

SPIN-AI: A Deep Learning Model That Identifies Spatially Predictive Genes


 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Packages
2.2. Data Pre-Processing
2.3. Cellular Subpopulation and Cluster Analysis
2.4. SPIN-AI Training Procedure
2.5. Model Validation
2.6. Identification of Spatially Predictive Genes (SPGs)
2.7. Enrichment Analyses
3. Results
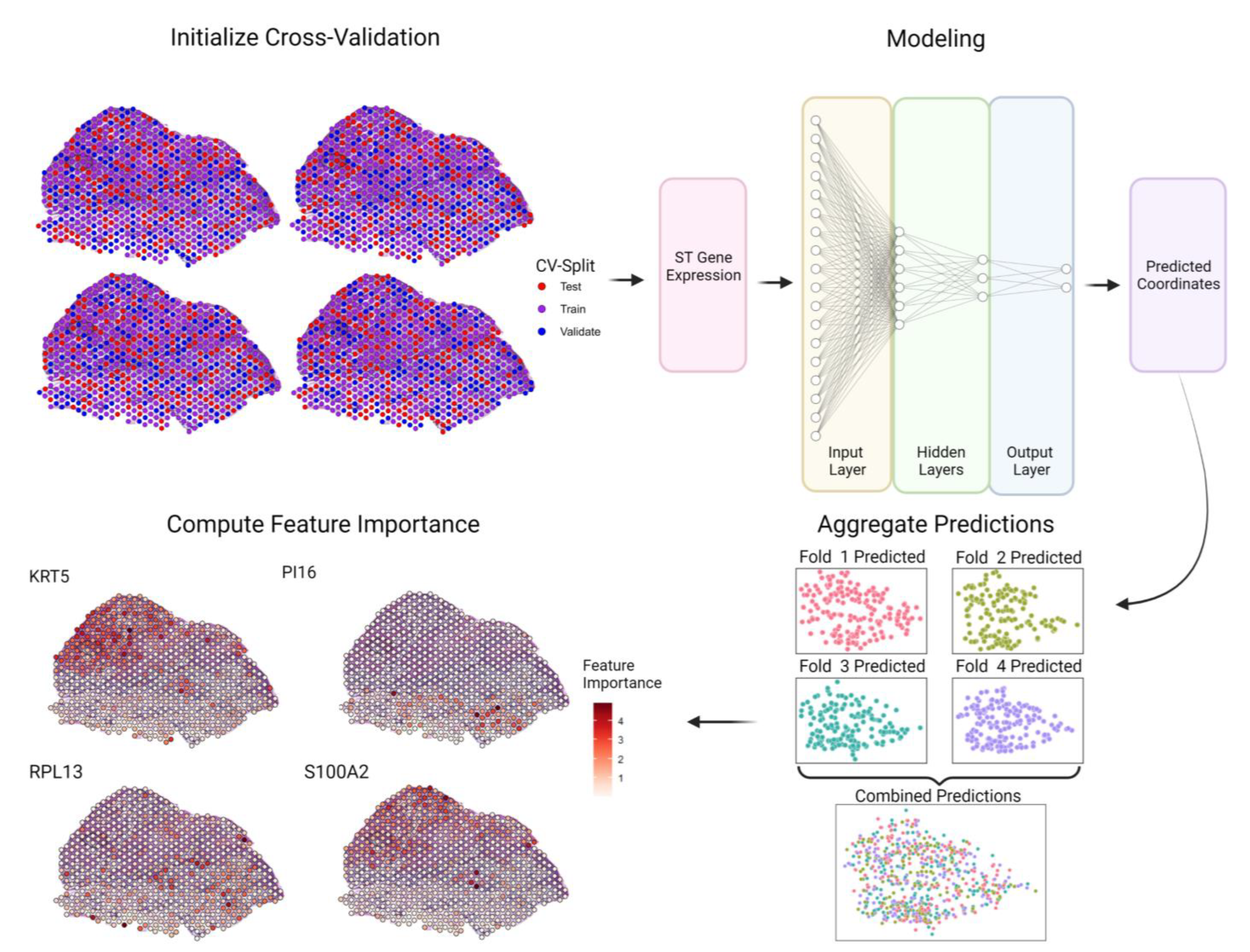
3.1. Design of the Spatially Informed Artificial Intelligence (SPIN-AI) Platform
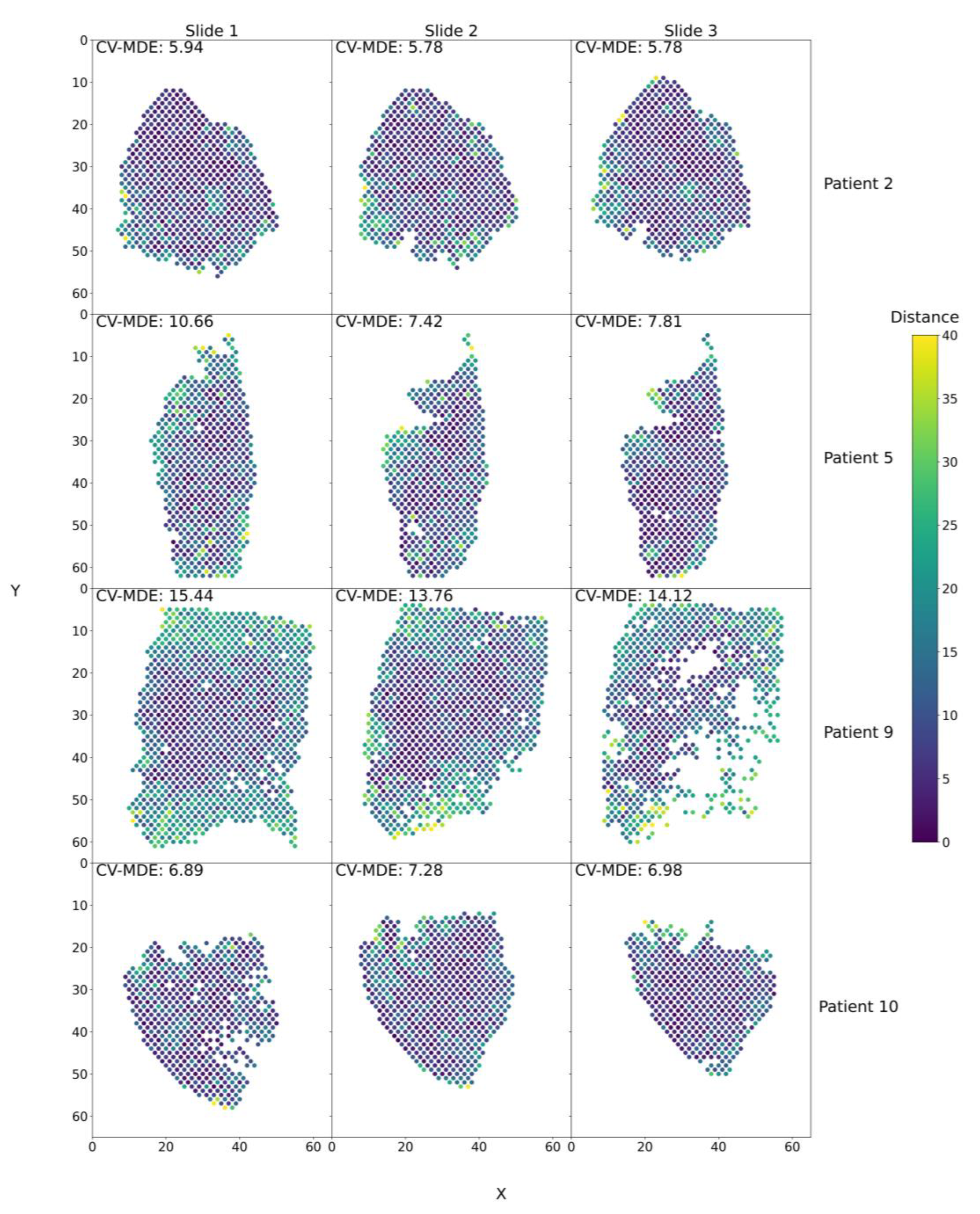
3.2. Trained SPIN-AI Models Can Predict Spatial Organization of Spots on Slide Per Patient
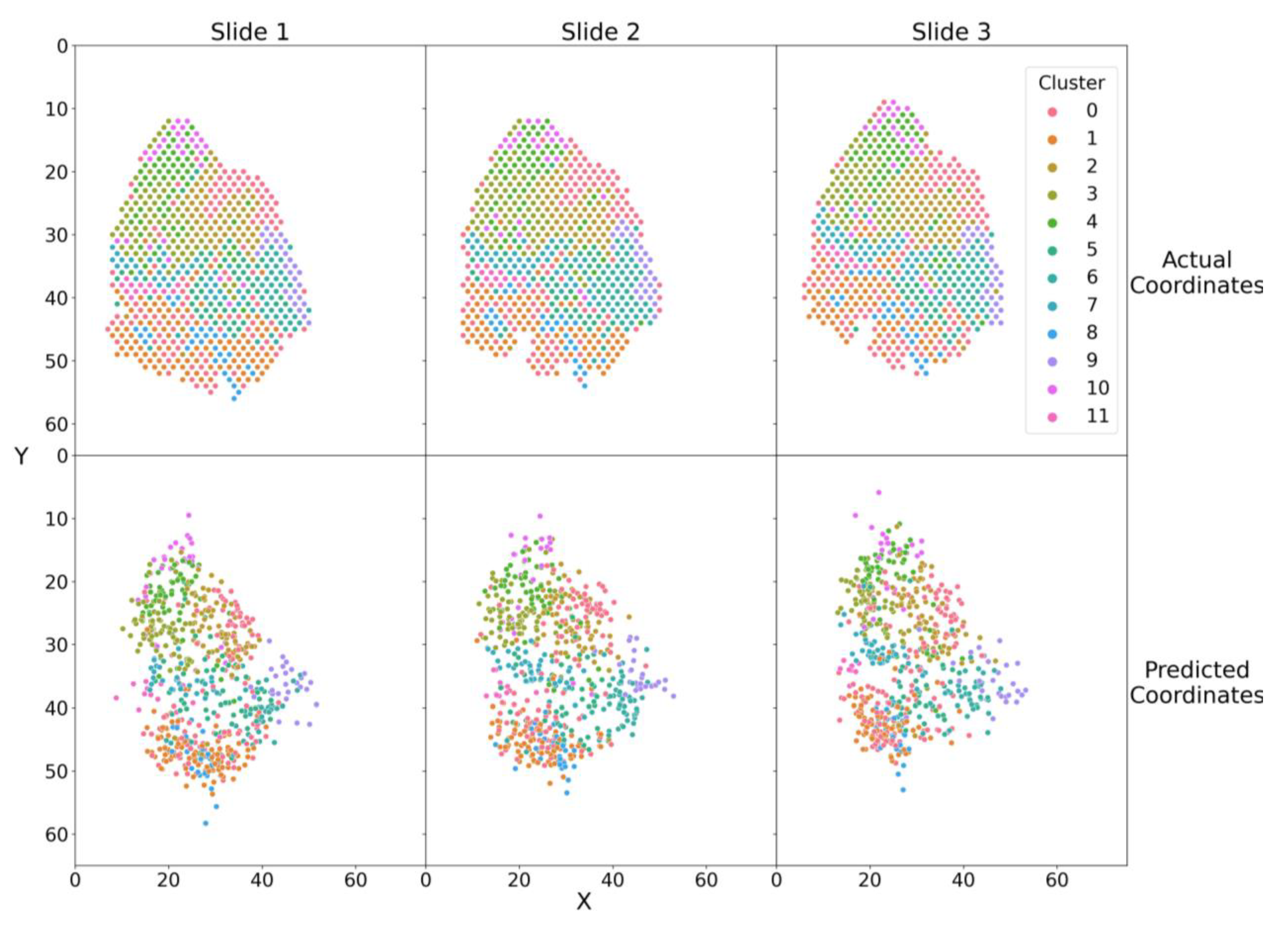
3.3. SPIN-AI Recovers the Spatial Distribution of Gene Expression Clusters
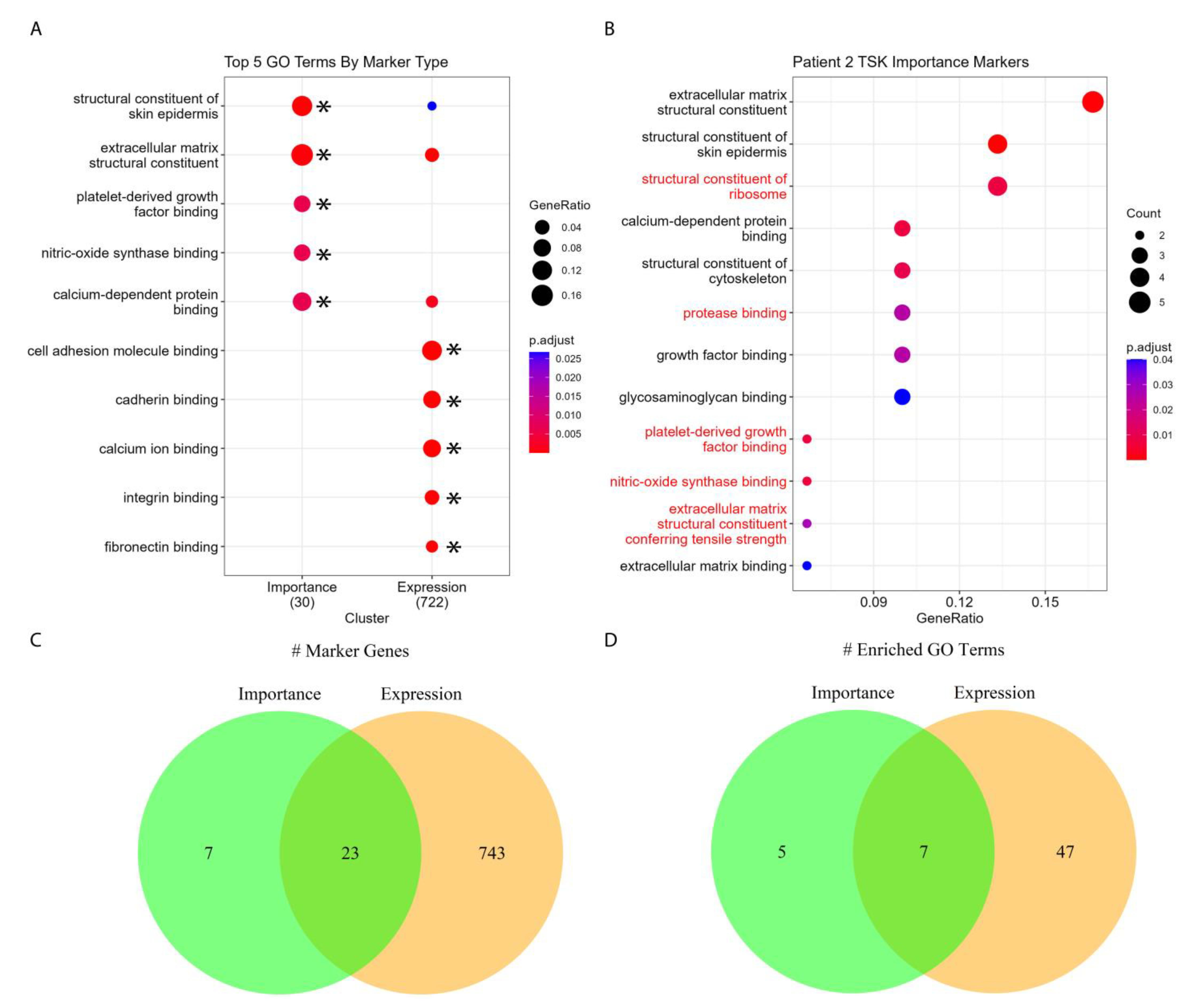
3.4. SPIN-AI Identifies SPGs
3.5. SPGs Recapitulate the Biology of Squamous Carcinoma and Its Cellular Microenvironment
3.6. SPGs Discover Unique Biology
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hwang, B.; Lee, J.H.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef]
- duVerle, D.A.; Yotsukura, S.; Nomura, S.; Aburatani, H.; Tsuda, K. CellTree: An R/bioconductor package to infer the hierarchical structure of cell populations from single-cell RNA-seq data. BMC Bioinform. 2016, 17, 363. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.; Lennon, N.J.; Livak, K.J.; Mikkelsen, T.S.; Rinn, J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014, 32, 381–386. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Stubbington, M.J.T.; Rozenblatt-Rosen, O.; Regev, A.; Teichmann, S.A. Single-cell transcriptomics to explore the immune system in health and disease. Science 2017, 358, 58–63. [Google Scholar] [CrossRef]
- Medaglia, C.; Giladi, A.; Stoler-Barak, L.; De Giovanni, M.; Salame, T.M.; Biram, A.; David, E.; Li, H.; Iannacone, M.; Shulman, Z.; et al. Spatial reconstruction of immune niches by combining photoactivatable reporters and scRNA-seq. Science 2017, 358, 1622–1626. [Google Scholar] [CrossRef]
- Boisset, J.C.; Vivie, J.; Grun, D.; Muraro, M.J.; Lyubimova, A.; van Oudenaarden, A. Mapping the physical network of cellular interactions. Nat. Methods 2018, 15, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.H.; Boettiger, A.N.; Moffitt, J.R.; Wang, S.; Zhuang, X. RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 2015, 348, aaa6090. [Google Scholar] [CrossRef]
- Stahl, P.L.; Salmen, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef]
- Rodriques, S.G.; Stickels, R.R.; Goeva, A.; Martin, C.A.; Murray, E.; Vanderburg, C.R.; Welch, J.; Chen, L.M.; Chen, F.; Macosko, E.Z. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science 2019, 363, 1463–1467. [Google Scholar] [CrossRef] [PubMed]
- Burgess, D.J. Spatial transcriptomics coming of age. Nat. Rev. Genet. 2019, 20, 317. [Google Scholar] [CrossRef] [PubMed]
- Asp, M.; Bergenstrahle, J.; Lundeberg, J. Spatially Resolved Transcriptomes-Next Generation Tools for Tissue Exploration. Bioessays 2020, 42, e1900221. [Google Scholar] [CrossRef]
- Ortiz, C.; Navarro, J.F.; Jurek, A.; Martin, A.; Lundeberg, J.; Meletis, K. Molecular atlas of the adult mouse brain. Sci. Adv. 2020, 6, eabb3446. [Google Scholar] [CrossRef] [PubMed]
- Asp, M.; Giacomello, S.; Larsson, L.; Wu, C.; Furth, D.; Qian, X.; Wardell, E.; Custodio, J.; Reimegard, J.; Salmen, F.; et al. A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart. Cell 2019, 179, 1647–1660 e19. [Google Scholar] [CrossRef]
- Arnol, D.; Schapiro, D.; Bodenmiller, B.; Saez-Rodriguez, J.; Stegle, O. Modeling Cell-Cell Interactions from Spatial Molecular Data with Spatial Variance Component Analysis. Cell Rep. 2019, 29, 202–211 e6. [Google Scholar] [CrossRef]
- Moriel, N.; Senel, E.; Friedman, N.; Rajewsky, N.; Karaiskos, N.; Nitzan, M. NovoSpaRc: Flexible spatial reconstruction of single-cell gene expression with optimal transport. Nat. Protoc. 2021, 16, 4177–4200. [Google Scholar] [CrossRef]
- Edsgard, D.; Johnsson, P.; Sandberg, R. Identification of spatial expression trends in single-cell gene expression data. Nat. Methods 2018, 15, 339–342. [Google Scholar] [CrossRef]
- Svensson, V.; Teichmann, S.A.; Stegle, O. SpatialDE: Identification of spatially variable genes. Nat. Methods 2018, 15, 343–346. [Google Scholar] [CrossRef]
- Sun, S.; Zhu, J.; Zhou, X. Statistical analysis of spatial expression patterns for spatially resolved transcriptomic studies. Nat. Methods 2020, 17, 193–200. [Google Scholar] [CrossRef]
- Hao, M.; Hua, K.; Zhang, X. SOMDE: A scalable method for identifying spatially variable genes with self-organizing map. Bioinformatics 2021, 37, 4392–4398. [Google Scholar] [CrossRef] [PubMed]
- Ji, A.L.; Rubin, A.J.; Thrane, K.; Jiang, S.; Reynolds, D.L.; Meyers, R.M.; Guo, M.G.; George, B.M.; Mollbrink, A.; Bergenstrahle, J.; et al. Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. Cell 2020, 182, 497–514.e22. [Google Scholar] [CrossRef]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Bergenstrahle, J.; Larsson, L.; Lundeberg, J. Seamless integration of image and molecular analysis for spatial transcriptomics workflows. BMC Genom. 2020, 21, 482. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Delving Deep Into Rectifiers: Surpassing Human-Level Performance on ImageNet Classification. arXiv 2015, arXiv:1502.01852v1. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Shrikumar, A.; Greenside, P.; Kundaje, A. Learning Important Features through Propagating Activation Differences. In Proceedings of the Machine Learning Research—International Conference on Machine Learning, Sydney, Australia, 6–11 August 2017; pp. 3145–3153. [Google Scholar]
- Greener, J.G.; Kandathil, S.M.; Moffat, L.; Jones, D.T. A guide to machine learning for biologists. Nat. Rev. Mol. Cell Biol. 2022, 23, 40–55. [Google Scholar] [CrossRef]
- van Dorst, E.B.; van Muijen, G.N.; Litvinov, S.V.; Fleuren, G.J. The limited difference between keratin patterns of squamous cell carcinomas and adenocarcinomas is explicable by both cell lineage and state of differentiation of tumour cells. J. Clin. Pathol. 1998, 51, 679–684. [Google Scholar] [CrossRef]
- Ali, A.A.; Al-Jandan, B.A.; Suresh, C.S. The importance of ctokeratins in the early detection of oral squamous cell carcinoma. J. Oral Maxillofac. Pathol. 2018, 22, 441. [Google Scholar] [CrossRef]
- Ratushny, V.; Gober, M.D.; Hick, R.; Ridky, T.W.; Seykora, J.T. From keratinocyte to cancer: The pathogenesis and modeling of cutaneous squamous cell carcinoma. J. Clin. Investig. 2012, 122, 464–472. [Google Scholar] [CrossRef]
- Mito, I.; Takahashi, H.; Kawabata-Iwakawa, R.; Ida, S.; Tada, H.; Chikamatsu, K. Comprehensive analysis of immune cell enrichment in the tumor microenvironment of head and neck squamous cell carcinoma. Sci. Rep. 2021, 11, 16134. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, N.; Mizuno-Kamiya, M. The Role of Immune Modulatory Cytokines in the Tumor Microenvironments of Head and Neck Squamous Cell Carcinomas. Cancers 2022, 14, 2884. [Google Scholar] [CrossRef] [PubMed]
- Choudhari, S.K.; Chaudhary, M.; Bagde, S.; Gadbail, A.R.; Joshi, V. Nitric oxide and cancer: A review. World J. Surg. Oncol. 2013, 11, 118. [Google Scholar] [CrossRef] [PubMed]
- Sangle, V.A.; Chaware, S.J.; Kulkarni, M.A.; Ingle, Y.C.; Singh, P.; Pooja, V.K. Elevated tissue nitric oxide in oral squamous cell carcinoma. J. Oral Maxillofac. Pathol. 2018, 22, 35–39. [Google Scholar] [CrossRef]
- Pelletier, J.; Thomas, G.; Volarevic, S. Ribosome biogenesis in cancer: New players and therapeutic avenues. Nat. Rev. Cancer 2018, 18, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Pecoraro, A.; Pagano, M.; Russo, G.; Russo, A. Ribosome Biogenesis and Cancer: Overview on Ribosomal Proteins. Int. J. Mol. Sci. 2021, 22, 5496. [Google Scholar] [CrossRef] [PubMed]
- Emmott, E.; Jovanovic, M.; Slavov, N. Ribosome Stoichiometry: From Form to Function. Trends Biochem. Sci. 2019, 44, 95–109. [Google Scholar] [CrossRef]
- Fusco, C.M.; Desch, K.; Dorrbaum, A.R.; Wang, M.; Staab, A.; Chan, I.C.W.; Vail, E.; Villeri, V.; Langer, J.D.; Schuman, E.M. Neuronal ribosomes exhibit dynamic and context-dependent exchange of ribosomal proteins. Nat. Commun. 2021, 12, 6127. [Google Scholar] [CrossRef] [PubMed]
- Parks, M.M.; Kurylo, C.M.; Dass, R.A.; Bojmar, L.; Lyden, D.; Vincent, C.T.; Blanchard, S.C. Variant ribosomal RNA alleles are conserved and exhibit tissue-specific expression. Sci. Adv. 2018, 4, eaao0665. [Google Scholar] [CrossRef]
- Slavov, N.; Semrau, S.; Airoldi, E.; Budnik, B.; van Oudenaarden, A. Differential Stoichiometry among Core Ribosomal Proteins. Cell Rep. 2015, 13, 865–873. [Google Scholar] [CrossRef]
- Xue, S.; Barna, M. Specialized ribosomes: A new frontier in gene regulation and organismal biology. Nat. Rev. Mol. Cell Biol. 2012, 13, 355–369. [Google Scholar] [CrossRef]
- Giesen, C.; Wang, H.A.; Schapiro, D.; Zivanovic, N.; Jacobs, A.; Hattendorf, B.; Schuffler, P.J.; Grolimund, D.; Buhmann, J.M.; Brandt, S.; et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 2014, 11, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Ornatsky, O.I.; Siddiqui, I.; Loboda, A.; Baranov, V.I.; Hedley, D.W. Imaging Mass Cytometry. Cytom. A 2017, 91, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Angelo, M.; Bendall, S.C.; Finck, R.; Hale, M.B.; Hitzman, C.; Borowsky, A.D.; Levenson, R.M.; Lowe, J.B.; Liu, S.D.; Zhao, S.; et al. Multiplexed ion beam imaging of human breast tumors. Nat. Med. 2014, 20, 436–442. [Google Scholar] [CrossRef]
- Almet, A.A.; Cang, Z.; Jin, S.; Nie, Q. The landscape of cell-cell communication through single-cell transcriptomics. Curr. Opin. Syst. Biol. 2021, 26, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Longo, S.K.; Guo, M.G.; Ji, A.L.; Khavari, P.A. Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics. Nat. Rev. Genet. 2021, 22, 627–644. [Google Scholar] [CrossRef] [PubMed]
- Abdelaal, T.; Mourragui, S.; Mahfouz, A.; Reinders, M.J.T. SpaGE: Spatial Gene Enhancement using scRNA-seq. Nucleic Acids Res. 2020, 48, e107. [Google Scholar] [CrossRef]
- Welch, J.D.; Kozareva, V.; Ferreira, A.; Vanderburg, C.; Martin, C.; Macosko, E.Z. Single-Cell Multi-omic Integration Compares and Contrasts Features of Brain Cell Identity. Cell 2019, 177, 1873–1887.e1817. [Google Scholar] [CrossRef]
- Li, K.; Yan, C.; Li, C.; Chen, L.; Zhao, J.; Zhang, Z.; Bao, S.; Sun, J.; Zhou, M. Computational elucidation of spatial gene expression variation from spatially resolved transcriptomics data. Mol. Ther. Nucleic Acids 2022, 27, 404–411. [Google Scholar] [CrossRef]
- Ren, X.; Zhong, G.; Zhang, Q.; Zhang, L.; Sun, Y.; Zhang, Z. Reconstruction of cell spatial organization from single-cell RNA sequencing data based on ligand-receptor mediated self-assembly. Cell Res. 2020, 30, 763–778. [Google Scholar] [CrossRef]
- Lander, A.D. How cells know where they are. Science 2013, 339, 923–927. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Stanojevic, S.; Garmire, L.X. Emerging artificial intelligence applications in Spatial Transcriptomics analysis. Comput. Struct. Biotechnol. J. 2022, 20, 2895–2908. [Google Scholar] [CrossRef]
- Zhang, K.; Feng, W.; Wang, P. Identification of spatially variable genes with graph cuts. Nat. Commun. 2022, 13, 5488. [Google Scholar] [CrossRef]
- Dong, K.; Zhang, S. Deciphering spatial domains from spatially resolved transcriptomics with an adaptive graph attention auto-encoder. Nat. Commun. 2022, 13, 1739. [Google Scholar] [CrossRef]
- Hu, J.; Li, X.; Coleman, K.; Schroeder, A.; Ma, N.; Irwin, D.J.; Lee, E.B.; Shinohara, R.T.; Li, M. SpaGCN: Integrating gene expression, spatial location and histology to identify spatial domains and spatially variable genes by graph convolutional network. Nat. Methods 2021, 18, 1342–1351. [Google Scholar] [CrossRef]
- Xu, Y.; McCord, R.P. CoSTA: Unsupervised convolutional neural network learning for spatial transcriptomics analysis. BMC Bioinform. 2021, 22, 397. [Google Scholar] [CrossRef]
- Yuan, Y.; Bar-Joseph, Z. GCNG: Graph convolutional networks for inferring gene interaction from spatial transcriptomics data. Genome Biol. 2020, 21, 300. [Google Scholar] [CrossRef]
- Tanevski, J.; Flores, R.O.R.; Gabor, A.; Schapiro, D.; Saez-Rodriguez, J. Explainable multiview framework for dissecting spatial relationships from highly multiplexed data. Genome Biol. 2022, 23, 97. [Google Scholar] [CrossRef] [PubMed]
- Bergenstrahle, L.; He, B.; Bergenstrahle, J.; Abalo, X.; Mirzazadeh, R.; Thrane, K.; Ji, A.L.; Andersson, A.; Larsson, L.; Stakenborg, N.; et al. Super-resolved spatial transcriptomics by deep data fusion. Nat. Biotechnol. 2022, 40, 476–479. [Google Scholar] [CrossRef]
- Monjo, T.; Koido, M.; Nagasawa, S.; Suzuki, Y.; Kamatani, Y. Efficient prediction of a spatial transcriptomics profile better characterizes breast cancer tissue sections without costly experimentation. Sci. Rep. 2022, 12, 4133. [Google Scholar] [CrossRef] [PubMed]
- Maseda, F.; Cang, Z.; Nie, Q. DEEPsc: A Deep Learning-Based Map Connecting Single-Cell Transcriptomics and Spatial Imaging Data. Front. Genet. 2021, 12, 636743. [Google Scholar] [CrossRef] [PubMed]
- Shengquan, C.; Boheng, Z.; Xiaoyang, C.; Xuegong, Z.; Rui, J. stPlus: A reference-based method for the accurate enhancement of spatial transcriptomics. Bioinformatics 2021, 37 (Suppl. 1), i299–i307. [Google Scholar] [CrossRef] [PubMed]
- El Khoury, W.; Nasr, Z. Deregulation of ribosomal proteins in human cancers. Biosci. Rep. 2021, 41, BSR20211577. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Liu, Y.; Yu, X.Y.; Pan, X.; Zhang, Y.; Tu, J.; Song, Y.H.; Li, Y. Ribosome biogenesis in disease: New players and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 15. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meng-Lin, K.; Ung, C.-Y.; Zhang, C.; Weiskittel, T.M.; Wisniewski, P.; Zhang, Z.; Tan, S.-H.; Yeo, K.-S.; Zhu, S.; Correia, C.; et al. SPIN-AI: A Deep Learning Model That Identifies Spatially Predictive Genes. Biomolecules 2023, 13, 895. https://doi.org/10.3390/biom13060895
Meng-Lin K, Ung C-Y, Zhang C, Weiskittel TM, Wisniewski P, Zhang Z, Tan S-H, Yeo K-S, Zhu S, Correia C, et al. SPIN-AI: A Deep Learning Model That Identifies Spatially Predictive Genes. Biomolecules. 2023; 13(6):895. https://doi.org/10.3390/biom13060895
Chicago/Turabian StyleMeng-Lin, Kevin, Choong-Yong Ung, Cheng Zhang, Taylor M. Weiskittel, Philip Wisniewski, Zhuofei Zhang, Shyang-Hong Tan, Kok-Siong Yeo, Shizhen Zhu, Cristina Correia, and et al. 2023. "SPIN-AI: A Deep Learning Model That Identifies Spatially Predictive Genes" Biomolecules 13, no. 6: 895. https://doi.org/10.3390/biom13060895
APA StyleMeng-Lin, K., Ung, C.-Y., Zhang, C., Weiskittel, T. M., Wisniewski, P., Zhang, Z., Tan, S.-H., Yeo, K.-S., Zhu, S., Correia, C., & Li, H. (2023). SPIN-AI: A Deep Learning Model That Identifies Spatially Predictive Genes. Biomolecules, 13(6), 895. https://doi.org/10.3390/biom13060895