, A.A.; Vollmayr, B.; Popoli, M. Synaptoproteomics of Learned Helpless Rats Involve Energy Metabolism and Cellular Remodeling Pathways in Depressive-like Behavior and Antidepressant Response. Neuropharmacology 2011, 60, 1243–1253. [Google Scholar] [CrossRef]
- Vollmayr, B.; Henn, F.A. Learned Helplessness in the Rat: Improvements in Validity and Reliability. Brain Res. Brain Res. Protoc. 2001, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Katz, R.J.; Roth, K.A.; Carroll, B.J. Acute and Chronic Stress Effects on Open Field Activity in the Rat: Implications for a Model of Depression. Neurosci. Biobehav. Rev. 1981, 5, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Porsolt, R.D.; Bertin, A.; Jalfre, M. Behavioral Despair in Mice: A Primary Screening Test for Antidepressants. Arch. Int. Pharmacodyn. Ther. 1977, 229, 327–336. [Google Scholar]
- Landgraf, D.; Long, J.; Der-Avakian, A.; Streets, M.; Welsh, D.K. Dissociation of Learned Helplessness and Fear Conditioning in Mice: A Mouse Model of Depression. PLoS ONE 2015, 10, e0125892. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, M.M.; Ladd, C.O.; Plotsky, P.M. Early Adverse Experience as a Developmental Risk Factor for Later Psychopathology: Evidence from Rodent and Primate Models. Dev. Psychopathol. 2001, 13, 419–449. [Google Scholar] [CrossRef]
- Becker, M.; Pinhasov, A.; Ornoy, A. Animal Models of Depression: What Can They Teach Us about the Human Disease? Diagnostics 2021, 11, 123. [Google Scholar] [CrossRef]
- Steru, L.; Chermat, R.; Thierry, B.; Simon, P. The Tail Suspension Test: A New Method for Screening Antidepressants in Mice. Psychopharmacology 1985, 85, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Nestler, E.J.; Gould, E.; Manji, H.; Buncan, M.; Duman, R.S.; Greshenfeld, H.K.; Hen, R.; Koester, S.; Lederhendler, I.; Meaney, M.; et al. Preclinical Models: Status of Basic Research in Depression. Biol. Psychiatry 2002, 52, 503–528. [Google Scholar] [CrossRef] [PubMed]
- Calvo, S.E.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An Updated Inventory of Mammalian Mitochondrial Proteins. Nucleic Acids Res. 2016, 44, D1251–D1257. [Google Scholar] [CrossRef]
- Pei, L.; Wallace, D.C. Mitochondrial Etiology of Neuropsychiatric Disorders. Biol. Psychiatry 2018, 83, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Martín-Jiménez, R.; Lurette, O.; Hebert-Chatelain, E. Damage in Mitochondrial DNA Associated with Parkinson’s Disease. DNA Cell Biol. 2020, 39, 1421–1430. [Google Scholar] [CrossRef]
- Fattal, O.; Link, J.; Quinn, K.; Cohen, B.H.; Franco, K. Psychiatric Comorbidity in 36 Adults with Mitochondrial Cytopathies. CNS Spectr. 2007, 12, 429–438. [Google Scholar] [CrossRef]
- Gardner, A.; Johansson, A.; Wibom, R.; Nennesmo, I.; von Döbeln, U.; Hagenfeldt, L.; Hällström, T. Alterations of Mitochondrial Function and Correlations with Personality Traits in Selected Major Depressive Disorder Patients. J. Affect. Disord. 2003, 76, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-C.; Jou, S.-H.; Lin, T.-T.; Lai, T.-J.; Liu, C.-S. Mitochondria DNA Change and Oxidative Damage in Clinically Stable Patients with Major Depressive Disorder. PLoS ONE 2015, 10, e0125855. [Google Scholar] [CrossRef] [PubMed]
- Brymer, K.J.; Fenton, E.Y.; Kalynchuk, L.E.; Caruncho, H.J. Peripheral Etanercept Administration Normalizes Behavior, Hippocampal Neurogenesis, and Hippocampal Reelin and GABAA Receptor Expression in a Preclinical Model of Depression. Front. Pharmacol. 2018, 9, 121. [Google Scholar] [CrossRef]
- Wang, J.; Hodes, G.E.; Zhang, H.; Zhang, S.; Zhao, W.; Golden, S.A.; Bi, W.; Menard, C.; Kana, V.; Leboeuf, M.; et al. Epigenetic Modulation of Inflammation and Synaptic Plasticity Promotes Resilience against Stress in Mice. Nat. Commun. 2018, 9, 477. [Google Scholar] [CrossRef]
- Adzic, M.; Brkic, Z.; Bulajic, S.; Mitic, M.; Radojcic, M.B. Antidepressant Action on Mitochondrial Dysfunction in Psychiatric Disorders. Drug Dev. Res. 2016, 77, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Inczedy-Farkas, G.; Trampush, J.W.; Perczel Forintos, D.; Beech, D.; Andrejkovics, M.; Varga, Z.; Remenyi, V.; Bereznai, B.; Gal, A.; Molnar, M.J. Mitochondrial DNA Mutations and Cognition: A Case-Series Report. Arch. Clin. Neuropsychol. 2014, 29, 315–321. [Google Scholar] [CrossRef]
- Munkholm, K.; Peijs, L.; Vinberg, M.; Kessing, L.V. A Composite Peripheral Blood Gene Expression Measure as a Potential Diagnostic Biomarker in Bipolar Disorder. Transl. Psychiatry 2015, 5, e614. [Google Scholar] [CrossRef]
- Ceylan, D.; Tuna, G.; Kirkali, G.; Tunca, Z.; Can, G.; Arat, H.E.; Kant, M.; Dizdaroglu, M.; Özerdem, A. Oxidatively-Induced DNA Damage and Base Excision Repair in Euthymic Patients with Bipolar Disorder. DNA Repair 2018, 65, 64–72. [Google Scholar] [CrossRef]
- Wang, Q.; Dwivedi, Y. Transcriptional Profiling of Mitochondria Associated Genes in Prefrontal Cortex of Subjects with Major Depressive Disorder. World J. Biol. Psychiatry 2017, 18, 592–603. [Google Scholar] [CrossRef]
- Shyn, S.I.; Shi, J.; Kraft, J.B.; Potash, J.B.; Knowles, J.A.; Weissman, M.M.; Garriock, H.A.; Yokoyama, J.S.; McGrath, P.J.; Peters, E.J.; et al. Novel Loci for Major Depression Identified by Genome-Wide Association Study of Sequenced Treatment Alternatives to Relieve Depression and Meta-Analysis of Three Studies. Mol. Psychiatry 2011, 16, 202–215. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Suleiman, J.; Almannai, M.; Scaglia, F. Mitochondrial Dynamics: Biological Roles, Molecular Machinery, and Related Diseases. Mol. Genet. Metab. 2018, 125, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Scifo, E.; Pabba, M.; Kapadia, F.; Ma, T.; Lewis, D.A.; Tseng, G.C.; Sibille, E. Sustained Molecular Pathology Across Episodes and Remission in Major Depressive Disorder. Biol. Psychiatry 2018, 83, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Villa, R.F.; Ferrari, F.; Bagini, L.; Gorini, A.; Brunello, N.; Tascedda, F. Mitochondrial Energy Metabolism of Rat Hippocampus after Treatment with the Antidepressants Desipramine and Fluoxetine. Neuropharmacology 2017, 121, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Saia-Cereda, V.M.; Cassoli, J.S.; Martins-de-Souza, D.; Nascimento, J.M. Psychiatric Disorders Biochemical Pathways Unraveled by Human Brain Proteomics. Eur. Arch. Psychiatry Clin. Neurosci. 2017, 267, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Zubenko, G.S.; Hughes III, H.B.; Jordan, R.M.; Lyons-Weiler, J.; Cohen, B.M. Differential Hippocampal Gene Expression and Pathway Analysis in an Etiology-Based Mouse Model of Major Depressive Disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2014, 165, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Carboni, L. The Contribution of Proteomic Studies in Humans, Animal Models, and after Antidepressant Treatments to Investigate the Molecular Neurobiology of Major Depression. PROTEOMICS–Clin. Appl. 2015, 9, 889–898. [Google Scholar] [CrossRef] [PubMed]
- Beasley, C.L.; Pennington, K.; Behan, A.; Wait, R.; Dunn, M.J.; Cotter, D. Proteomic Analysis of the Anterior Cingulate Cortex in the Major Psychiatric Disorders: Evidence for Disease-Associated Changes. PROTEOMICS 2006, 6, 3414–3425. [Google Scholar] [CrossRef]
- Martins-de-Souza, D. Proteomics, Metabolomics, and Protein Interactomics in the Characterization of the Molecular Features of Major Depressive Disorder. Dialogues Clin. Neurosci. 2014, 16, 63–73. [Google Scholar] [CrossRef]
- Karry, R.; Klein, E.; Ben Shachar, D. Mitochondrial Complex i Subunits Expression Is Altered in Schizophrenia: A Postmortem Study. Biol. Psychiatry 2004, 55, 676–684. [Google Scholar] [CrossRef]
- Martins-de-Souza, D.; Guest, P.C.; Vanattou-Saifoudine, N.; Rahmoune, H.; Bahn, S. Phosphoproteomic Differences in Major Depressive Disorder Postmortem Brains Indicate Effects on Synaptic Function. Eur. Arch. Psychiatry Clin. Neurosci. 2012, 262, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Rappeneau, V.; Wilmes, L.; Touma, C. Molecular Correlates of Mitochondrial Dysfunctions in Major Depression: Evidence from Clinical and Rodent Studies. Mol. Cell. Neurosci. 2020, 109, 103555. [Google Scholar] [CrossRef] [PubMed]
- Johnston-Wilson, N.L.; Sims, C.D.; Hofmann, J.-P.; Anderson, L.; Shore, A.D.; Torrey, E.F.; Yolken, R.H. Disease-Specific Alterations in Frontal Cortex Brain Proteins in Schizophrenia, Bipolar Disorder, and Major Depressive Disorder. Mol. Psychiatry 2000, 5, 142–149. [Google Scholar] [CrossRef]
- Taylor, M.J.; Freemantle, N.; Geddes, J.R.; Bhagwagar, Z. Early Onset of Selective Serotonin Reuptake Inhibitor Antidepressant Action: Systematic Review and Meta-Analysis. Arch. Gen. Psychiatry 2006, 63, 1217–1223. [Google Scholar] [CrossRef]
- Filipović, D.; Costina, V.; Perić, I.; Stanisavljević, A.; Findeisen, P. Chronic Fluoxetine Treatment Directs Energy Metabolism towards the Citric Acid Cycle and Oxidative Phosphorylation in Rat Hippocampal Nonsynaptic Mitochondria. Brain Res. 2017, 1659, 41–54. [Google Scholar] [CrossRef]
- Moretti, A.; Gorini, A.; Villa, R.F. Affective Disorders, Antidepressant Drugs and Brain Metabolism. Mol. Psychiatry 2003, 8, 773–785. [Google Scholar] [CrossRef]
- Sherman, A.D.; Sacquitne, J.L.; Petty, F. Specificity of the Learned Helplessness Model of Depression. Pharmacol. Biochem. Behav. 1982, 16, 449–454. [Google Scholar] [CrossRef]
- Kondo, D.G.; Hellem, T.L.; Sung, Y.-H.; Kim, N.; Jeong, E.-K.; DelMastro, K.K.; Shi, X.; Renshaw, P.F. Review: Magnetic Resonance Spectroscopy Studies of Pediatric Major Depressive Disorder. Depress. Res. Treat. 2011, 2011, 650450. [Google Scholar] [CrossRef]
- Ahmad, A.; Rasheed, N.; Banu, N.; Palit, G. Alterations in Monoamine Levels and Oxidative Systems in Frontal Cortex, Striatum, and Hippocampus of the Rat Brain during Chronic Unpredictable Stress. Stress Amst. Neth. 2010, 13, 355–364. [Google Scholar] [CrossRef]
- Tobe, E.H. Mitochondrial Dysfunction, Oxidative Stress, and Major Depressive Disorder. Neuropsychiatr. Dis. Treat. 2013, 9, 567–573. [Google Scholar] [CrossRef]
- Wen, L.; Jin, Y.; Li, L.; Sun, S.; Cheng, S.; Zhang, S.; Zhang, Y.; Svenningsson, P. Exercise Prevents Raphe Nucleus Mitochondrial Overactivity in a Rat Depression Model. Physiol. Behav. 2014, 132, 57–65. [Google Scholar] [CrossRef]
- Nathan, C.; Cunningham-Bussel, A. Beyond Oxidative Stress: An Immunologist’s Guide to Reactive Oxygen Species. Nat. Rev. Immunol. 2013, 13, 349–361. [Google Scholar] [CrossRef]
- Smith, R.A.J.; Hartley, R.C.; Cochemé, H.M.; Murphy, M.P. Mitochondrial Pharmacology. Trends Pharmacol. Sci. 2012, 33, 341–352. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Czarny, P.; Kwiatkowski, D.; Kacperska, D.; Kawczyńska, D.; Talarowska, M.; Orzechowska, A.; Bielecka-Kowalska, A.; Szemraj, J.; Gałecki, P.; Śliwiński, T. Elevated Level of DNA Damage and Impaired Repair of Oxidative DNA Damage in Patients with Recurrent Depressive Disorder. Med. Sci. Monit. 2015, 21, 412–418. [Google Scholar] [CrossRef]
- Ullah, R.; Khan, M.; Shah, S.A.; Saeed, K.; Kim, M.O. Natural Antioxidant Anthocyanins—A Hidden Therapeutic Candidate in Metabolic Disorders with Major Focus in Neurodegeneration. Nutrients 2019, 11, 1195. [Google Scholar] [CrossRef]
- Palta, P.; Samuel, L.J.; Miller, E.R.; Szanton, S.L. Depression and Oxidative Stress: Results from a Meta-Analysis of Observational Studies. Psychosom. Med. 2014, 76, 12–19. [Google Scholar] [CrossRef]
- Black, C.N.; Bot, M.; Scheffer, P.G.; Cuijpers, P.; Penninx, B.W.J.H. Is Depression Associated with Increased Oxidative Stress? A Systematic Review and Meta-Analysis. Psychoneuroendocrinology 2015, 51, 164–175. [Google Scholar] [CrossRef]
- Ben-Shachar, D.; Karry, R. Neuroanatomical Pattern of Mitochondrial Complex I Pathology Varies between Schizophrenia, Bipolar Disorder and Major Depression. PLoS ONE 2008, 3, e3676. [Google Scholar] [CrossRef]
- Talarowska, M.; Orzechowska, A.; Szemraj, J.; Su, K.-P.; Maes, M.; Gałecki, P. Manganese Superoxide Dismutase Gene Expression and Cognitive Functions in Recurrent Depressive Disorder. Neuropsychobiology 2014, 70, 23–28. [Google Scholar] [CrossRef]
- Anderson, G. Linking the Biological Underpinnings of Depression: Role of Mitochondria Interactions with Melatonin, Inflammation, Sirtuins, Tryptophan Catabolites, DNA Repair and Oxidative and Nitrosative Stress, with Consequences for Classification and Cognition. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 80, 255–266. [Google Scholar] [CrossRef]
- Madrigal, J.L.; Olivenza, R.; Moro, M.A.; Lizasoain, I.; Lorenzo, P.; Rodrigo, J.; Leza, J.C. Glutathione Depletion, Lipid Peroxidation and Mitochondrial Dysfunction Are Induced by Chronic Stress in Rat Brain. Neuropsychopharmacology 2001, 24, 420–429. [Google Scholar] [CrossRef]
- Almeida, R.F.D.; Ganzella, M.; Machado, D.G.; Loureiro, S.O.; Leffa, D.; Quincozes-Santos, A.; Pettenuzzo, L.F.; Duarte, M.M.M.F.; Duarte, T.; Souza, D.O. Olfactory Bulbectomy in Mice Triggers Transient and Long-Lasting Behavioral Impairments and Biochemical Hippocampal Disturbances. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 76, 1–11. [Google Scholar] [CrossRef]
- Holzmann, I.; da Silva, L.M.; Corrêa da Silva, J.A.; Steimbach, V.M.B.; de Souza, M.M. Antidepressant-like Effect of Quercetin in Bulbectomized Mice and Involvement of the Antioxidant Defenses, and the Glutamatergic and Oxidonitrergic Pathways. Pharmacol. Biochem. Behav. 2015, 136, 55–63. [Google Scholar] [CrossRef]
- Filiou, M.D.; Zhang, Y.; Teplytska, L.; Reckow, S.; Gormanns, P.; Maccarrone, G.; Frank, E.; Kessler, M.S.; Hambsch, B.; Nussbaumer, M.; et al. Proteomics and Metabolomics Analysis of a Trait Anxiety Mouse Model Reveals Divergent Mitochondrial Pathways. Biol. Psychiatry 2011, 70, 1074–1082. [Google Scholar] [CrossRef]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA Damage: Mechanisms, Mutation, and Disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as Sensors and Regulators of Calcium Signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Sagua, R.; Parra, V.; López-Crisosto, C.; Díaz, P.; Quest, A.F.G.; Lavandero, S. Calcium Transport and Signaling in Mitochondria. In Comprehensive Physiology; John Wiley & Sons, Ltd: New York, NY, USA, 2017; pp. 623–634. ISBN 978-0-470-65071-4. [Google Scholar]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A Mutual Interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef]
- Giorgi, C.; Marchi, S.; Pinton, P. The Machineries, Regulation and Cellular Functions of Mitochondrial Calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef]
- Belosludtsev, K.N.; Dubinin, M.V.; Belosludtseva, N.V.; Mironova, G.D. Mitochondrial Ca2+ Transport: Mechanisms, Molecular Structures, and Role in Cells. Biochem. Mosc. 2019, 84, 593–607. [Google Scholar] [CrossRef]
- Vollmayr, B.; Bachteler, D.; Vengeliene, V.; Gass, P.; Spanagel, R.; Henn, F. Rats with Congenital Learned Helplessness Respond Less to Sucrose but Show No Deficits in Activity or Learning. Behav. Brain Res. 2004, 150, 217–221. [Google Scholar] [CrossRef]
- Rygula, R.; Abumaria, N.; Flügge, G.; Fuchs, E.; Rüther, E.; Havemann-Reinecke, U. Anhedonia and Motivational Deficits in Rats: Impact of Chronic Social Stress. Behav. Brain Res. 2005, 162, 127–134. [Google Scholar] [CrossRef]
- Kabir, Z.D.; Lee, A.S.; Burgdorf, C.E.; Fischer, D.K.; Rajadhyaksha, A.M.; Mok, E.; Rizzo, B.; Rice, R.C.; Singh, K.; Ota, K.T.; et al. Cacna1c in the Prefrontal Cortex Regulates Depression-Related Behaviors via REDD1. Neuropsychopharmacology 2017, 42, 2032–2042. [Google Scholar] [CrossRef]
- Ebert, D.H.; Greenberg, M.E. Activity-Dependent Neuronal Signalling and Autism Spectrum Disorder. Nature 2013, 493, 327–337. [Google Scholar] [CrossRef]
- Bhat, S.; Dao, D.T.; Terrillion, C.E.; Arad, M.; Smith, R.J.; Soldatov, N.M.; Gould, T.D. CACNA1C (Cav1.2) in the Pathophysiology of Psychiatric Disease. Prog. Neurobiol. 2012, 99, 1–14. [Google Scholar] [CrossRef]
- Kabir, Z.D.; Lee, A.S.; Rajadhyaksha, A.M. L-Type Ca2+ Channels in Mood, Cognition and Addiction: Integrating Human and Rodent Studies with a Focus on Behavioural Endophenotypes. J. Physiol. 2016, 594, 5823–5837. [Google Scholar] [CrossRef]
- Dao, D.T.; Mahon, P.B.; Cai, X.; Kovacsics, C.E.; Blackwell, R.A.; Arad, M.; Shi, J.; Zandi, P.P.; O’Donnell, P.; Knowles, J.A.; et al. Mood Disorder Susceptibility Gene CACNA1C Modifies Mood-Related Behaviors in Mice and Interacts with Sex to Influence Behavior in Mice and Diagnosis in Humans. Biol. Psychiatry 2010, 68, 801–810. [Google Scholar] [CrossRef]
- Cross-Disorder Group of the Psychiatric Genomics Consortium Identification of Risk Loci with Shared Effects on Five Major Psychiatric Disorders: A Genome-Wide Analysis. Lancet Lond. Engl. 2013, 381, 1371–1379. [CrossRef]
- Dedic, N.; Pöhlmann, M.L.; Richter, J.S.; Mehta, D.; Czamara, D.; Metzger, M.W.; Dine, J.; Bedenk, B.T.; Hartmann, J.; Wagner, K.V.; et al. Cross-Disorder Risk Gene CACNA1C Differentially Modulates Susceptibility to Psychiatric Disorders during Development and Adulthood. Mol. Psychiatry 2018, 23, 533–543. [Google Scholar] [CrossRef]
- Michels, S.; Ganjam, G.K.; Martins, H.; Schratt, G.M.; Wöhr, M.; Schwarting, R.K.W.; Culmsee, C. Downregulation of the Psychiatric Susceptibility Gene Cacna1c Promotes Mitochondrial Resilience to Oxidative Stress in Neuronal Cells. Cell Death Discov. 2018, 4. [Google Scholar] [CrossRef]
- Pariante, C.M. Why Are Depressed Patients Inflamed? A Reflection on 20 Years of Research on Depression, Glucocorticoid Resistance and Inflammation. Eur. Neuropsychopharmacol. 2017, 27, 554–559. [Google Scholar] [CrossRef]
- Fries, G.R.; Saldana, V.A.; Finnstein, J.; Rein, T. Molecular Pathways of Major Depressive Disorder Converge on the Synapse. Mol. Psychiatry 2022, 1–14. [Google Scholar] [CrossRef]
- Steptoe, A.; Hamer, M.; Chida, Y. The Effects of Acute Psychological Stress on Circulating Inflammatory Factors in Humans: A Review and Meta-Analysis. Brain. Behav. Immun. 2007, 21, 901–912. [Google Scholar] [CrossRef]
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctôt, K.L. A Meta-Analysis of Cytokines in Major Depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef]
- Miller, A.H.; Maletic, V.; Raison, C.L. Inflammation and Its Discontents: The Role of Cytokines in the Pathophysiology of Major Depression. Biol. Psychiatry 2009, 65, 732–741. [Google Scholar] [CrossRef]
- Leonard, B.; Maes, M. Mechanistic Explanations How Cell-Mediated Immune Activation, Inflammation and Oxidative and Nitrosative Stress Pathways and Their Sequels and Concomitants Play a Role in the Pathophysiology of Unipolar Depression. Neurosci. Biobehav. Rev. 2012, 36, 764–785. [Google Scholar] [CrossRef]
- Song, C.; Halbreich, U.; Han, C.; Leonard, B.E.; Luo, H. Imbalance between Pro- and Anti-Inflammatory Cytokines, and between Th1 and Th2 Cytokines in Depressed Patients: The Effect of Electroacupuncture or Fluoxetine Treatment. Pharmacopsychiatry 2009, 42, 182–188. [Google Scholar] [CrossRef]
- Shizuya, K.; Komori, T.; Fujiwara, R.; Miyahara, S.; Ohmori, M.; Nomura, J. The Influence of Restraint Stress on the Expression of MRNAs for IL-6 and the IL-6 Receptor in the Hypothalamus and Midbrain of the Rat. Life Sci. 1997, 61, PL135–PL140. [Google Scholar] [CrossRef]
- Shintani, F.; Nakaki, T.; Kanba, S.; Sato, K.; Yagi, G.; Shiozawa, M.; Aiso, S.; Kato, R.; Asai, M. Involvement of Interleukin-1 in Immobilization Stress-Induced Increase in Plasma Adrenocorticotropic Hormone and in Release of Hypothalamic Monoamines in the Rat. J. Neurosci. Off. J. Soc. Neurosci. 1995, 15, 1961–1970. [Google Scholar] [CrossRef]
- Kaster, M.P.; Gadotti, V.M.; Calixto, J.B.; Santos, A.R.S.; Rodrigues, A.L.S. Depressive-like Behavior Induced by Tumor Necrosis Factor-α in Mice. Neuropharmacology 2012, 62, 419–426. [Google Scholar] [CrossRef]
- Valerio, A.; Cardile, A.; Cozzi, V.; Bracale, R.; Tedesco, L.; Pisconti, A.; Palomba, L.; Cantoni, O.; Clementi, E.; Moncada, S.; et al. TNF-Alpha Downregulates ENOS Expression and Mitochondrial Biogenesis in Fat and Muscle of Obese Rodents. J. Clin. Investig. 2006, 116, 2791–2798. [Google Scholar] [CrossRef]
- Najjar, S.; Pearlman, D.M.; Alper, K.; Najjar, A.; Devinsky, O. Neuroinflammation and Psychiatric Illness. J. Neuroinflamm. 2013, 10, 816. [Google Scholar] [CrossRef]
- Maes, M.; Ruckoanich, P.; Chang, Y.S.; Mahanonda, N.; Berk, M. Multiple Aberrations in Shared Inflammatory and Oxidative & Nitrosative Stress (IO&NS) Pathways Explain the Co-Association of Depression and Cardiovascular Disorder (CVD), and the Increased Risk for CVD and Due Mortality in Depressed Patients. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 769–783. [Google Scholar] [CrossRef]
- Samavati, L.; Lee, I.; Mathes, I.; Lottspeich, F.; Hüttemann, M. Tumor Necrosis Factor α Inhibits Oxidative Phosphorylation through Tyrosine Phosphorylation at Subunit I of Cytochrome c Oxidase. J. Biol. Chem. 2008, 283, 21134–21144. [Google Scholar] [CrossRef] [PubMed]
- Adzic, M.; Djordjevic, J.; Mitic, M.; Brkic, Z.; Lukic, I.; Radojcic, M. The Contribution of Hypothalamic Neuroendocrine, Neuroplastic and Neuroinflammatory Processes to Lipopolysaccharide-Induced Depressive-like Behaviour in Female and Male Rats: Involvement of Glucocorticoid Receptor and C/EBP-β. Behav. Brain Res. 2015, 291, 130–139. [Google Scholar] [CrossRef]
- Chen, W.-J.; Du, J.-K.; Hu, X.; Yu, Q.; Li, D.-X.; Wang, C.-N.; Zhu, X.-Y.; Liu, Y.-J. Protective Effects of Resveratrol on Mitochondrial Function in the Hippocampus Improves Inflammation-Induced Depressive-like Behavior. Physiol. Behav. 2017, 182, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Brkic, Z.; Zivanovic, A.; Adzic, M. Sex-Specific Effects of Lipopolysaccharide on Hippocampal Mitochondrial Processes in Neuroinflammatory Model of Depression. Neuroscience 2020, 451, 174–183. [Google Scholar] [CrossRef]
- Haj-Mirzaian, A.; Amiri, S.; Amini-Khoei, H.; Hosseini, M.-J.; Haj-Mirzaian, A.; Momeny, M.; Rahimi-Balaei, M.; Dehpour, A.R. Anxiety- and Depressive-Like Behaviors Are Associated with Altered Hippocampal Energy and Inflammatory Status in a Mouse Model of Crohn’s Disease. Neuroscience 2017, 366, 124–137. [Google Scholar] [CrossRef] [PubMed]

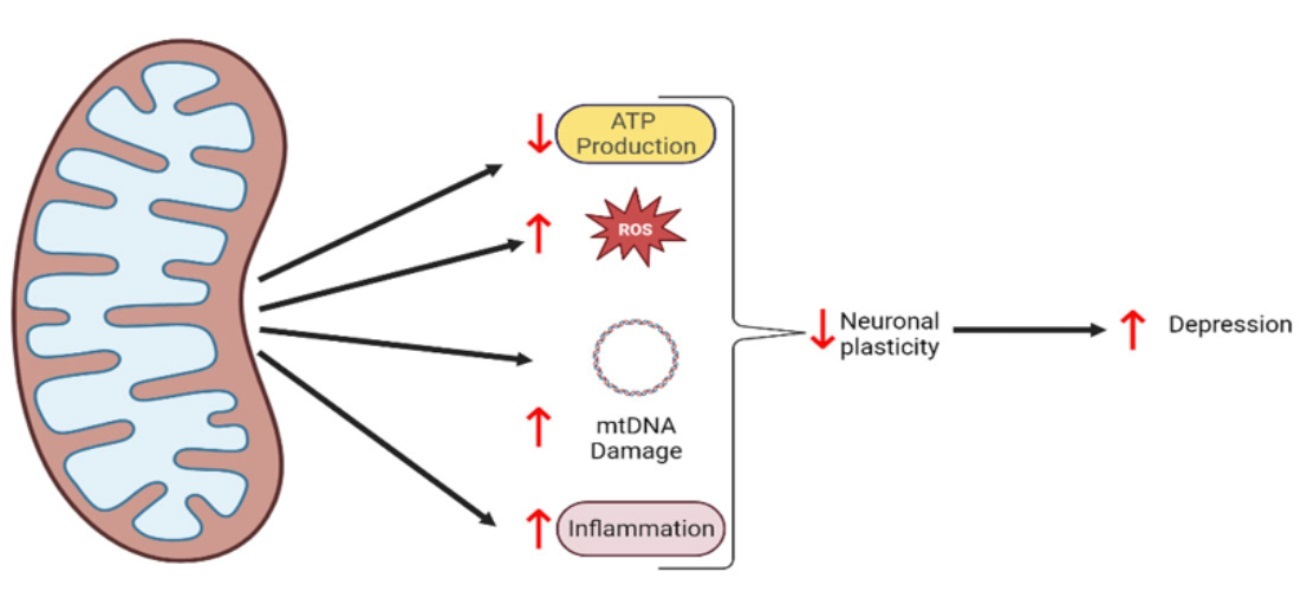{kind=link}
| Depression Protocol | Depression-like Behavior | Mitochondrial Alteration | Ref. |
|---|---|---|---|
| Prenatal stress | Not analyzed | Different expression of TOM70 and ATP5F1C | [91] |
| Maternal separation | Increased immobilization time in the forced swim test | Upregulation of GLUD1, ATP5F1A and ATP5PD, and downregulation of IDH3A | [92,93] |
| Reduced appetite, anhedonia | Downregulation of fumarate hydratase and IDH; upregulation of citrate synthase | [94,95] | |
| Not analyzed | Downregulation of ATP5F1B, complex III subunit I, PDHE1-B | [96] | |
| Chronic mild stress | Anhedonia | Modified expression of Hsp60 | [97] |
| Anhedonia | Upregulation of ATP5F1C, IDH3B, complex I 75 kDa subunit | [98] | |
| Anhedonia | Upregulation of NDUFB7, COX5A, COX5B | [99] | |
| Anhedonia | Downregulation of MDH | [100] | |
| Anhedonia | Upregulation of COXVa, downregulation of IDH3A and ACO2 | [101] | |
| Anhedonia, reduced weight and decreased locomotion | Downregulation of OGDH, NDUFS3 | [102] | |
| Anhedonia, no weight gain | Inhibition of Complex I, III and IV activities in cerebral cortex and cerebellum but not in hippocampus, prefrontal cortex and striatum | [103] | |
| Anhedonia, reduced body weight, increased immobility in the tail suspension test | Reduced O2 consumption, mitochondrial depolarization in hippocampus, cortex and hypothalamus | [104] | |
| Unpredictable chronic mild stress | Anhedonia, increased immobilization time in the forced swim test | Upregulation of NDUFA4, COXVIb-1, complex III subunit 1, MDH; downregulation of SDHA and SOD2 | [105] |
| Anhedonia, increased immobilization time in the forced swim test | Downregulation of MFN1, MFN2 and DOD2; reduced activity of complex I and IV, mitochondrial depolarization | [106] | |
| Social defeat stress | Reduced locomotion in the open field test | Upregulation of ACO1 and GDH; downregulation of MDH and GRP75 | [107] |
| Learned helplessness | Reduced active avoidance | Downregulation of ATP5F1A, fumarate hydratase, HSP70, PDHE1-B, EF-Tu, GDH, MDH and aconitase | [108,109] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, M.; Baussan, Y.; Hebert-Chatelain, E. Connecting Dots between Mitochondrial Dysfunction and Depression. Biomolecules 2023, 13, 695. https://doi.org/10.3390/biom13040695
Khan M, Baussan Y, Hebert-Chatelain E. Connecting Dots between Mitochondrial Dysfunction and Depression. Biomolecules. 2023; 13(4):695. https://doi.org/10.3390/biom13040695
Chicago/Turabian StyleKhan, Mehtab, Yann Baussan, and Etienne Hebert-Chatelain. 2023. "Connecting Dots between Mitochondrial Dysfunction and Depression" Biomolecules 13, no. 4: 695. https://doi.org/10.3390/biom13040695
APA StyleKhan, M., Baussan, Y., & Hebert-Chatelain, E. (2023). Connecting Dots between Mitochondrial Dysfunction and Depression. Biomolecules, 13(4), 695. https://doi.org/10.3390/biom13040695